
A Case of Inherited t(4;10)(q26;q26.2) Chromosomal Translocation Elucidated by Multiple Chromosomal and Molecular Analyses. Case Report and Review of the Literature

 , , , , ,
, , , , ,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Clinical Evaluation
2.2. Cytogenetic Analyses
2.3. MLPA Technique
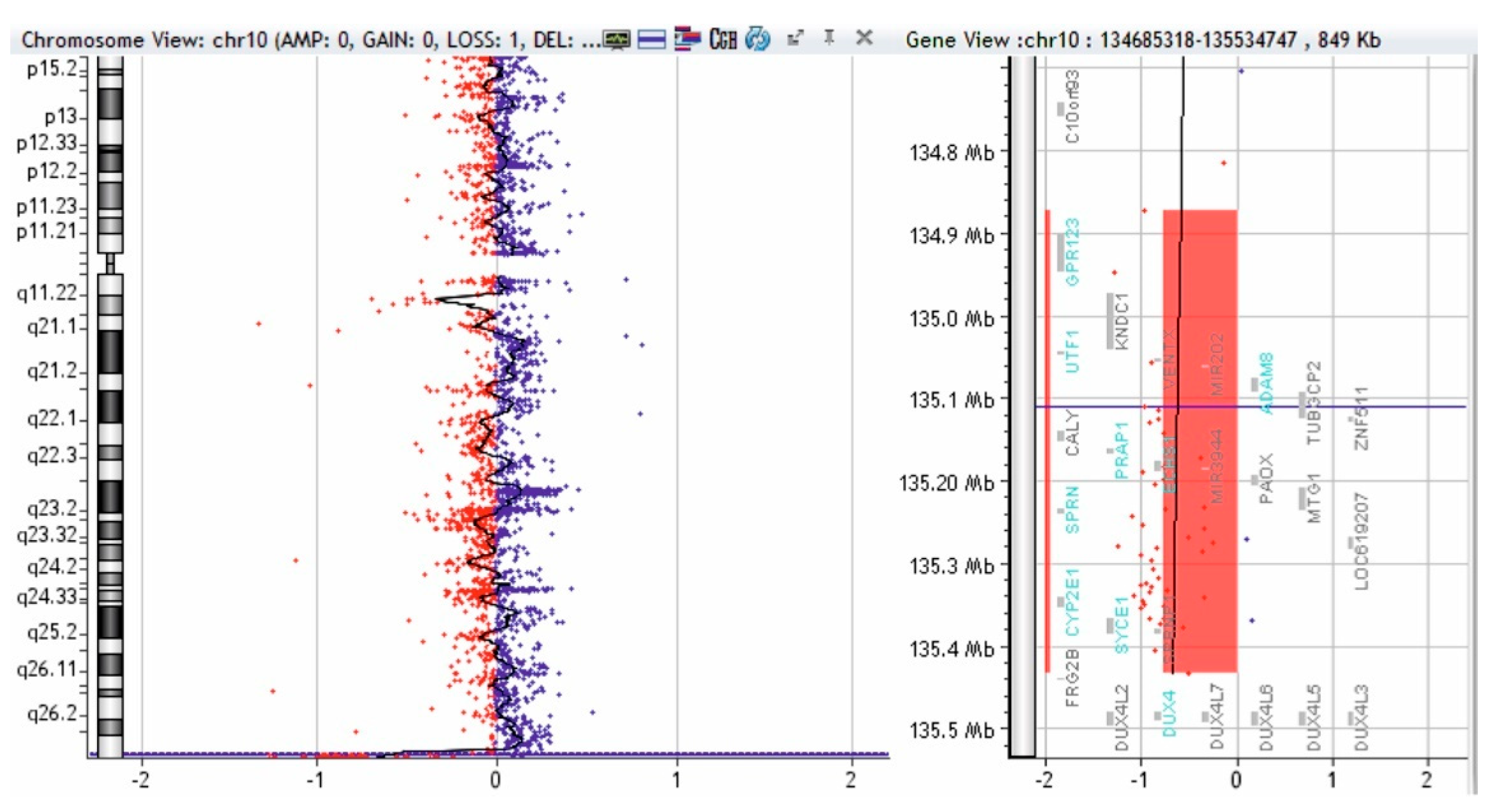
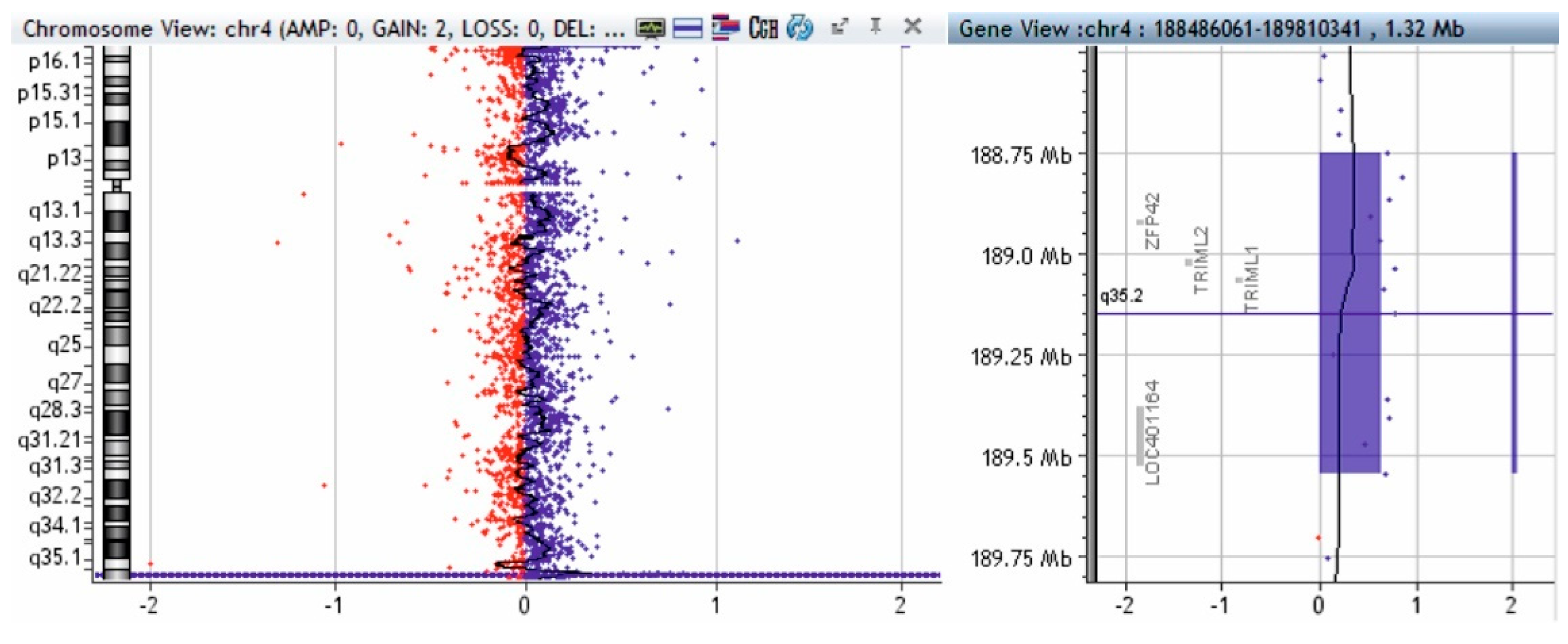
