
Optical Genome Mapping in Routine Human Genetic Diagnostics—Its Advantages and Limitations

Abstract
1. Introduction
2. Materials and Methods
2.1. Samples
2.2. DNA Extraction
2.3. DNA Labeling and Further Processing for OGM
2.4. Assembly of OGM-Data and Quality Metrics
2.5. Routine Diagnostic Methods
3. Results
3.1. Samples
3.2. OGM Quality Metrics
3.3. Confirmation of Numerical and Structural Chromosomal Aberrations
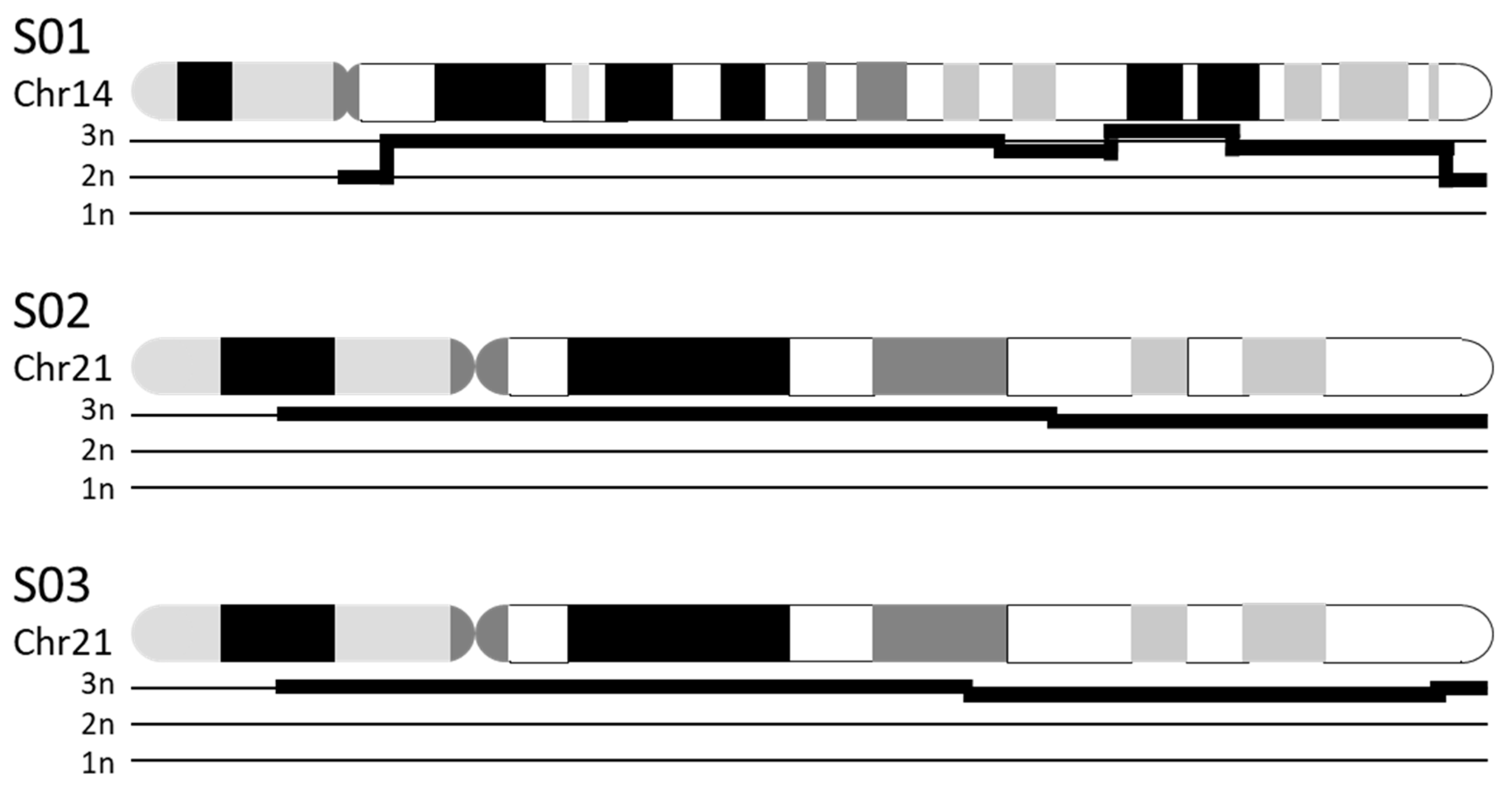
3.3.1. Chromosomal Numerical Aberrations
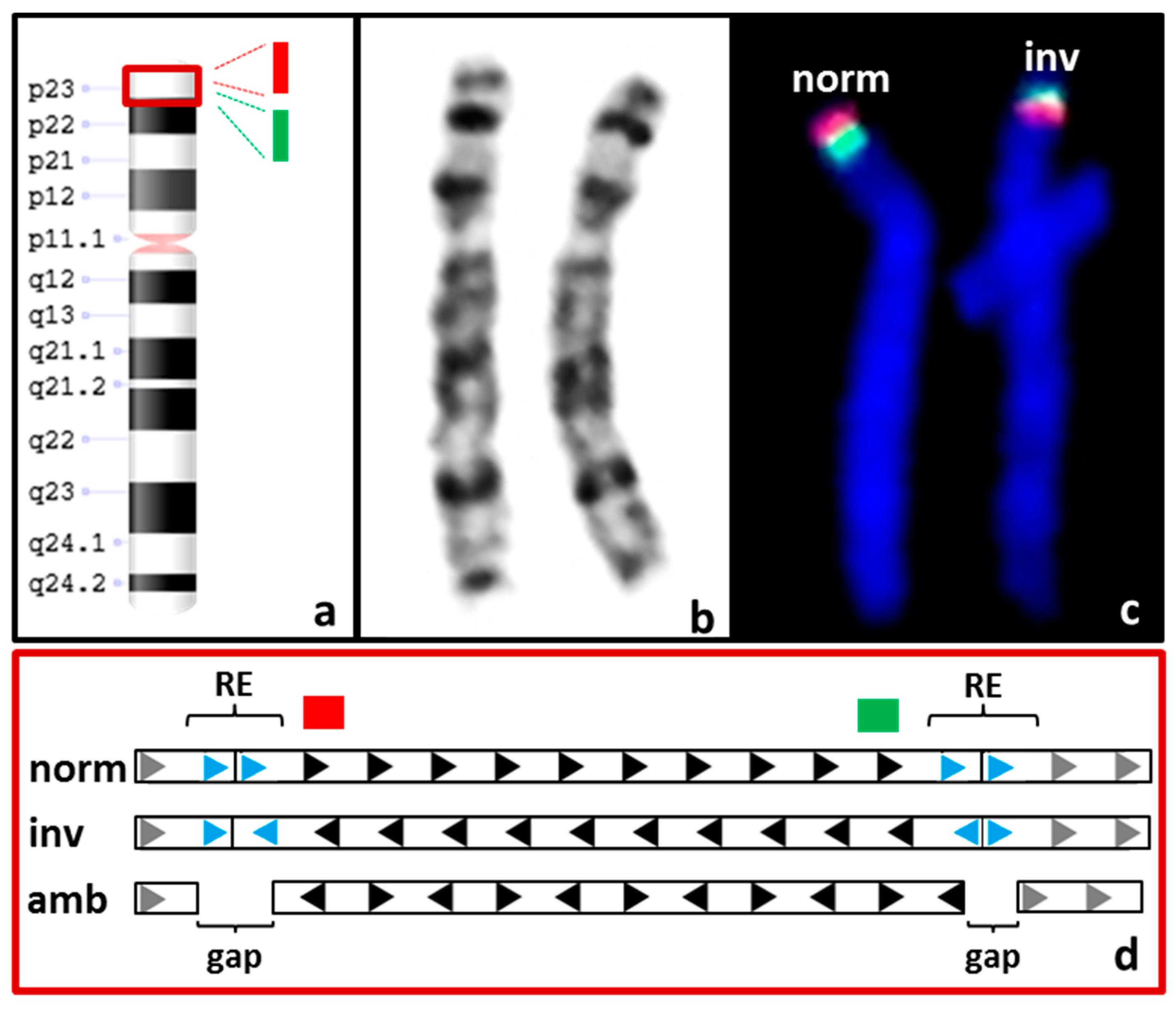
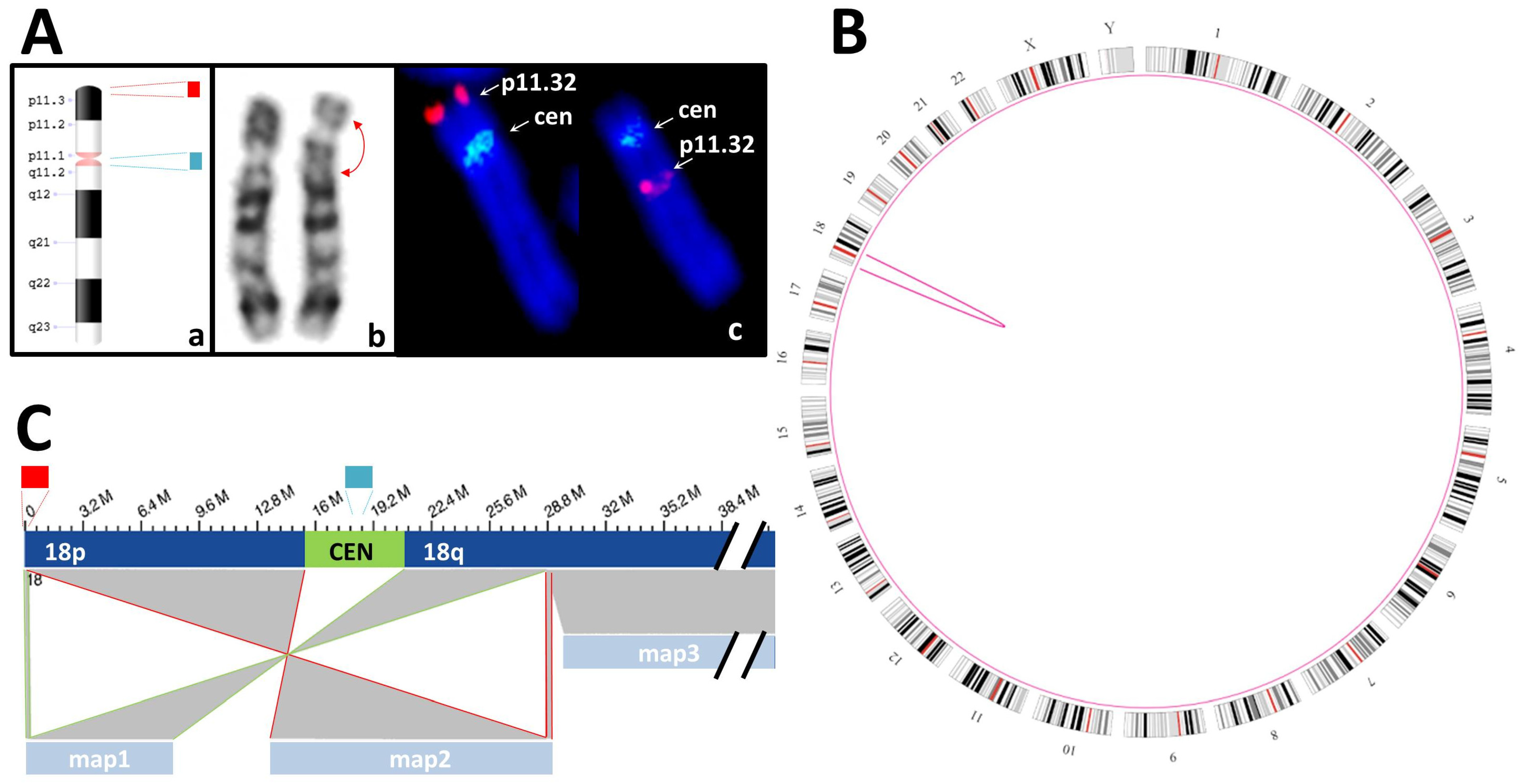
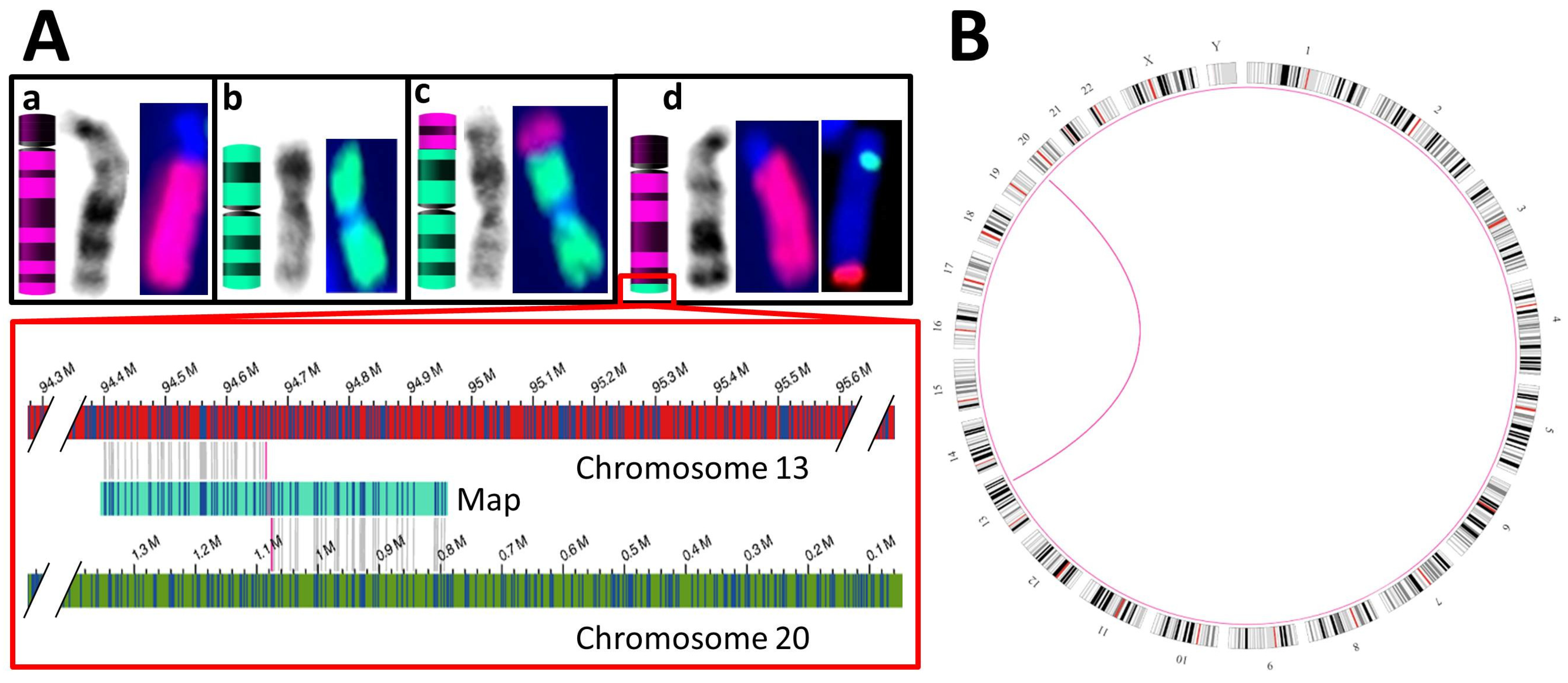
3.3.2. Balanced SVs
3.3.3. Large Unbalanced SVs
3.3.4. Small Unbalanced SVs
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Tyson, C.; Harvard, C.; Locker, R.; Friedman, J.M.; Langlois, S.; Lewis, M.E.; Van Allen, M.; Somerville, M.; Arbour, L.; Clarke, L.; et al. Submicroscopic deletions and duplications in individuals with intellectual disability detected by array-CGH. Am. J. Med. Genet A 2005, 139, 173–185. [Google Scholar] [CrossRef] [PubMed]
- Ishihara, T.; Kikuchi, Y.; Sandberg, A.A. Chromosomes of twenty cancer effusions: Correlation of karyotypic, clinical, and pathologic aspects. J. Natl. Cancer Inst. 1963, 30, 1303–1361. [Google Scholar]
- Shaffer, L.G.; Bejjani, B.A. A cytogeneticist’s perspective on genomic microarrays. Hum. Reprod. Update 2004, 10, 221–226. [Google Scholar] [CrossRef] [PubMed]
- Bednar, M. DNA microarray technology and application. Med. Sci. Monit. 2000, 6, 796–800. [Google Scholar] [PubMed]
- Kosugi, S.; Momozawa, Y.; Liu, X.; Terao, C.; Kubo, M.; Kamatani, Y. Comprehensive evaluation of structural variation detection algorithms for whole genome sequencing. Genome Biol. 2019, 20, 117. [Google Scholar] [CrossRef]
