
Gonosomal Mosaicism for a Novel COL5A1 Pathogenic Variant in Classic Ehlers-Danlos Syndrome

 , ,
, , {kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Family Enrollment and Sample Preparation
2.2. NGS Analysis
2.3. Conservation of the Variant
2.4. Sanger Sequencing
2.5. Digital Droplet PCR
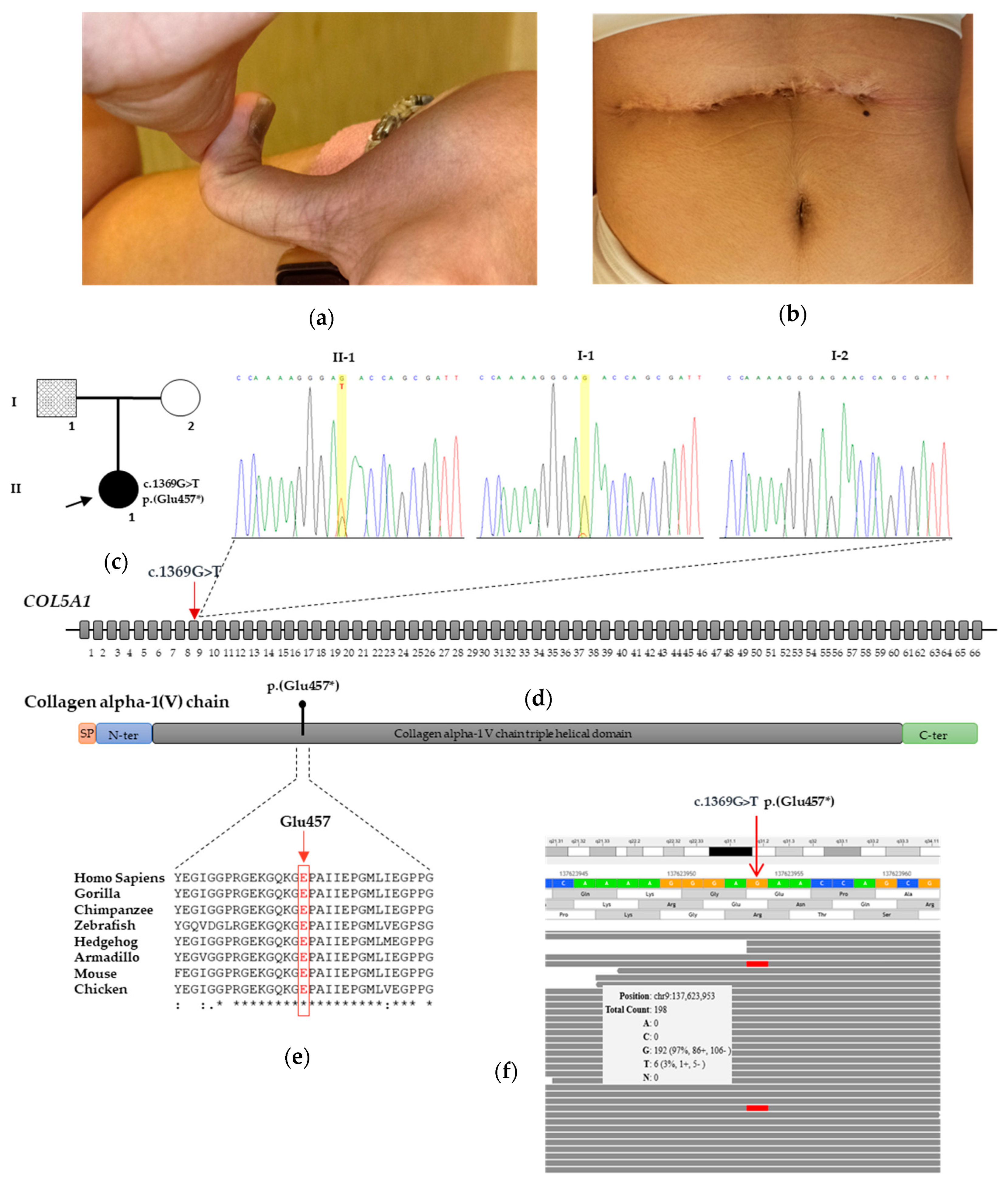
3. Results
3.1. Case Report
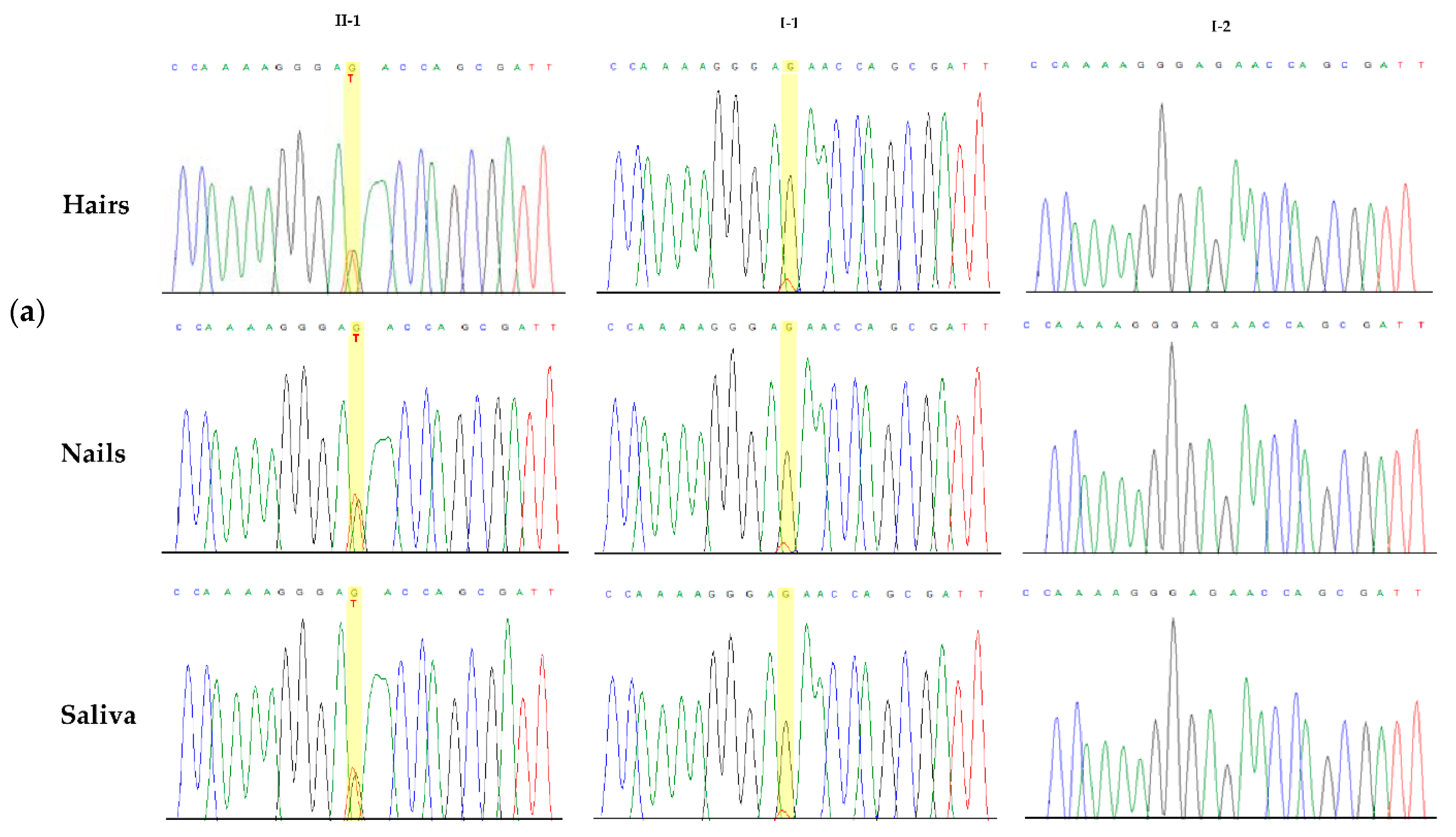
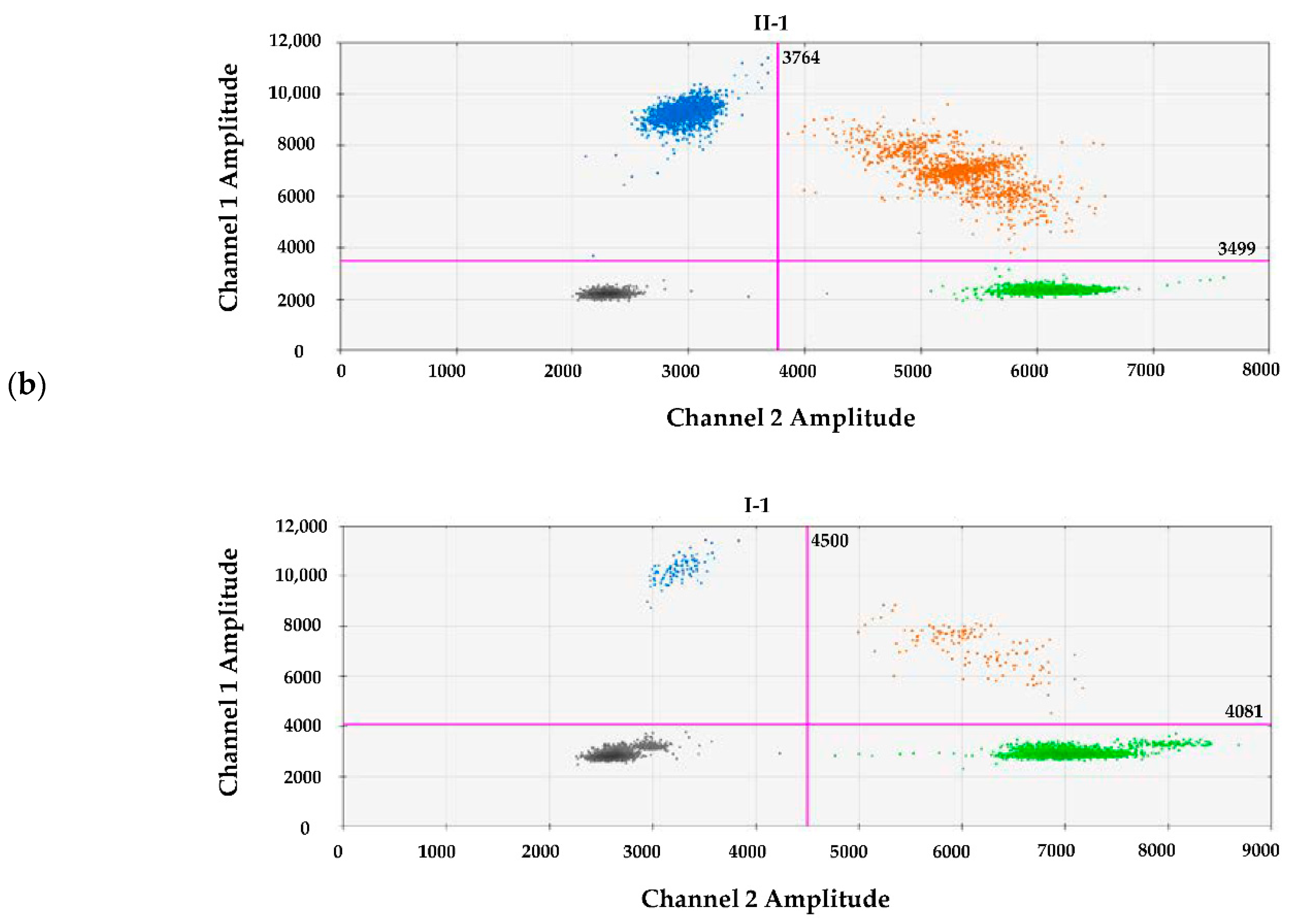
3.2. Molecular Findings
4. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Malfait, F.; Castori, M.; Francomano, C.A.; Giunta, C.; Kosho, T.; Byers, P.H. The Ehlers-Danlos syndromes. Nat. Rev. Dis. Primers 2020, 6, 64. [Google Scholar] [CrossRef] [PubMed]
- Blackburn, P.R.; Xu, Z.; Tumelty, K.E.; Zhao, R.W.; Monis, W.J.; Harris, K.G.; Gass, J.M.; Cousin, M.A.; Boczek, N.J.; Mitkov, M.V.; et al. Bi-allelic Alterations in AEBP1 Lead to Defective Collagen Assembly and Connective Tissue Structure Resulting in a Variant of Ehlers-Danlos Syndrome. Am. J. Hum. Genet. 2018, 102, 696–705. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Malfait, F.; Francomano, C.; Byers, P.; Belmont, J.; Berglund, B.; Black, J.; Bloom, L.; Bowen, J.M.; Brady, A.F.; Burrows, N.P.; et al. The 2017 international classification of the Ehlers-Danlos syndromes. Am. J. Med. Genet. C Semin. Med. Genet. 