
Heritability and Genomic Architecture of Episodic Exercise-Induced Collapse in Border Collies

 , ,
, , 
Abstract
:1. Introduction
2. Materials and Methods
2.1. Sample Collection and Criteria
2.2. Genotype Data Processing and Imputation
2.3. Model Analysis
2.4. Estimation of Heritability
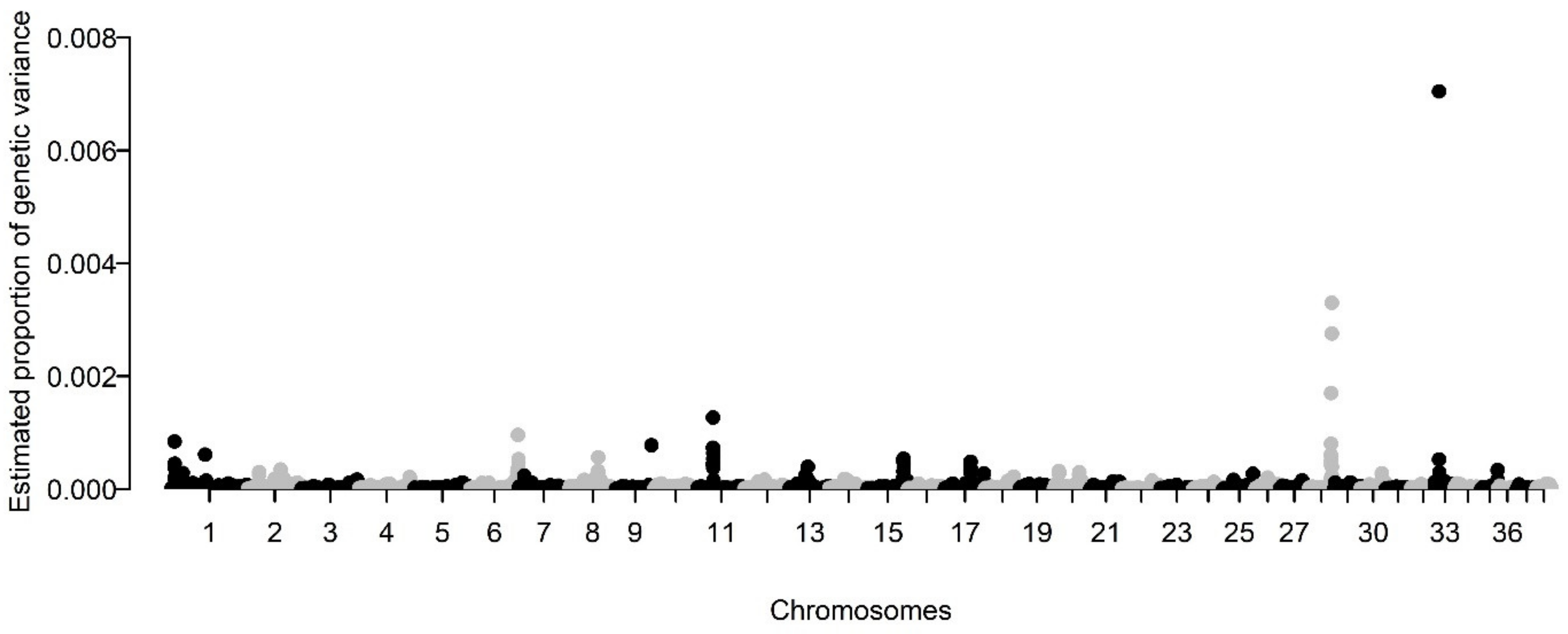
2.5. Determination of Genomic Architecture
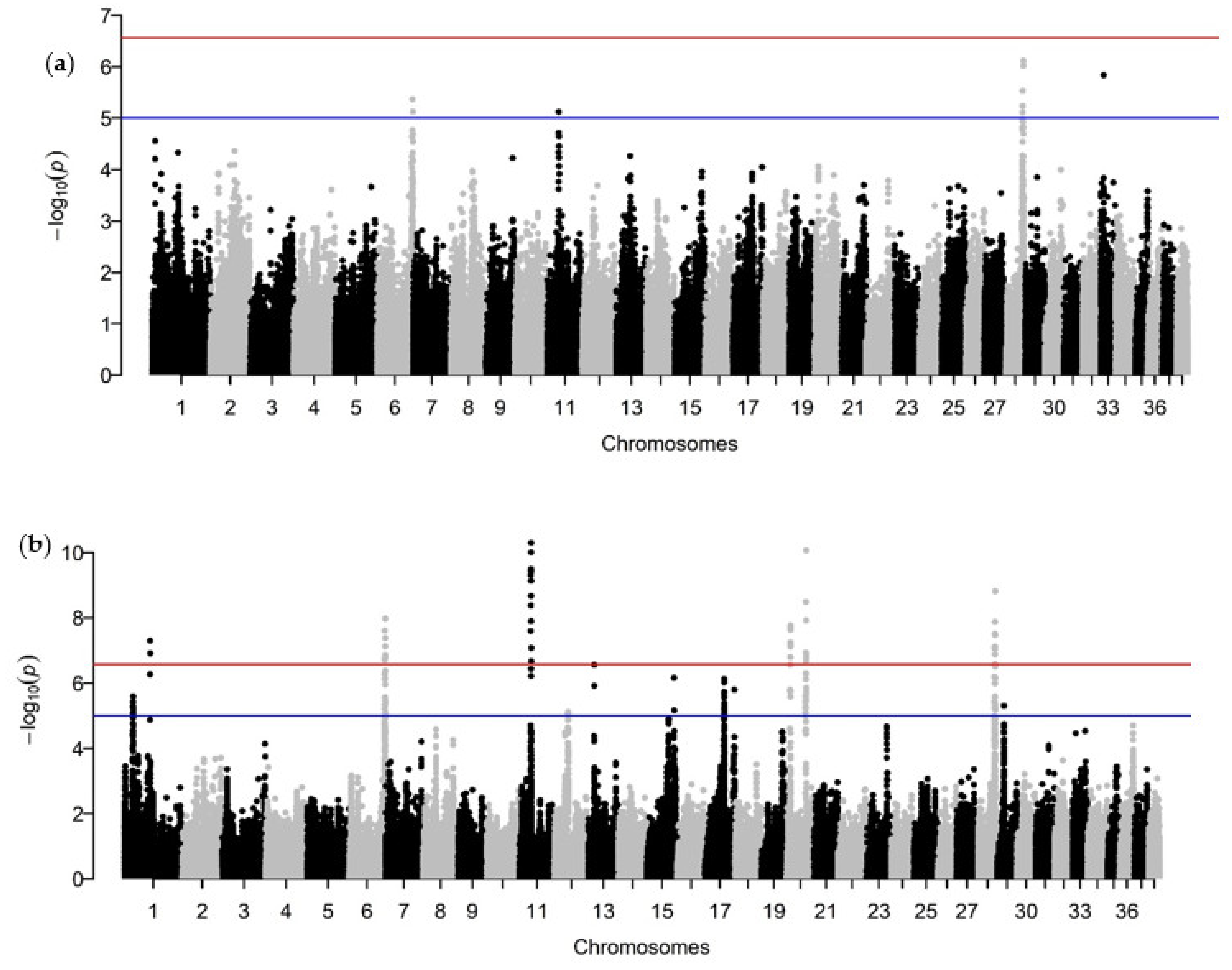
2.6. Genome-Wide Association Analysis
3. Results
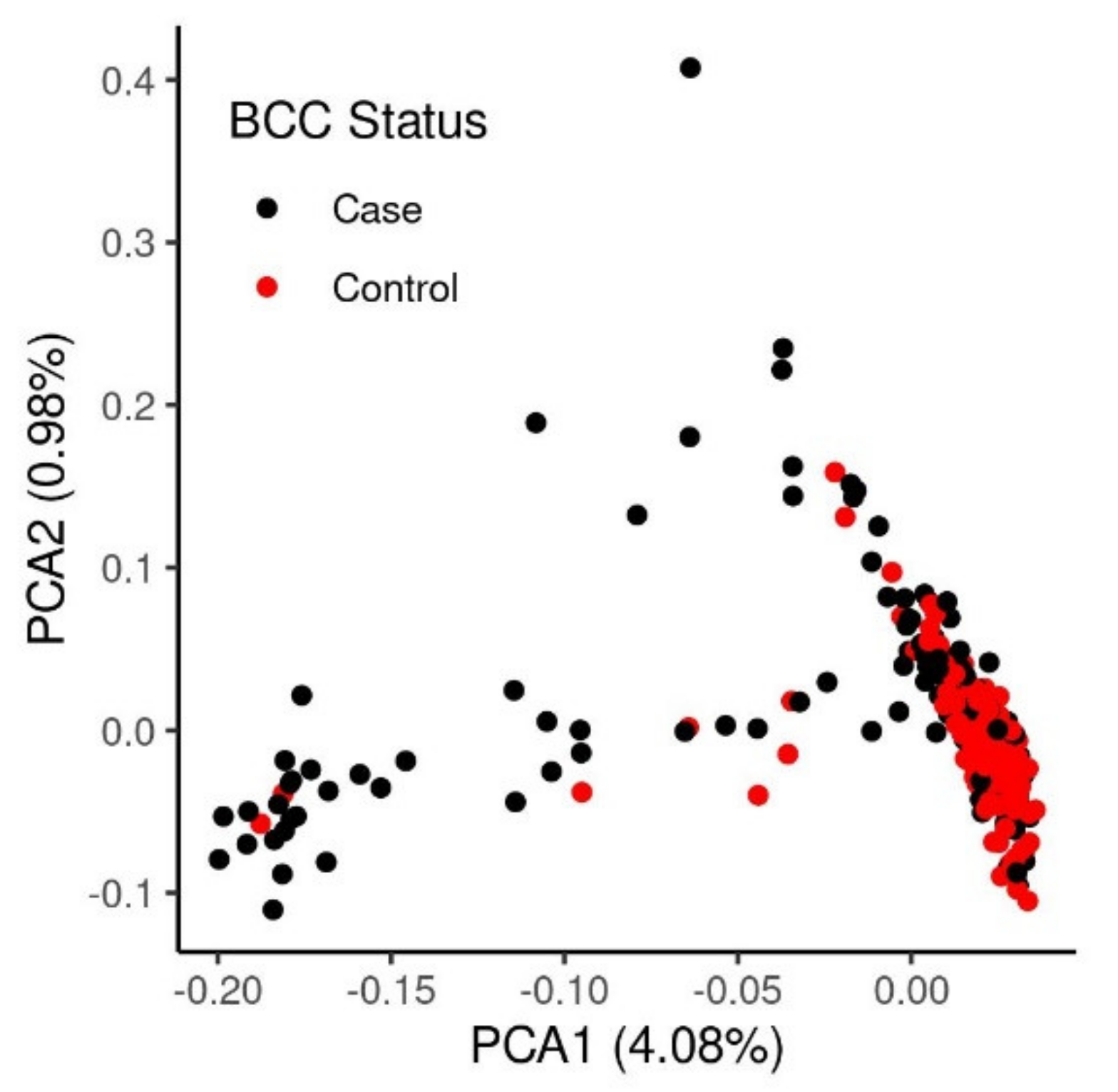
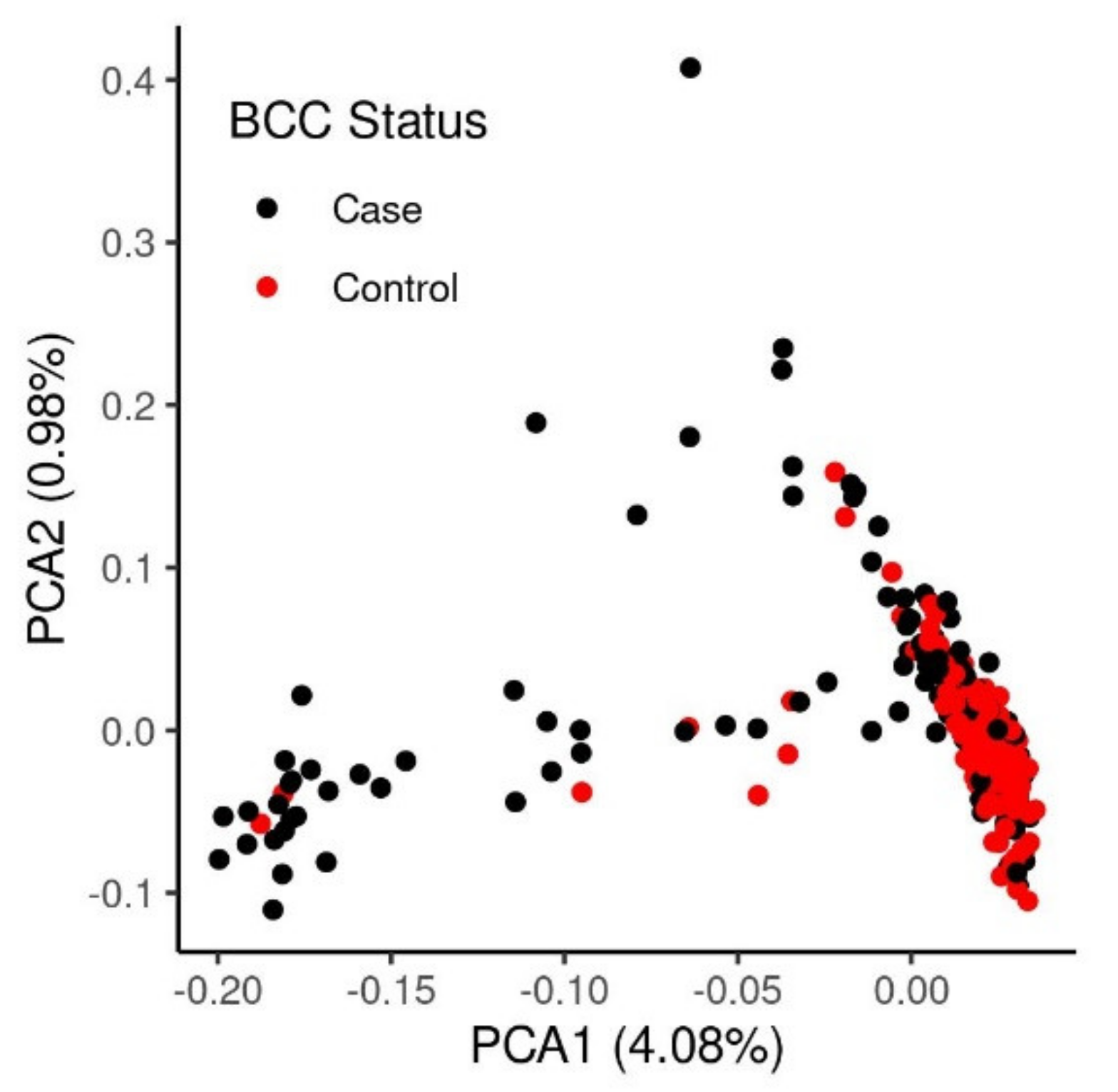
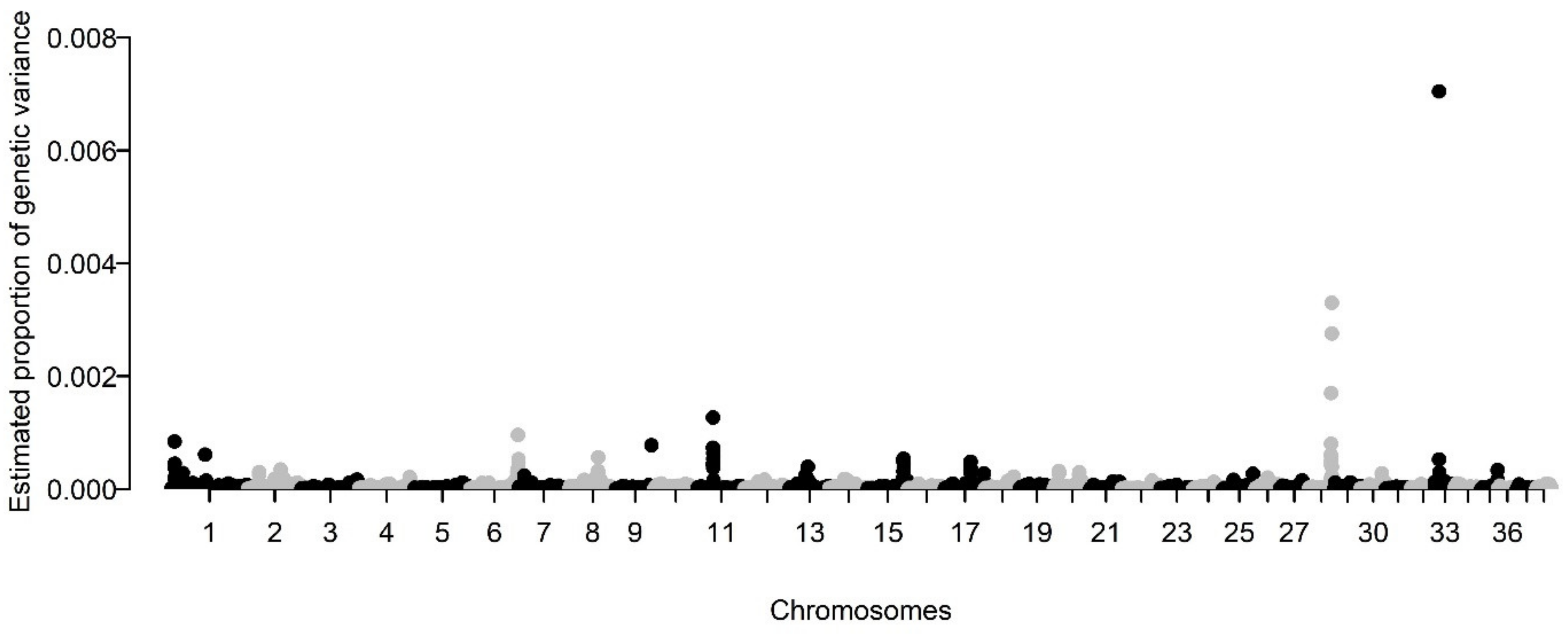
3.1. Assessment of the Heritability and Genetic Architecture of BCC
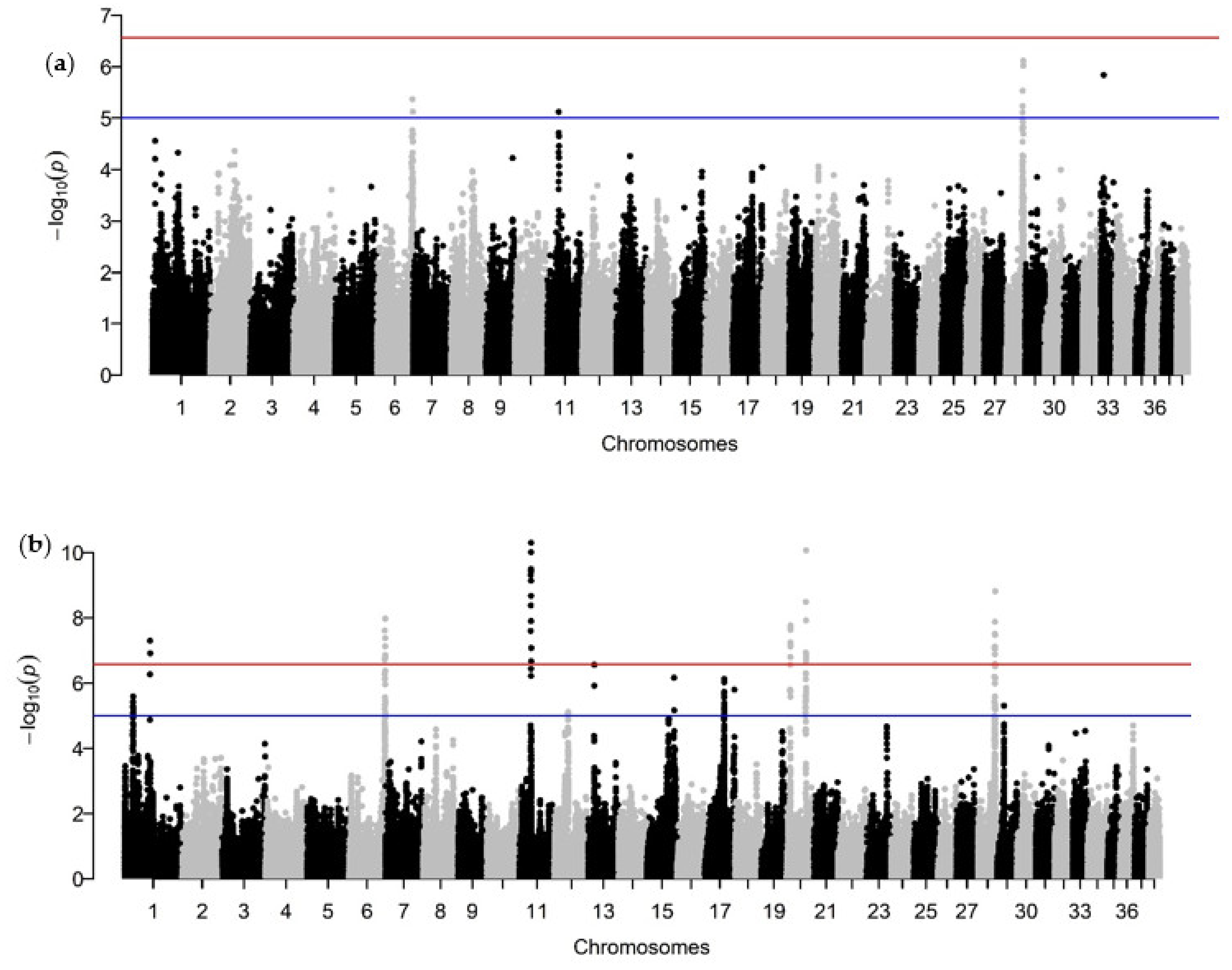