- Dong, Z.; Wang, H.; Chen, H.; Jiang, H.; Yuan, J.; Yang, Z.; Wang, W.J.; Xu, F.; Guo, X.; Cao, Y.; et al. Identification of balanced chromosomal rearrangements previously unknown among participants in the 1000 Genomes Project: Implications for interpretation of structural variation in genomes and the future of clinical cytogenetics. Genet. Med. 2018, 20, 697–707. [Google Scholar] [CrossRef]
- Dennis, M.Y.; Eichler, E.E. Human adaptation and evolution by segmental duplication. Curr. Opin. Genet. Dev. 2016, 41, 44–52. [Google Scholar] [CrossRef] [PubMed]
- Rudd, M.K.; Keene, J.; Bunke, B.; Kaminsky, E.B.; Adam, M.P.; Mulle, J.G.; Ledbetter, D.H.; Martin, C.L. Segmental duplications mediate novel, clinically relevant chromosome rearrangements. Hum. Mol. Genet. 2009, 18, 2957–2962. [Google Scholar] [CrossRef]
- Ardui, S.; Ameur, A.; Vermeesch, J.R.; Hestand, M.S. Single molecule real-time (SMRT) sequencing comes of age: Applications and utilities for medical diagnostics. Nucleic Acids Res. 2018, 46, 2159–2168. [Google Scholar] [CrossRef] [PubMed]
- Jain, M.; Koren, S.; Miga, K.H.; Quick, J.; Rand, A.C.; Sasani, T.A.; Tyson, J.R.; Beggs, A.D.; Dilthey, A.T.; Fiddes, I.T.; et al. Nanopore sequencing and assembly of a human genome with ultra-long reads. Nat. Biotechnol. 2018, 36, 338–345. [Google Scholar] [CrossRef] [PubMed]
- Bionano Genomics. Bionano Solve Theory of Operation: Structural Variant Calling; Bionano Genomics: San Diego, CA, USA, 2020. [Google Scholar]
- Sahajpal, N.S.; Barseghyan, H.; Kolhe, R.; Hastie, A.; Chaubey, A. Optical Genome Mapping as a Next-Generation Cytogenomic Tool for Detection of Structural and Copy Number Variations for Prenatal Genomic Analyses. Genes 2021, 12, 398. [Google Scholar] [CrossRef]
- Dai, Y.; Li, P.; Wang, Z.; Liang, F.; Yang, F.; Fang, L.; Huang, Y.; Huang, S.; Zhou, J.; Wang, D.; et al. Single-molecule optical mapping enables quantitative measurement of D4Z4 repeats in facioscapulohumeral muscular dystrophy (FSHD). J. Med. Genet. 2020, 57, 109–120. [Google Scholar] [CrossRef]