2017, 175, 8–26. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Symoens, S.; Syx, D.; Malfait, F.; Callewaert, B.; De Backer, J.; Vanakker, O.; Coucke, P.; De Paepe, A. Comprehensive molecular analysis demonstrates type V collagen mutations in over 90% of patients with classic EDS and allows to refine diagnostic criteria. Hum. Mutat. 2012, 33, 1485–1493. [Google Scholar] [CrossRef] [PubMed]
- Colman, M.; Syx, D.; De Wandele, I.; Dhooge, T.; Symoens, S.; Malfait, F. Clinical and molecular characteristics of 168 probands and 65 relatives with a clinical presentation of classical Ehlers-Danlos syndrome. Hum. Mutat. 2021, 42, 1294–1306. [Google Scholar] [CrossRef] [PubMed]
- Mak, K.M.; Png, C.Y.; Lee, D.J. Type V Collagen in Health, Disease, and Fibrosis. Anat. Rec. 2016, 299, 613–629. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Steiner, R.D.; Basel, D. COL1A1/2 Osteogenesis Imperfecta. In GeneReviews((R)); Adam, M.P., Ardinger, H.H., Pagon, R.A., Wallace, S.E., Bean, L.J.H., Mirzaa, G., Amemiya, A., Eds.; University of Washington: Seattle, WA, USA, 1993. [Google Scholar]
- Chesneau, B.; Plancke, A.; Rolland, G.; Chassaing, N.; Coubes, C.; Brischoux-Boucher, E.; Edouard, T.; Dulac, Y.; Aubert-Mucca, M.; Lavabre-Bertrand, T.; et al. Parental mosaicism in Marfan and Ehlers-Danlos syndromes and related disorders. Eur. J. Hum. Genet. 