3.2. Genome-Wide Association Analysis
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Taylor, S.M.; Shmon, C.L.; Su, L.; Minor, K.M.; Patterson, E.E.; Mickelson, J.R.; Shelton, G.D. Effects of a standardized sheep herding strenuous exercise protocol on clinical and laboratory parameters in healthy border collies and dogs with border collie collapse. J. Vet. Intern. Med. 2011, 25, 764. [Google Scholar]
- Taylor, S.M. Preliminary investigations of an exercise intolerance syndrome in Border collies. In Proceedings of the European Veterinary Conference—Voorjaarsdagen, Amsterdam, The Netherlands, 29 April 2011; pp. 93–95. [Google Scholar]
- Taylor, S.M. Preliminary Investigations of an Exercise Intolerance Syndrome in Border Collies; Control and Therapy Series; Centre for Veterinary Education: Sydney, Australia, December 2011; p. 33. [Google Scholar]
- Taylor, S.; Minor, K.; Shmon, C.L.; Shelton, G.D.; Patterson, E.E.; Mickelson, J. Border Collie Collapse: Owner Survey Results and Veterinary Description of Videotaped Episodes. J. Am. Anim. Hosp. Assoc. 2016, 52, 364–370. [Google Scholar] [CrossRef]
- Taylor, S.; Shmon, C.; Su, L.; Epp, T.; Minor, K.; Mickelson, J.; Patterson, E.; Shelton, G.D. Evaluation of Dogs with Border Collie Collapse, Including Response to Two Standardized Strenuous Exercise Protocols. J. Am. Anim. Hosp. Assoc. 2016, 52, 281–290. [Google Scholar] [CrossRef]