- Wang, H.; Jia, Z.; Mao, A.; Xu, B.; Wang, S.; Wang, L.; Liu, S.; Zhang, H.; Zhang, X.; Yu, T.; et al. Analysis of balanced reciprocal translocations in patients with subfertility using single-molecule optical mapping. J. Assist. Reprod. Genet. 2020, 37, 509–516. [Google Scholar] [CrossRef] [PubMed]
- Ng, J.; Sams, E.; Baldridge, D.; Kremitzki, M.; Wegner, D.J.; Lindsay, T.; Fulton, R.; Cole, F.S.; Turner, T.N. Precise breakpoint detection in a patient with 9p-syndrome. Mol. Case Stud. 2020, 6, a005348. [Google Scholar] [CrossRef]
- Neveling, K.; Mantere, T.; Vermeulen, S.; Oorsprong, M.; van Beek, R.; Kater-Baats, E.; Pauper, M.; van der Zande, G.; Smeets, D.; Weghuis, D.O.; et al. Next generation cytogenetics: Comprehensive assessment of 48 leukemia genomes by genome imaging. bioRxiv 2020. [CrossRef]
- Mantere, T.; Neveling, K.; Pebrel-Richard, C.; Benoist, M.; van der Zande, G.; Kater-Baats, E.; Baatout, I.; van Beek, R.; Yammine, T.; Oorsprong, M.; et al. Next generation cytogenetics: Genome-imaging enables comprehensive structural variant detection for 100 constitutional chromosomal aberrations in 85 samples. bioRxiv 2020. [CrossRef]
- Bionano Genomics. Bionano Access®: Assembly Report Guidelines; Bionano Genomics: San Diego, CA, USA, 2019. [Google Scholar]
- McGowan-Jordan, J.; Hastings, R.J.; Moore, S. ISCN 2020: An International System for Human Cytogenomic Nomenclature (2020); Karger: Basel, Switzerland, 2020. [Google Scholar]
- Salm, M.P.; Horswell, S.D.; Hutchison, C.E.; Speedy, H.E.; Yang, X.; Liang, L.; Schadt, E.E.; Cookson, W.O.; Wierzbicki, A.S.; Naoumova, R.P.; et al. The origin, global distribution, and functional impact of the human 8p23 inversion polymorphism. Genome Res. 2012, 22, 1144–1153. [Google Scholar] [CrossRef] [PubMed]