2021, 29, 771–779. [Google Scholar] [CrossRef] [PubMed]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 2015, 17, 405–424. [Google Scholar] [CrossRef] [PubMed]
- Grummer-Strawn, L.; Krebs, N.F.; Reinold, C.M. Use of World Health Organization and CDC growth charts for children aged 0-59 months in the United States. MMWR Recomm. Rep. 2010, 59, 1–15. [Google Scholar] [PubMed]
- Ritelli, M.; Dordoni, C.; Venturini, M.; Chiarelli, N.; Quinzani, S.; Traversa, M.; Zoppi, N.; Vascellaro, A.; Wischmeijer, A.; Manfredini, E.; et al. Clinical and molecular characterization of 40 patients with classic Ehlers-Danlos syndrome: Identification of 18 COL5A1 and 2 COL5A2 novel mutations. Orphanet J. Rare Dis. 2013, 8, 58. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bowen, J.M.; Sobey, G.J.; Burrows, N.P.; Colombi, M.; Lavallee, M.E.; Malfait, F.; Francomano, C.A. Ehlers-Danlos syndrome, classical type. Am. J. Med. Genet. C Semin. Med. Genet. 2017, 175, 27–39. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Biesecker, L.G.; Spinner, N.B. A genomic view of mosaicism and human disease. Nat. Rev. Genet. 2013, 14, 307–320. [Google Scholar] [CrossRef] [PubMed]