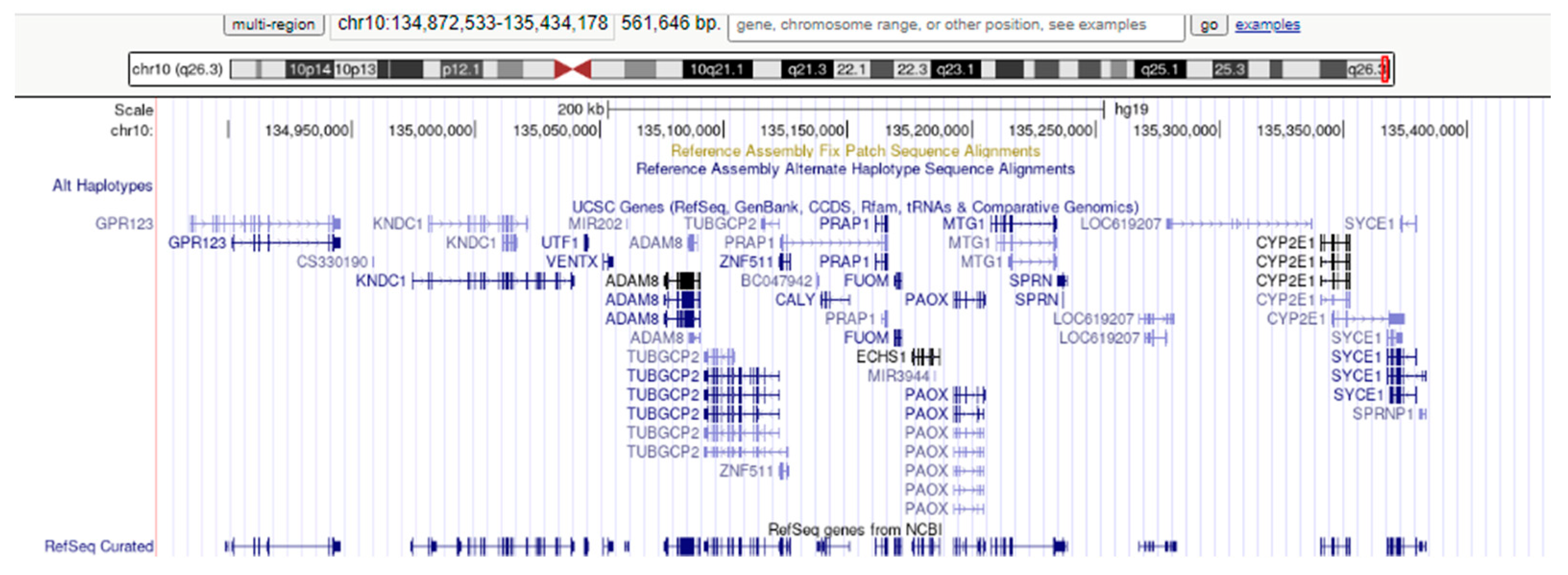
2.4. Array-CGH Technique
3. Results
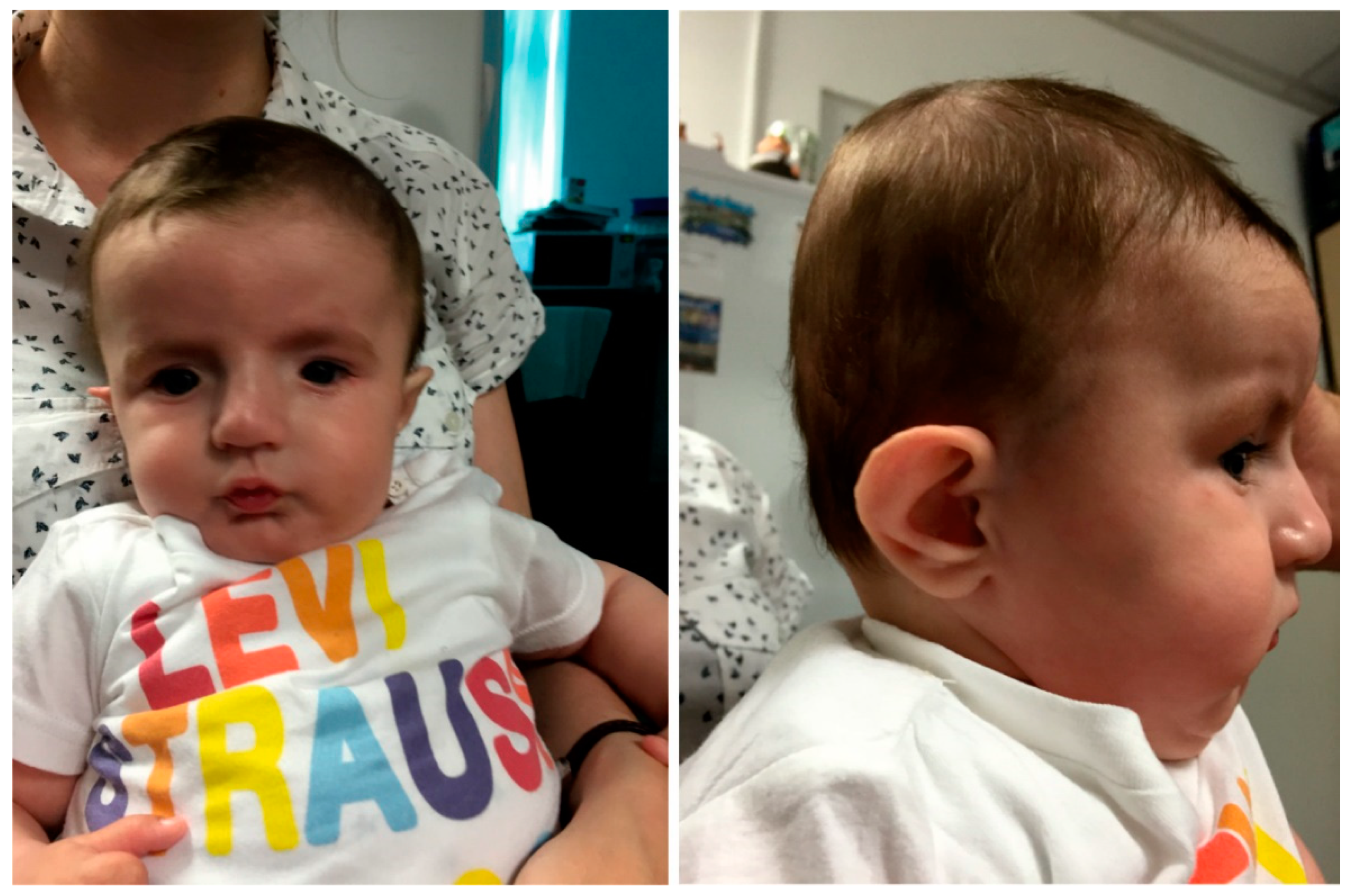
Clinical Presentation
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Conflicts of Interest
References
- World Health Organization. Congenital Anomalies. Available online: https://www.who.int/health-topics/congenital-anomalies#tab=tab (accessed on 12 February 2021).