- Antonacci, F.; Kidd, J.M.; Marques-Bonet, T.; Ventura, M.; Siswara, P.; Jiang, Z.; Eichler, E.E. Characterization of six human disease-associated inversion polymorphisms. Hum. Mol. Genet. 2009, 18, 2555–2566. [Google Scholar] [CrossRef] [PubMed]
- Giglio, S.; Broman, K.W.; Matsumoto, N.; Calvari, V.; Gimelli, G.; Neumann, T.; Ohashi, H.; Voullaire, L.; Larizza, D.; Giorda, R.; et al. Olfactory receptor-gene clusters, genomic-inversion polymorphisms, and common chromosome rearrangements. Am J. Hum. Genet. 2001, 68, 874–883. [Google Scholar] [CrossRef] [PubMed]
- Sugawara, H.; Harada, N.; Ida, T.; Ishida, T.; Ledbetter, D.H.; Yoshiura, K.; Ohta, T.; Kishino, T.; Niikawa, N.; Matsumoto, N. Complex low-copy repeats associated with a common polymorphic inversion at human chromosome 8p23. Genomics 2003, 82, 238–244. [Google Scholar] [CrossRef]
- Thienpont, B.; de Ravel, T.; Van Esch, H.; Van Schoubroeck, D.; Moerman, P.; Vermeesch, J.R.; Fryns, J.P.; Froyen, G.; Lacoste, C.; Badens, C.; et al. Partial duplications of the ATRX gene cause the ATR-X syndrome. Eur. J. Hum. Genet. 2007, 15, 1094–1097. [Google Scholar] [CrossRef]
- Lee, H.H. Chimeric CYP21P/CYP21 and TNXA/TNXB genes in the RCCX module. Mol. Genet. Metab. 2005, 84, 4–8. [Google Scholar] [CrossRef]
- Sahoo, T.; Cheung, S.W.; Ward, P.; Darilek, S.; Patel, A.; del Gaudio, D.; Kang, S.H.; Lalani, S.R.; Li, J.; McAdoo, S.; et al. Prenatal diagnosis of chromosomal abnormalities using array-based comparative genomic hybridization. Genet. Med. 2006, 8, 719–727. [Google Scholar] [CrossRef] [PubMed]
- Levy, B.; Burnside, R.D. Are all chromosome microarrays the same? What clinicians need to know. Prenat. Diagn. 2019, 39, 157–164. [Google Scholar] [CrossRef] [PubMed]
- Marin, D.; Zimmerman, R.; Tao, X.; Zhan, Y.; Scott, R.T., Jr.; Treff, N.R. Validation of a targeted next generation sequencing-based comprehensive chromosome screening platform for detection of triploidy in human blastocysts. Reprod. Biomed. Online 2018, 36, 388–395. [Google Scholar] [CrossRef]