- Martínez-Quintana, E.; Caballero-Sánchez, N.; Rodríguez-González, F.; Garay-Sánchez, P.; Tugores, A. Novel Marfan Syndrome-Associated Mutation in the FBN1 Gene Caused by Parental Mosaicism and Leading to Abnormal Limb Patterning. Mol. Syndromol. 2017, 8, 148–154. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Acuna-Hidalgo, R.; Bo, T.; Kwint, M.P.; Van De Vorst, M.; Pinelli, M.; Veltman, J.A.; Hoischen, A.; Vissers, L.E.; Gilissen, C. Post-zygotic Point Mutations Are an Underrecognized Source of De Novo Genomic Variation. Am. J. Hum. Genet. 2015, 97, 67–74. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moreno-García, M.; Arteche-López, A.R.; Álvarez-Mora, M.I.; Palma Milla, C.; Quesada Espinosa, J.F.; Lezana Rosales, J.M.; Sanchez Calvin, M.T.; Gomez Manjon, I.; Gómez Rodríguez, M.J.; Mendez-Guerrero, A.; et al. First patient with mosaic NOTCH3 gene pathogenic variant. Unrevealed mosaicisms and importance of their detection. Am. J. Med. Genet. A 2021, 185, 591–595. [Google Scholar] [CrossRef] [PubMed]
- Legrand, A.; Devriese, M.; Dupuis-Girod, S.; Simian, C.; Venisse, A.; Mazzella, J.M.; Auribault, K.; Adham, S.; Frank, M.; Albuisson, J.; et al. Frequency of de novo variants and parental mosaicism in vascular Ehlers-Danlos syndrome. Genet. Med. 2019, 21, 1568–1575. [Google Scholar] [CrossRef] [PubMed]