- Covic, M.; Ştefănescu, D.; Sandovici, I.; Gorduza, E.V. Genetică Medicală, 3rd ed.; Polirom: Iaşi, Romania, 2017. [Google Scholar]
- Shaffer, L.; Theisen, A. Disorders caused by chromosome abnormalities. Appl. Clin. Genet. 2010, 3, 159–174. [Google Scholar] [CrossRef] [Green Version]
- Francke, U. Quinacrine mustard fluorescence of human chromosomes: Characterization of unusual translocations. Am. J. Hum. Genet. 1972, 24, 189–213. [Google Scholar]
- Surana, R.B.; Conen, P.E. Partial trisomy 4 resulting from a 4-18 reciprocal translocation. Ann. Génét. 1972, 15, 191–194. [Google Scholar]
- Schrott, H.G.; Sakaguchi, S.; Francke, U.; Luzzatti, L.; Fialkow, P.J. Translocation, t(4q-;13q+), in three generations resulting in partial trisomy of the long arm of chromosome 4 in the fourth generation. J. Med. Genet. 1974, 11, 201–205. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.-L.; Dai, Y.; Tu, Z.-G.; Li, Q.-Y. ‘Pure’ Partial Trisomy 4q26?q35.2 Resulting from a Familial Unbalanced Translocation t(4;10)(q26;q26.3). Cytogenet. Genome Res. 2009, 127, 67–72. [Google Scholar] [CrossRef]
- Battaglia, A.; Chen, Z.; Brothman, A.; Morelli, S.; Palumbos, J.; Carey, J.; Hudgins, L.; Disteche, C. Karyotype/phenotype correlations in duplication 4q: Evidence for a critical region within 4q27-28 for preaxial defects. Am. J. Med. Genet. Part A 2005, 134A, 334–337. [Google Scholar] [CrossRef]
- Cernakova, I.; Kvasnicova, M.; Lovasova, Z.; Badova, N.; Drabek, J.; Bouchalova, K.; Trojanec, R.; Hajduch, M. A DUPLICATION dup(4)(q28q35.2) DE NOVO IN A NEWBORN. Biomed. Pap. 2006, 150, 113–116. [Google Scholar] [CrossRef] [Green Version]
- Lewandowski, R.C.; Kukolich, M.K.; Sears, J.W.; Mankinen, C.B. Partial deletion 10q. Qual. Life Res. 1978, 42, 339–343. [Google Scholar] [CrossRef]
- Irving, M.; Hanson, H.; Turnpenny, P.; Brewer, C.; Ogilvie, C.M.; Davies, A.; Berg, J. Deletion of the distal long arm of chromosome 10; is there a characteristic phenotype? A report of 15 de novo and familial cases. Am. J. Med. Genet. 2003, 123A, 153–163. [Google Scholar] [CrossRef]
- Scigliano, S.; Grégoire, M.; Schmitt, M.; Jonveaux, P.; LeHeup, B. Terminal deletion of the long arm of chromosome. Clin. Genet. 2004, 65, 294–298. [Google Scholar] [CrossRef] [PubMed]
- Kehrer-Sawatzki, H.; Daumiller, E.; Müller-Navia, J.; Kendziorra, H.; Rossier, E.; Du Bois, G.; Barbi, G. Interstitial deletion del(10)(q25.2q25.3∼26.11)—Case report and review of the literature. Prenat. Diagn. 2005, 25, 954–959. [Google Scholar] [CrossRef]
- Courtens, W.; Wuyts, W.; Rooms, L.; Pera, S.B.; Wauters, J. A subterminal deletion of the long arm of chromosome 10: A clinical report and review. Am. J. Med. Genet. Part A 2006, 140A, 402–409. [Google Scholar] [CrossRef] [PubMed]
- Yatsenko, S.; Kruer, M.; Bader, P.; Corzo, D.; Schuette, J.; Keegan, C.; Nowakowska, B.; Peacock, S.; Cai, W.; Peiffer, D.; et al. Identification of critical regions for clinical features of distal 10q deletion syndrome. Clin. Genet. 2009, 76, 54–62. [Google Scholar] [CrossRef]
- Lin, S.; Zhou, Y.; Fang, Q.; Wu, J.; Zhang, Z.; Ji, Y.; Luo, Y. Chromosome 10q26 deletion syndrome: Two new cases and a review of the literature. Mol. Med. Rep. 2016, 14, 5134–5140. [Google Scholar] [CrossRef] [Green Version]
- Celle, L.; Lee, L.; Rintoul, N.; Savani, R.C.; Long, W.; Mennuti, M.T.; Krantz, I.D. Duplication of chromosome region 4q28.3-qter in monozygotic twins with discordant phenotypes. Am. J. Med. Genet. 2000, 94, 125–140. [Google Scholar] [CrossRef]
- Mattei, M.-G.; Mattei, J.-F.; Bernard, R.; Giraud, F. Partial trisomy 4 resulting from a complex maternal rearrangement of chromosomes 2, 4, and 18 with interstitial translocation. Qual. Life Res. 1979, 51, 55–61. [Google Scholar] [CrossRef]
- Zollino, M.; Zampino, G.; Torrioli, G.; Pomponi, M.G.; Neri, G. Further contribution to the description of phenotypes associated with partial 4q duplication. Am. J. Med. Genet. 1995, 57, 69–73. [Google Scholar] [CrossRef] [PubMed]
- Halal, F.; Vekemans, M.; Chitayat, D. Interstitial tandem direct duplication of the long arm of chromosome 4 (q23–q27) and possible assignment of the structural gene encoding human aspartylglucosaminidase to this segment. Am. J. Med. Genet. 1991, 39, 418–421. [Google Scholar] [CrossRef]
- Jeziorowska, A.; Ciesla, W.; Houck, G.E., Jr.; Yao, X.L.; Harris, M.S.; Truszczak, B.; Skorski, M.; Jakubowski, L.; Jenkins, E.C.; Kaluzewski, B. Cytogenetic and molecular identification of a de novo direct duplication of the long arm of chromosome 4(q21.3→q31.3). Am. J. Med. Genet. 1993, 46, 83–87. [Google Scholar] [CrossRef] [PubMed]
- Fryns, J.P.; Berghe, H.V.D. Partial duplication of the long arm of chromosome. Ann. Génét. 1980, 23, 52–53. [Google Scholar]
- Vogel, W.; Siebers, J.W.; Gunkel, J.; Haas, B.; Knörr-Gärtner, H.; Niethammer, D.G.; Noel, B. Uneinheitlicher Phänotyp bei Partialtrisomie 4q. Qual. Life Res. 1975, 28, 103–112. [Google Scholar] [CrossRef]