- Hassold, T.; Hall, H.; Hunt, P. The origin of human aneuploidy: Where we have been, where we are going. Hum. Mol. Genet. 2007, 16, R203–R208. [Google Scholar] [CrossRef] [PubMed]
- McFadden, D.E.; Robinson, W.P. Phenotype of triploid embryos. J. Med. Genet. 2006, 43, 609–612. [Google Scholar] [CrossRef]
- Kriegova, E.; Fillerova, R.; Minarik, J.; Savara, J.; Manakova, J.; Petrackova, A.; Dihel, M.; Balcarkova, J.; Krhovska, P.; Pika, T.; et al. Whole-genome optical mapping of bone-marrow myeloma cells reveals association of extramedullary multiple myeloma with chromosome 1 abnormalities. Sci. Rep. 2021, 11, 14671. [Google Scholar] [CrossRef] [PubMed]
- Cope, H.; Barseghyan, H.; Bhattacharya, S.; Fu, Y.; Hoppman, N.; Marcou, C.; Walley, N.; Rehder, C.; Deak, K.; Alkelai, A.; et al. Detection of a mosaic CDKL5 deletion and inversion by optical genome mapping ends an exhaustive diagnostic odyssey. Mol. Genet. Genom. Med. 2021, 9, e1665. [Google Scholar] [CrossRef] [PubMed]
- Vejerslev, L.O.; Mikkelsen, M. The European collaborative study on mosaicism in chorionic villus sampling: Data from 1986 to 1987. Prenat. Diagn. 1989, 9, 575–588. [Google Scholar] [CrossRef] [PubMed]
- Grati, F.R.; Grimi, B.; Frascoli, G.; Di Meco, A.M.; Liuti, R.; Milani, S.; Trotta, A.; Dulcetti, F.; Grosso, E.; Miozzo, M.; et al. Confirmation of mosaicism and uniparental disomy in amniocytes, after detection of mosaic chromosome abnormalities in chorionic villi. Eur. J. Hum. Genet. 2006, 14, 282–288. [Google Scholar] [CrossRef]
- Taylor, T.H.; Gitlin, S.A.; Patrick, J.L.; Crain, J.L.; Wilson, J.M.; Griffin, D.K. The origin, mechanisms, incidence and clinical consequences of chromosomal mosaicism in humans. Hum. Reprod. Update 2014, 20, 571–581. [Google Scholar] [CrossRef] [PubMed]