- Yokoi, T.; Enomoto, Y.; Tsurusaki, Y.; Kurosawa, K. Siblings with vascular Ehlers-Danlos syndrome inherited via maternal mosaicism. Congenit. Anom. 2021, 61, 101–102. [Google Scholar] [CrossRef] [PubMed]
- Symoens, S.; Steyaert, W.; Demuynck, L.; De Paepe, A.; Diderich, K.E.; Malfait, F.; Coucke, P.J. Tissue-specific mosaicism for a lethal osteogenesis imperfecta COL1A1 mutation causes mild OI/EDS overlap syndrome. Am. J. Med. Genet. A 2017, 173, 1047–1050. [Google Scholar] [CrossRef] [PubMed]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Micale, L.; Foiadelli, T.; Russo, F.; Cinque, L.; Bassanese, F.; Granatiero, M.; Fusco, C.; Savasta, S.; Castori, M. Gonosomal Mosaicism for a Novel COL5A1 Pathogenic Variant in Classic Ehlers-Danlos Syndrome. Genes 2021, 12, 1928. https://doi.org/10.3390/genes12121928
Micale L, Foiadelli T, Russo F, Cinque L, Bassanese F, Granatiero M, Fusco C, Savasta S, Castori M. Gonosomal Mosaicism for a Novel COL5A1 Pathogenic Variant in Classic Ehlers-Danlos Syndrome. Genes. 2021; 12(12):1928. https://doi.org/10.3390/genes12121928
Chicago/Turabian StyleMicale, Lucia, Thomas Foiadelli, Federica Russo, Luigia Cinque, Francesco Bassanese, Matteo Granatiero, Carmela Fusco, Salvatore Savasta, and Marco Castori. 2021. "Gonosomal Mosaicism for a Novel COL5A1 Pathogenic Variant in Classic Ehlers-Danlos Syndrome" Genes 12, no. 12: 1928. https://doi.org/10.3390/genes12121928