- Dutrillaux, B.; Laurent, C.; Forabosco, A.; Noel, B.; Suerinc, E.; Biemont, M.C.; Cotton, J.B. La trisomie 4q partielle. Apropos de trois observations. Ann. Genet. 1975, 18, 21–27. [Google Scholar]
- Taylor, K.M.; Francke, U.; Brown, M.G.; George, D.L.; Kaufhold, M.; Opitz, J.M. Inverted tandem (“mirror”) duplications in human chromosomes: Inv dup 8p, 4q, 22q. Am. J. Med. Genet. 1977, 1, 3–19. [Google Scholar] [CrossRef]
- Goodman, B.K.; Capone, G.T.; Hennessey, J.; Thomas, G.H. Familial tandem duplication of band q31.1 to q32.3 on chromo-some 4 with mild phenotypic effect. Am. J. Med. Genet. 1997, 73, 119–124. [Google Scholar] [CrossRef]
- Muraki, K.; Katano, R.; Hiraki, Y.; Ueda, K.; Fujita, H. A case of an interstitial tandem direct duplication of long arm of chromosome 4: 46, XY, dup (4) (q25q31.3) de novo. Hiroshima J. Med. Sci. 1997, 46, 105–108. [Google Scholar]
- Shashi, V.; Berry, M.N.; Santos, C.; Pettenati, M.J. Partial duplication of 4q12q13 leads to a mild phenotype. Am. J. Med. Genet. 1999, 86, 51–53. [Google Scholar] [CrossRef]
- Navarro, E.G.; Romero, M.M.; Expósito, I.L.; Velasco, C.M.; Llamas, J.G.; Ramón, F.H.; Jimenez, R.D. De novo interstitial tandem dupli-cation of chromosome 4(q21–q28). Am. J. Med. Genet. 1996, 62, 297–299. [Google Scholar] [CrossRef]
- Otsuka, T.; Fujinaka, H.; Imamura, M.; Tanaka, Y.; Hayakawa, H.; Tomizawa, S. Duplication of chromosome 4q: Renal pathology of two siblings. Am. J. Med. Genet. Part A 2005, 134A, 330–333. [Google Scholar] [CrossRef]
- Lundin, C.; Zech, L.; Sjörs, K.; Wadelius, C.; Annerén, G. Trisomy 4q syndrome: Presentation of a new case and review of the literature. Ann. Génét. 2002, 45, 53–57. [Google Scholar] [CrossRef]
- Mikelsaar, R.V.; Lurie, I.W.; EIlus, T. “Pure” partial trisomy 4q25-qter owing to a de novo 4;22 translocation. J. Med. Genet. 1996, 33, 344–345. [Google Scholar] [CrossRef] [Green Version]
- Cui, Y.-X.; Wang, Y.-H.; Hao, L.-J.; Hou, L.; Li, W.; Huang, Y.-F. Partial trisomy 4q: A case report. Chin. Med. J. 2006, 119, 1136–1139. [Google Scholar] [CrossRef]
- Maltby, E.L.; Barnes, I.C.; Bennett, C.P. Duplication involving band 4q32 with minimal clinical effect. Am. J. Med. Genet. 1999, 83, 431. [Google Scholar] [CrossRef]
- Assawamakin, A.; Wattanasirichaigoon, D.; Tocharoentanaphol, C.; Waeteekul, S.; Tansatit, M.; Thongnoppakhun, W.; Limwongse, C. A novel maternally-derived insertional translocation resulting in partial trisomy 4q13.2-q22.1 with complex translocation t(8;20) in a family with intellectual disability. Am. J. Med. Genet. Part A 2012, 158, 901–908. [Google Scholar] [CrossRef]
- Angulo, M.A.; Castro-Magana, M.; Sherman, J.; Collipp, P.J.; Milson, J.; Trunca, C.; Derenoncourt, A.N. Endocrine abnormalities in a patient with partial trisomy 4q. J. Med. Genet. 1984, 21, 303–307. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Watanabe, G.; Kiyoi, Y.; Takeyama, I.; Kawana, S.; Yamamoto, M. Inherited chromosomal translocation in two families (t(4q-;13q+) and t(5q-;13q+)). Tohoku J. Exp. Med. 1977, 121, 179–184. [Google Scholar] [CrossRef] [Green Version]
- Jenkins, E.C.; Curcuru-Giordano, F.M.; Krishna, S.G.; Cantarella, J. De novo occurrence of 46,XX,t(4;13) (q31;q14) in a mentally retarded girl. Ann. de Génétique 1975, 18, 117–120. [Google Scholar]
- Horbinski, C.; Carter, E.M.; Heard, P.L.; Sathanoori, M.; Hu, J.; Vockley, J.; Gunn, S.; Hale, D.E.; Surti, U.; Cody, J.D. Molecular and clinical characterization of a recurrent cryptic unbalanced t(4q;18q) resulting in an 18q deletion and 4q duplication. Am. J. Med. Genet. Part A 2008, 146A, 2898–2904. [Google Scholar] [CrossRef] [Green Version]
- Rinaldi, R.; De Bernardo, C.; Assumma, M.; Grammatico, B.; Buffone, E.; Poscente, M.; Grammatico, P. Cytogenetic and molecular characterization of a de novo 4q24qter duplication and correlation to the associated phenotype. Am. J. Med. Genet. 2003, 118A, 122–126. [Google Scholar] [CrossRef]
- Kadotani, T.; Watanabe, Y.; Kiyuna, T.; Takeuchi, S. A case of partial trisomy 4q resulting from familial (4;9)(q23;p24) translocation. Proc. Jpn. Acad. Ser. B 1981, 57, 374–377. [Google Scholar] [CrossRef] [Green Version]
- Issa, M.; Potter, A.M.; Blank, C.E. Multiple congenital defects associated with trisomy for long arm of No. J. Med. Genet. 1976, 13, 326–329. [Google Scholar] [CrossRef] [Green Version]
- Colla, A. Duplicación parcial del cromosoma 4 asociada con coloboma ocular bilateral. Arch. Argent. Pediatr. 2012, 110, 59–62. [Google Scholar] [CrossRef] [PubMed]
- Baccichetti, C.; Tenconi, R.; Anglani, F.; Zacchello, F. Trisomy 4q32 leads to 4qter due to a maternal 4/21 translocation. J. Med. Genet. 1975, 12, 425–427. [Google Scholar] [CrossRef]
- Patel, V.; Raghuveer, T.; Persons, D.; Myers, S. Trisomy 4q Syndrome: A rare Syndrome Phenotypically Similar to Trisomy. Internet J. Pediatr. Neonatol. 2006, 7, 133–172. [Google Scholar]
- Cervenka, J.; Djavadi, G.R.; Gorlin, R.J. Partial trisomy 4q syndrome: Case report and review. Hum. Genet. 1976, 34, 1–7. [Google Scholar] [CrossRef]