- Kong, Y.; Berko, E.R.; Marcketta, A.; Maqbool, S.B.; Simoes-Pires, C.A.; Kronn, D.F.; Ye, K.Q.; Suzuki, M.; Auton, A.; Greally, J.M. Detecting, quantifying, and discriminating the mechanism of mosaic chromosomal aneuploidies using MAD-seq. Genome Res. 2018, 28, 1039–1052. [Google Scholar] [CrossRef] [PubMed]
- Hamerton, J.L.; Canning, N.; Ray, M.; Smith, S. A cytogenetic survey of 14,069 newborn infants. I. Incidence of chromosome abnormalities. Clin. Genet. 1975, 8, 223–243. [Google Scholar] [CrossRef] [PubMed]
- Scriven, P.N.; Flinter, F.A.; Braude, P.R.; Ogilvie, C.M. Robertsonian translocations--reproductive risks and indications for preimplantation genetic diagnosis. Hum. Reprod. 2001, 16, 2267–2273. [Google Scholar] [CrossRef]
- Bangs, C.D.; Donlon, T.A. Metaphase chromosome preparation from cultured peripheral blood cells. Curr. Protoc. Hum. Genet. 2005, 45, 4.1.1–4.1.19. [Google Scholar] [CrossRef]
- Jackson, L.; Gibas, L.M.; Barr, M.A. Preparation of metaphase spreads from chorionic villus samples. Curr. Protoc. Hum. Genet. 2001, 1, 8.3.1–8.3.8. [Google Scholar] [CrossRef]
- Philip, J.; Bryndorf, T.; Christensen, B. Prenatal aneuploidy detection in interphase cells by fluorescence in situ hybridization (FISH). Prenat. Diagn. 1994, 14, 1203–1215. [Google Scholar] [CrossRef] [PubMed]
- Ogilvie, C.M.; Donaghue, C.; Fox, S.P.; Docherty, Z.; Mann, K. Rapid prenatal diagnosis of aneuploidy using quantitative fluorescence-PCR (QF-PCR). J. Histochem. Cytochem. 2005, 53, 285–288. [Google Scholar] [CrossRef] [PubMed]
- Miron, P.M. Preparation, Culture, and Analysis of Amniotic Fluid Samples. Curr. Protoc. Hum. Genet. 2018, 98, e62. [Google Scholar] [CrossRef] [PubMed]
- Redin, C.; Brand, H.; Collins, R.L.; Kammin, T.; Mitchell, E.; Hodge, J.C.; Hanscom, C.; Pillalamarri, V.; Seabra, C.M.; Abbott, M.A.; et al. The genomic landscape of balanced cytogenetic abnormalities associated with human congenital anomalies. Nat. Genet. 2017, 49, 36–45. [Google Scholar] [CrossRef] [PubMed]
- Talkowski, M.E.; Rosenfeld, J.A.; Blumenthal, I.; Pillalamarri, V.; Chiang, C.; Heilbut, A.; Ernst, C.; Hanscom, C.; Rossin, E.; Lindgren, A.M.; et al. Sequencing chromosomal abnormalities reveals neurodevelopmental loci that confer risk across diagnostic boundaries. Cell 2012, 149, 525–537. [Google Scholar] [CrossRef] [PubMed]
- Porat, Y.; Lev, O.; Einhorn, M.; Shani, O.; Vilain, E.; Barseghyan, H.; Bhattacharya, S.; Paz-Yaacov, N. Human genome meeting 2016: Houston, TX, USA. 28 February–2 March 2016. Hum. Genom. 2016, 10, 1–40. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| ID | Results of Routine Methods | Results of OGM |
|---|---|---|
| S01 | K 1: 47,XX,+14 | CNV 2: duplication of chr14:19,922,034–104,122,329 (i.e., majority of chr14) |
| S02 | K: 47,XX,+21 | CNV: duplication of chr21:13,033,053–46,697,230 (i.e., majority of chr21) |
| S03 | K: 47,XY,+21 | CNV: duplication of chr21:12,406,577–45,259,300 (i.e., majority of chr21) |
| S04 | F 3: 46,XX.ish inv(8)(p23.1)(p23.1)(RP11-399J23+)(p23.1)(RP11-589N15+) | CNV + SV 4: not called and not detectable upon manual inspection |
| S05 | K + F: 46,XY,inv(18)(p11.3q12?).ish inv(18)(p11q11)(D18Z1+)(18)(p11.3)(D18S1244+) | SV: intrachromosomal translocations from chr18:191,456 to chr18:28,753,623 and from chr18:192,099 to chr18:28,753,623 |
| S06 | K + F: 46,XX,t(13;20)(q32;p13) & aCGH 5 of a relative: duplication of chr13:94,677,014–114,327,680 and deletion of chr20:80,100–1,076,209 | SV: interchromosomal translocations from chr13:94,664,378 to chr20:1,076,206 and vice versa |
| S07 | aCGH: duplication of chrX:89,800,893–97,156,872 | CNV: duplication of chrX:89,708,306–97,160,715 SV: not called but detectable upon manual inspection |
| S08 | aCGH: duplication of chr14:188,966,428–190,415,619 and chrX:71,670,725–77,748,054 | CNV + SV: duplication of chr4:188,157,784–189,603,002 and chrX:71,651,551–77,757,633 |