- Biederman, B.; Bowen, P. Partial trisomy 4q due to familial 2/4 translocation. Qual. Life Res. 1976, 33, 147–153. [Google Scholar] [CrossRef]
- Bonfante, A.; Stella, M.; Rossi, G. Partial trisomy 4q: Two cases resulting from a familial translocation t(4;18)(q27;p11). Qual. Life Res. 1979, 52, 85–90. [Google Scholar] [CrossRef]
- Skrlec, I.; Wagner, J.; Puseljić, S.; Heffer, M.; Stipoljev, F. Partial monosomy 2p and partial trisomy 4q due to paternal trans-location t(2;4)(p25.1;q31.3). Coll. Antropol. 2014, 38, 759–762. [Google Scholar]
- Sag, S.O.; Gorukmez, O.; Ture, M.; Gulten, T.; Yakut, T. De novo partial trisomy distal 4q: A case report. Genet. Couns. 2014, 25, 423–428. [Google Scholar]
- Machuca, S.T.V. Duplicación parcial 4q en un neonato originado por reordenamiento der (20), t(4;20) (q21;q13.1)mat. Rev. Peru. Investig. Matern. Perinat. 2016, 5, 75–81. [Google Scholar] [CrossRef] [Green Version]
- El-Ruby, M.; Hemly, N.A.; Zaki, M.S. Maternal balanced translocation (4;21) leading to an offspring with partial duplica-tion of 4q and 21q without phenotypic manifestations of Down syndrome. Genet. Couns. 2007, 18, 217–226. [Google Scholar]
- Annerén, G.; Lübeck, P.-O. Trisomy 4q31 →qter due to a maternal 4/8 translocation. Hereditas 2008, 100, 45–49. [Google Scholar] [CrossRef]
- Romero, M.C.; Mialdea, O.G.; Company, A.V.; Tapia, M.C.; Piñera, J.G. Duplicación parcial del cromosoma 4q (q31, q35): Síndrome aurículo-acro-renal. In Anales de Pediatría; Elsevier: Amsterdam, The Netherlands, 2008; Volume 68, pp. 361–364. [Google Scholar] [CrossRef]
- Bellucco, F.T.; Fock, R.A.; de Oliveira-Júnior, H.R.; Perez, A.B.; Melaragno, M.I. Complex Small Supernumerary Marker Chromosome Leading to Partial 4q/21q Duplications: Clinical Implication and Review of the Literature. Cytogenet Genome Res 2018, 156, 173–178. [Google Scholar] [CrossRef]
- Lin, S.; Rawlins, L.; Turner, C.; Doyle, E.; Sleep, T. Novel ocular findings with 5p deletion and partial trisomy of distal 4q. Can. J. Ophthalmol. 2017, 53, e89–e90. [Google Scholar] [CrossRef]
- Mohamed, A.M.; El-Bassyouni, H.T.; El-Gerzawy, A.M.; Hammad, S.A.; Helmy, N.A.; Kamel, A.K.; Ismail, S.I.; Issa, M.Y.; Eid, O.; Zaki, M.S. Cytogenomic characterization of 1q43q44 deletion associated with 4q32.1q35.2 duplication and phenotype correlation. Mol. Cytogenet. 2018, 11, 57. [Google Scholar] [CrossRef]
- Shenoy, R.D.; Shenoy, V.; Shetty, V. Chromosomal Abnormalities in Syndromic Orofacial Clefts: Report of Three Children. Case Rep. Genet. 2018, 2018, 1928918. [Google Scholar] [CrossRef] [Green Version]
- Thapa, M.; Asamoah, A.; Gowans, G.C.; Platky, K.C.; Barch, M.J.; Mouchrani, P.; Rajakaruna, C.; Hersh, J.H. Molecular characterization of distal 4q duplication in two patients using oligonucleotide array-based comparative genomic hybridization (oaCGH) analysis. Am. J. Med. Genet. Part A 2014, 164, 1069–1074. [Google Scholar] [CrossRef] [PubMed]
- Zaki, M.S.; Eid, O.M.; Eid, M.M.; Mohamed, A.M.; Sayed, I.S.; Abdel-Hamid, M.S.; Abdel-Salam, G.M. Bilateral Calcification of Basal Ganglia in a Patient with Duplication of Both 11q13.1q22.1 and 4q35.2 with New Phenotypic Features. Cytogenet. Genome Res. 2019, 159, 130–136. [Google Scholar] [CrossRef]
- Bonnet, C.; Zix, C.; Grégoire, M.-J.; Brochet, K.; Duc, M.; Rousselet, F.; Philippe, C.; Jonveaux, P. Characterization of mosaic supernumerary ring chromosomes by array-CGH: Segmental aneusomy for proximal 4q in a child with tall stature and obesity. Am. J. Med. Genet. Part A 2006, 140, 233–237. [Google Scholar] [CrossRef]
- Matoso, E.; Melo, J.B.; Ferreira, S.I.; Jardim, A.; Castelo, T.M.; Weise, A.; Carreira, I.M. Insertional translocation leading to a 4q13 duplication including theEPHA5gene in two siblings with attention-deficit hyperactivity disorder. Am. J. Med. Genet. Part A 2013, 161, 1923–1928. [Google Scholar] [CrossRef]
- Mehta, L.; Duckett, D.P.; Young, I.D. Behaviour disorder in monosomy 10qter. J. Med. Genet. 1987, 24, 185–186. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tanabe, S.; Akiba, T.; Katoh, M.; Satoh, T. Terminal deletion of chromosome 10q: Clinical features and literature review. Pediatr. Int. 1999, 41, 565–567. [Google Scholar] [CrossRef]
- Miller, N.D.; Nance, M.A.; Wohler, E.S.; Hoover-Fong, J.E.; Lisi, E.; Thomas, G.H.; Pevsner, J. Molecular (SNP) analyses of overlapping hemizygous deletions of 10q25.3 to 10qter in four patients: Evidence for HMX2 and HMX3 as candidate genes in hearing and vestibular function. Am. J. Med. Genet. Part A 2009, 149A, 669–680. [Google Scholar] [CrossRef] [Green Version]
- Sangu, N.; Okamoto, N.; Shimojima, K.; Ondo, Y.; Nishikawa, M.; Yamamoto, T. A de novo microdeletion in a patient with inner ear abnormalities suggests that the 10q26.13 region contains the responsible gene. Hum. Genome Var. 2016, 3, 16008. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Online Mendelian Inheritance in Man (OMIMTM). Available online: http://www.ncbi.nlm.nih.gov/omim/ (accessed on 29 November 2021).
- Martin, C.L.; Waggoner, D.J.; Wong, A.; Uhrig, S.; A Roseberry, J.; Hedrick, J.F.; Pack, S.; Russell, K.L.; Zackai, E.H.; Dobyns, W.; et al. “Molecular rulers” for calibrating phenotypic effects of telomere imbalance. J. Med. Genet. 2002, 39, 734–740. [Google Scholar] [CrossRef]
- Ravnan, J.B.; Tepperberg, J.H.; Papenhausen, P.; Lamb, A.N.; Hedrick, J.; Eash, D.; Ledbetter, D.H.; Martin, C.L. Subtelomere FISH analysis of 11 688 cases: An evaluation of the frequency and pattern of subtelomere rearrangements in individuals with developmental disabilities. J. Med. Genet. 2006, 43, 478–489. [Google Scholar] [CrossRef] [Green Version]
- Riegel, M.; Baumer, A.; Jamar, M.; Delbecque, K.; Herens, C.; Verloes, A.; Schinzel, A. Submicroscopic terminal deletions and duplications in retarded patients with unclassified malformation syndromes. Qual. Life Res. 2001, 109, 286–294. [Google Scholar] [CrossRef] [PubMed]
- Yan, Y.; Wu, Q.; Zhang, L.; Wang, X.; Dan, S.; Deng, D.; Sun, L.; Yao, L.; Ma, Y.; Wang, L. Detection of submicroscopic chromosomal aberrations by array-based comparative genomic hybridization in fetuses with congenital heart disease. Ultrasound Obstet. Gynecol. 2013, 43, 404–412. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kowalczyk, M.; Srebniak, M.; Tomaszewska, A. Chromosome abnormalities without phenotypic consequences. J. Appl. Genet. 2007, 48, 157–166. [Google Scholar] [CrossRef] [PubMed]
- Navarro, P.; Oldfield, A.; Legoupi, J.; Festuccia, N.; Dubois, A.; Attia, M.; Schoorlemmer, J.; Rougeulle, C.; Chambers, I.; Avner, P. Molecular coupling of Tsix regulation and pluripotency. Nat. Cell Biol. 2010, 468, 457–460. [Google Scholar] [CrossRef]
- Tian, L.; Wu, X.; Lin, Y.; Liu, Z.; Xiong, F.; Han, Z.; Zhou, Y.; Zeng, Q.; Wang, Y.; Deng, J.; et al. Characterization and potential function of a novel pre-implantation embryo-specific RING finger protein: TRIML. Mol. Reprod. Dev. 2009, 76, 656–664. [Google Scholar] [CrossRef]
- Ji, Y.; Eichler, E.E.; Schwartz, S.; Nicholls, R.D. Structure of Chromosomal Duplicons and their Role in Mediating Human Genomic Disorders. Genome Res. 2000, 10, 597–610. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peoples, R.; Franke, Y.; Wang, Y.-K.; Pérez-Jurado, L.; Paperna, T.; Cisco, M.; Francke, U. A Physical Map, Including a BAC/PAC Clone Contig, of the Williams-Beuren Syndrome–Deletion Region at 7q11. Am. J. Hum. Genet. 2000, 66, 47–68. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Potocki, L.; Chen, K.-S.; Park, S.S.; Osterholm, D.E.; Withers, M.A.; Kimonis, V.; Summers, A.M.; Meschino, W.S.; Anyane-Yeboa, K.; Kashork, C.D.; et al. Molecular mechanism for duplication 17p11.2—The homologous recombination reciprocal of the Smith-Magenis microdeletion. Nat. Genet. 2000, 24, 84–87. [Google Scholar] [CrossRef] [PubMed]
- Yobb, T.M.; Somerville, M.J.; Willatt, L.; Firth, H.V.; Harrison, K.; MacKenzie, J.; Gallo, N.; Morrow, B.E.; Shaffer, L.G.; Babcock, M.; et al. Microduplication and Triplication of 22q11.2: A Highly Variable Syndrome. Am. J. Hum. Genet. 2005, 76, 865–876. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weckselblatt, B.; Rudd, M.K. Human Structural Variation: Mechanisms of Chromosome Rearrangements. Trends Genet. 2015, 31, 587–599. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, C.-P.; Lin, M.-H.; Chern, S.-R.; Chen, Y.-T.; Wu, P.-S.; Kuo, Y.-L.; Lee, M.-S.; Wang, W. Directly transmitted 4.5-Mb triplication of 4q12-q13.1: Prenatal diagnosis and molecular cytogenetic characterization. Taiwan. J. Obstet. Gynecol. 2014, 53, 123–125. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, C.-P.; Su, Y.-N.; Chen, Y.-T.; Chen, W.-L.; Hsu, L.J.; Wang, W. Prenatal diagnosis of directly transmitted benign 4q12-q13.1 quadruplication associated with tandem segmental amplifications of the LPHN3 gene. Taiwan. J. Obstet. Gynecol. 2011, 50, 401–404. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bertelsen, B.; Melchior, L.; Jensen, L.R.; Groth, C.; Glenthøj, B.; Rizzo, R.; Debes, N.M.; Skov, L.; Brøndum-Nielsen, K.; Paschou, P.; et al. Intragenic deletions affecting two alternative transcripts of the IMMP2L gene in patients with Tourette syndrome. Eur. J. Hum. Genet. 2014, 22, 1283–1289. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pope, B.; Tsumagari, K.; Battaglia, D.; Ryba, T.; Hiratani, I.; Ehrlich, M.; Gilbert, D.M. DNA Replication Timing Is Maintained Genome-Wide in Primary Human Myoblasts Independent of D4Z4 Contraction in FSH Muscular Dystrophy. PLoS ONE 2011, 6, e27413. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Matsumura, T.; Goto, K.; Yamanaka, G.; Lee, J.H.; Zhang, C.; Hayashi, Y.K.; Arahata, K. Chromosome 4q;10q translocations; Comparison with different ethnic populations and FSHD patients. BMC Neurol. 2002, 2, 7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gug, C.; Rațiu, A.; Navolan, D.; DrĂgan, I.; Groza, I.-M.; Păpurică, M.; Vaida, M.-A.; Mozoș, I.; Jurcă, M.C. Incidence and Spectrum of Chromosome Abnormalities in Miscarriage Samples: A Retrospective Study of 330 Cases. Cytogenet. Genome Res. 2019, 158, 171–183. [Google Scholar] [CrossRef] [PubMed]