| S09 | n/a, father of S08 | unremarkable |
| S10 | n/a, mother of S08 | CNV + SV: duplication of chr4:188,157,784–189,603,002 and chrX:71,651,551–77,757,633 |
| S11 | aCGH: deletion of chrX:352,452–446,323 and duplication of chrX:124,255,330–140,379,126 | deleted region is not covered by OGM maps CNV: duplication of chrX:124,262,540–140,175,735 SV: duplication not called but detectable upon manual inspection |
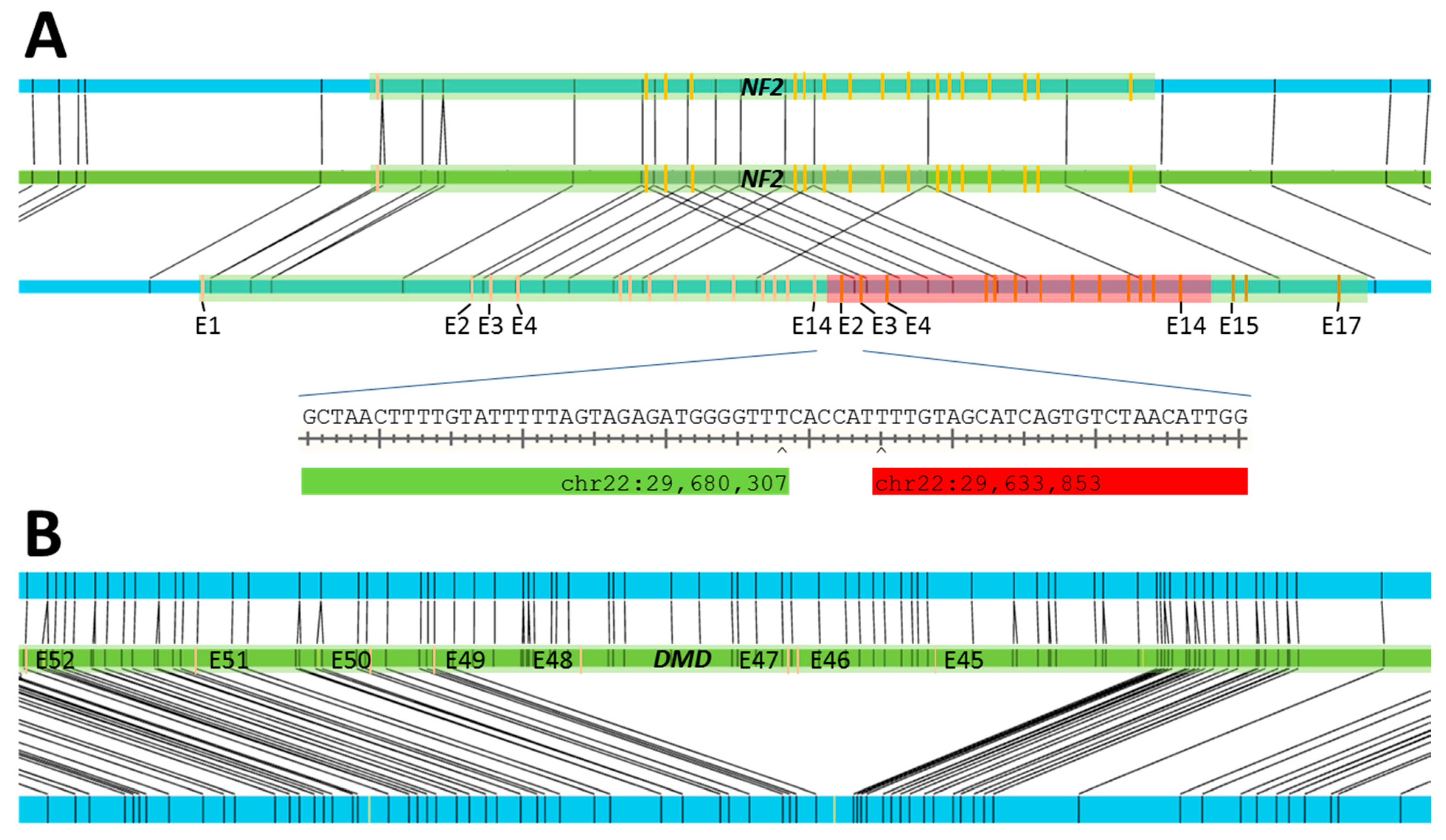
| S12 | MLPA 6: duplication of exons 2–14 of NF2 | SV: 34.6 kbp duplication at chr22:29,636,202–29,670,823 |
| S13 | MLPA: deletion of exons 45–48 of DMD | SV: 174 kbp deletion at chrX:31,841,660–32,025,691 |
| S14 | MLPA: deletion of at least exons 3–7 of CYP21A2 and deletion of at least exons 35–45 of TNXB | SV: 32.8 kbp deletion at chr6:32,012,952–32,045,806, found in 26.3% of samples in database |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dremsek, P.; Schwarz, T.; Weil, B.; Malashka, A.; Laccone, F.; Neesen, J. Optical Genome Mapping in Routine Human Genetic Diagnostics—Its Advantages and Limitations. Genes 2021, 12, 1958. https://doi.org/10.3390/genes12121958
Dremsek P, Schwarz T, Weil B, Malashka A, Laccone F, Neesen J. Optical Genome Mapping in Routine Human Genetic Diagnostics—Its Advantages and Limitations. Genes. 2021; 12(12):1958. https://doi.org/10.3390/genes12121958
Chicago/Turabian StyleDremsek, Paul, Thomas Schwarz, Beatrix Weil, Alina Malashka, Franco Laccone, and Jürgen Neesen. 2021. "Optical Genome Mapping in Routine Human Genetic Diagnostics—Its Advantages and Limitations" Genes 12, no. 12: 1958. https://doi.org/10.3390/genes12121958
APA StyleDremsek, P., Schwarz, T., Weil, B., Malashka, A., Laccone, F., & Neesen, J. (2021). Optical Genome Mapping in Routine Human Genetic Diagnostics—Its Advantages and Limitations. Genes, 12(12), 1958. https://doi.org/10.3390/genes12121958