- Gug, C.; Stoicanescu, D.; Mozos, I.; Nussbaum, L.; Cevei, M.; Stambouli, D.; Pavel, A.G.; Doros, G. De novo 8p21.3→p23.3 Duplication With t(4;8)(q35;p21.3) Translocation Associated With Mental Retardation, Autism Spectrum Disorder, and Congenital Heart Defects: Case Report With Literature Review. Front. Pediatr. 2020, 8, 375. [Google Scholar] [CrossRef] [PubMed]
- Gug, C.; Huțanu, D.; Vaida, M.; Doroş, G.; Popa, C.; Stroescu, R.; Furău, G.; Furău, C.; Grigoriță, L.; Mozos, I. De novo unbalanced translocation t(15;22)(q26.2;q12) with velo-cardio-facial syndrome: A case report and review of the literature. Exp. Ther. Med. 2018, 16, 3589–3595. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stene, J.; Stengel-Rutkowski, S. Genetic risks of familial reciprocal and Robertsonian translocation carriers. In The Cytogenetics of Mammalian Autosomal Rearran-Gements; Daniel, A., Ed.; Alan R Liss, Inc.: New York, NY, USA, 1988; pp. 1–54. [Google Scholar]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| No | Reference | Chromosome 4 Region | Type of Anomaly |
|---|---|---|---|
| 1 | Mattei et al., 1979 [18] | q12–q13 | IH, t(2;4) mat |
| 2 | Zollino et al., 1995 [19] | q13.3–q22.2 | DN, dup(4)(q13.3q22.2) |
| 3 | Halal et al., 1991 [20] | q23–q27 | DN, dup(4)(q23q27) |
| 4 | Jeziorowska et al., 1993 [21] | q21.3–q31.3 | DN, dup(4)(q21.3q31.3) |
| 5 | Fryns, 1980 [22] | q25–q31 | DN, dup(4)(q25q31) |
| 6 | Vogel et al., 1975 [23] | q22–q34 | DN, dup(4)(q22q34) |
| 7 | Dutrillaux et al., 1975 [24] | q22–q34 | DN, dup(4)(q22q34) |
| 8 | Taylor et al., 1977 [25] | q26–q35 | DN, dup(4)(q26q35) |
| 9 | Goodman et al., 1997 [26] | q31.1–q32.3 | IH, dup(4)(q31.1q32.3) |
| 10 | Muraki et al., 1997 [27] | q25–q31.3 | DN, dup(4)(q25q31.3) |
| 11 | Shashi et al., 1999 [28] | q12–q13 | DN, dup(4)(q12q13) |
| 12 | Guillen Navarro et al., 1996 [29] | q21–q28 | DN, dup(4)(q21q28) |
| 13 | Otsuka et al., Case 1, 2005 [30] | q31.22–q35.2 | IH, dup(4)(q31.22q35.2) |
| 14 | Otsuka et al., Case 2, 2005 [30] | q31.22–q35.2 | IH, dup(4)(q31.22q35.2) |
| 15 | Lundin et al., 2002 [31] | q27–q35 | DN, t(4;7)(q27;p22) |
| 16 | Mikelsaar et al., 1996 [32] | q25–qter | DN, t(4;22)(q25;p11) |
| 17 | Cui et al., 2006 [33] | q27–q35 | DN, t(4;5)(q27;q35) |
| 18 | Maltby et al., 1999 [34] | q31–q33 | IH, dup(4)(q31q33) |
| 19 | Assawamakin et al., 2012 [35] | q13.2–q22.1 | IH, der(8)(20qter–>20q12::4q22.1–>q21.21::4q13.3–>4q13.2::8q22.1–>8p11.12::8q22.3–>qter),der(20)(20pter –>20q12::4q13.3–>q21.21::8q22.3–>q22.1::8p11.12–>pter) |
| 20 | Angulo et al., 1984 [36] | q31–qter | IH, t(4;12)(q31;q24) |
| 21 | Watanabe et al., 1977 [37] | q33–qter | IH, t(4;13)(q33;q33) |
| 22 | Schrott et al., 1974 [6] | q26–qter | IH, t(4;13)(q26;q34) |
| 23 | Jenkins et al., 1975 [38] | q31–qter | DN, t(4;13)(q31;q14) |
| 24 | Zhang et al., 2009 [7] | q25–qter | IH, t(4;10)(q26;q26.3) |
| 25 | Cernakova et al., 2006 [9] | q28–q35.2 | DN, dup(4)(q28q35.2) |
| 26 | Horbinski et al., 2008 [39] | q35–qter | DN, t(4;18)(q35;q23) |
| 27 | Rinaldi et al., 2005 [40] | q24–q35 | DN, t(4;14)(q24;p12) |
| 28 | Kadotani et al., 1981 [41] | q32–qter | IH, t(4;9)(q23;p24) |
| 29 | Issa et al., 1976 [42] | q31–qter | IH, t(4;9)(q31;q34) |
| 30 | Collia et al., 2012 [43] | q12–q22 | DN, dup(4)(q11q22) |
| 31 | Baccichetti et al., 1975 [44] | q32–qter | IH, t(4;21)(q32;q22) |
| 32 | Patel et al., 2006 [45] | q25–qter | IH, t(4;18)(q25;q22) |
| 33 | Cervenka et al., 1976 [46] | q25–qter | IH, t(X;4)(q27;q25) |
| 34 | Biederman and Bowen 1976 [47] | q21–qter | IH, t(2;4)(p25;q21) |
| 35 | Bonfante et al., case A, 1979 [48] | q27–qter | IH, t(4;18)(q27;p11) |
| 36 | Bonfante et al., case B, 1979 [48] | q27–qter | IH, t(4;18)(q27;p11) |
| 37 | Škrlec et al., 2014 [49] | q31.3–qter | IH, t(2;4)(p25.1;q31.3) |
| 38 | Gorukmez et al., 2014 [50] | q21–q35 | DN, dup(4)(q21q35) |
| 39 | Vargas Machuca et al., 2016 [51] | q21–qter | IH, t(4;20)(q21;q13.1) |
| 40 | El-Ruby et al., 2007 [52] | q25–qter | IH, t(4;21)(q25;q22) |
| 41 | Anneren et al., 1984 [53] | q31–qter | IH, t(4;8)(q31;p23) |
| 42 | Carrascosa Romero et al., 2008 [54] | q31–q35 | DN, dup(4)( q31q35) |
| 43 | Bellucco et al., 2018 [55] | q32.1–q35.2 | 47,XX,+der(21)t(4; 21)(q32.1;q21.2)mat. arr[GRCh37/hg19] 4q32. 1q35.2 (158907036_190957460)x3,21q11.2q21.2(15016486_25605895)x3 |
| 44 | Lin et al., 2018 [56] | q32.3–qter | ND, t(4;5)(q32.3;p14.2) |
| 45 | Mohamed et al., 2018 [57] | q32.1–q35.2 | DN 46,XX,add1(q44) |
| 46 | Shenoy et al., 2018 [58] | q27q35.2 | IH, t(4;21)(q27;q22) |
| 47 | Thapa et al., case A, 2014 [59] | q32.1–q35.2 | ND, dup(4)(q32.1q35.2) |
| 48 | Thapa et al., case B, 2014 [59] | q32.2–q34.3 | ND,dup(4)(q32.2q34.3) |
| 49 | Zaki et al., 2019 [60] | q35.2 | DN, dup(4)(q35.2) |
| 50 | This case | q26–qter | IH, t(4;10)(q26;q26.3) |
| No | Reference | Chromosome 4 Region | a | b | c | d | e | f | g | h | i | j | k |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | Mattei et al., 1979 [18] | q12–q13 | + | + | + | + | − | + | − | − | − | − | − |
| 2 | Zollino et al., 1995 [19] | q13.3–q22.2 | − | + | − | − | − | − | + | − | − | − | − |
| 3 | Halal et al., 1991 [20] | q23–q27 | + | + | + | + | − | − | − | − | − | + | − |
| 4 | Jeziorowska et al., 1993 [21] | q21.3–q31.3 | + | + | + | + | + | + | + | + | + | − | + |
| 5 | Fryns, 1980 [22] | q25–q31 | + | + | − | − | + | + | − | + | − | − | − |
| 6 | Vogel et al., 1975 [23] | q22–q34 | + | + | − | + | + | + | + | + | + | − | + |
| 7 | Dutrillaux et al., 1975 [24] | q22–q34 | + | + | + | + | + | + | + | + | + | − | + |
| 8 | Taylor et al., 1977 [25] | q26–q35 | + | + | + | + | + | + | + | + | − | + | ND |
| 9 | Goodman et al., 1997 [26] | q31.1–q32.3 | + | + | − | + | − | ND | ND | − | − | + | − |
| 10 | Muraki et al., 1997 [27] | q25–q31.3 | + | + | − | − | − | − | − | + | − | − | − |
| 11 | Shashi et al., 1999 [28] | q12–q13 | + | + | + | + | + | − | − | + | − | − | − |
| 12 | Guillen Navarro et al., 1996 [29] | q21–q28 | + | + | − | + | + | ND | + | + | + | − | − |
| 13 | Otsuka et al., Case 1, 2005 [30] | q31.22–q35.2 | + | + | + | + | + | + | + | + | + | − | + |
| 14 | Otsuka et al., Case 2, 2005 [30] | q31.22–q35.2 | + | + | + | + | + | + | + | + | + | − | + |
| 15 | Lundin et al., 2002 [31] | q27–q35 | + | + | + | + | + | ND | − | − | + | − | − |
| 16 | Mikelsaar et al., 1996 [32] | q25–qter | + | + | + | + | + | + | − | + | − | − | − |
| 17 | Cui et al., 2006 [33] | q27–q35 | + | + | − | + | + | ND | − | − | + | − | − |
| 18 | Maltby et al., 1999 [34] | q31–q33 | − | − | − | − | − | ND | − | − | − | − | − |
| 19 | Assawamakin et al., 2012 [35] | q13.2–q22.1 | − | + | + | − | − | ND | − | − | − | + | − |
| 20 | Angulo et al., 1984 [36] | q31–qter | + | + | − | + | + | + | ND | + | − | − | + |
| 21 | Watanabe et al., 1977 [37] | q33–qter | + | + | + | − | + | ND | − | + | ND | − | + |
| 22 | Schrott et al., 1974 [6] | q26–qter | + | + | + | − | − | ND | + | − | ND | − | + |
| 23 | Jenkins et al., 1975 [38] | q31–qter | + | + | − | + | + | ND | − | + | ND | + | − |
| 24 | Zhang et al., 2009 [7] | q25–qter | + | + | + | + | + | + | + | + | − | − | − |
| 25 | Cernakova et al., 2006 [9] | q28–q35.2 | + | + | + | + | + | − | + | + | − | + | − |
| 26 | Horbinski et al., 2008 [39] | q35–qter | + | + | + | + | + | − | − | + | − | + | − |
| 27 | Rinaldi et al., 2005 [40] | q24–q35 | + | + | + | ND | + | ND | + | + | + | + | + |
| 28 | Kadotani et al., 1981 [41] | q32–qter | + | ND | + | ND | + | ND | + | + | + | ND | ND |
| 29 | Issa et al., 1976 [42] | q31–qter | + | + | − | ND | + | ND | − | + | − | − | ND |
| 30 | Collia et al., 2012 [43] | q12–q22 | − | − | − | + | + | ND | − | + | − | − | − |
| 31 | Baccichetti et al., 1975 [44] | q32–qter | + | + | + | − | + | − | + | + | − | ND | − |
| 32 | Patel et al., 2006 [45] | q25–qter | + | + | + | ND | + | ND | + | + | + | − | + |
| 33 | Cervenka et al., 1976 [46] | q25–qter | + | + | + | + | + | − | − | + | + | − | + |
| 34 | Biederman and Bowen, 1976 [47] | q21–qter | + | + | + | ND | + | + | + | + | + | − | ND |
| 35 | Bonfante et al., case A, 1979 [48] | q27–qter | + | + | − | − | − | − | + | + | − | + | − |
| 36 | Bonfante et al., case B, 1979 [48] | q27–qter | + | + | − | + | + | − | − | + | − | − | − |
| 37 | Škrlec et al., 2014 [49] | q31.3–qter | − | + | − | + | − | ND | ND | + | − | − | − |
| 38 | Gorukmez et al., 2014 [50] | q21–q35 | + | + | + | − | + | + | + | + | + | + | + |
| 39 | Vargas Machuca et al., 2016 [51] | q21–qter | + | ND | + | ND | + | ND | + | + | − | + | − |
| 40 | El-Ruby et al., 2007 [52] | q25–qter | + | + | + | − | + | − | − | + | + | − | − |
| 41 | Anneren et al., 1984 [53] | q31–qter | + | + | + | + | + | ND | − | + | − | − | − |
| 42 | Carrascosa Romero et al., 2008 [54] | q31–q35 | − | + | + | ND | + | + | + | + | − | − | + |
| 43 | Bellucco et al., 2018 [55] | q32.1–q35.2 | + | + | + | − | + | − | − | − | − | − | − |
| 44 | Lin et al., 2018 [56] | q32.3–qter | ND | + | ND | ND | ND | ND | ND | ND | + | + | + |
| 45 | Mohamed et al., 2018 [57] | q32.1–q35.2 | + | + | + | − | − | ND | + | + | − | − | − |
| 46 | Shenoy et al., 2018 [58] | q27q35.2 | + | ND | + | ND | ND | ND | + | + | ND | + | + |
| 47 | Thapa et al., case A, 2014 [59] | q32.1–q35.2 | + | + | ND | − | + | + | + | + | − | ND | − |
| 48 | Thapa et al., case B, 2014 [59] | q32.2–q34.3 | + | + | ND | − | ND | ND | − | + | + | ND | − |
| 49 | Zaki et al., 2019 [60] | q35.2 | − | + | + | ND | ND | ND | ND | + | − | − | − |
| 50 | This case | q26–qter | + | + | + | + | + | + | + | + | − | + | − |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Popescu, R.; Grămescu, M.; Caba, L.; Pânzaru, M.-C.; Butnariu, L.; Braha, E.; Popa, S.; Rusu, C.; Cardos, G.; Zeleniuc, M.; et al. A Case of Inherited t(4;10)(q26;q26.2) Chromosomal Translocation Elucidated by Multiple Chromosomal and Molecular Analyses. Case Report and Review of the Literature. Genes 2021, 12, 1957. https://doi.org/10.3390/genes12121957
Popescu R, Grămescu M, Caba L, Pânzaru M-C, Butnariu L, Braha E, Popa S, Rusu C, Cardos G, Zeleniuc M, et al. A Case of Inherited t(4;10)(q26;q26.2) Chromosomal Translocation Elucidated by Multiple Chromosomal and Molecular Analyses. Case Report and Review of the Literature. Genes. 2021; 12(12):1957. https://doi.org/10.3390/genes12121957
Chicago/Turabian StylePopescu, Roxana, Mihaela Grămescu, Lavinia Caba, Monica-Cristina Pânzaru, Lăcrămioara Butnariu, Elena Braha, Setalia Popa, Cristina Rusu, Georgeta Cardos, Monica Zeleniuc, and et al. 2021. "A Case of Inherited t(4;10)(q26;q26.2) Chromosomal Translocation Elucidated by Multiple Chromosomal and Molecular Analyses. Case Report and Review of the Literature" Genes 12, no. 12: 1957. https://doi.org/10.3390/genes12121957