- Steiss, J.; Ahmad, H.A.; Cooper, P.; Ledford, C. Physiologic responses in healthy Labrador retrievers during field trial training and competition. J. Vet. Int. Med. 2004, 18, 147–151. [Google Scholar] [CrossRef]
- Matwichuk, C.L.; Taylor, S.; Shmon, C.L.; Kass, P.H.; Shelton, G.D. Changes in rectal temperature and hematologic, biochemical, blood gas, and acid-base values in healthy Labrador Retrievers before and after strenuous exercise. Am. J. Vet. Res. 1999, 60, 88–92. [Google Scholar] [PubMed]
- Taylor, S.M. Seizures and other paroxysmal events. In Small Animal Internal Medicine, 5th ed.; Elsevier: Amsterdam, The Netherlands, 2020; Chapter 62; pp. 1093–1108. [Google Scholar]
- Patterson, E.E.; Minor, K.M.; Tchernatynskaia, A.V.; Taylor, S.M.; Shelton, G.D.; Ekenstedt, K.J.; Mickelson, J.R. Dynamin 1 (DNM1) mutation is highly associated with the syndrome of exercise-induced collapse in the Labrador retriever dog. Nat. Genet. 2008, 40, 1235–1239. [Google Scholar] [CrossRef] [PubMed]
- Minor, K.M.; Patterson, E.E.; Keating, M.K.; Gross, S.D.; Ekenstedt, K.J.; Taylor, S.M.; Mickelson, J.R. Presence and Impact of the Exercise-Induced Collapse associated DNM1 mutation in Labrador retrievers and other breeds. Vet. J. 2011, 189, 214–219. [Google Scholar] [CrossRef] [PubMed]
- Timpson, N.J.; Greenwood, C.M.T.; Soranzo, N.; Lawson, D.J.; Richards, J.B. Genetic architecture: The shape of the genetic contribution to human traits and disease. Nat. Rev. Genet. 2018, 19, 110–124. [Google Scholar] [CrossRef]
- Visscher, P.M.; Hill, W.G.; Wray, N.R. Heritability in the genomics era—Concepts and misconceptions. Nat. Rev. Genet. 2008, 9, 255–266. [Google Scholar] [CrossRef] [PubMed]
- Available online: https://vetmed.umn.edu/research/labs/canine-genetics-lab/genetic-research/border-collie-collapse (accessed on 7 November 2021).
- Browning, S.R.; Browning, B.L. Rapid and accurate haplotype phasing and missing-data inference for whole-genome association studies by use of localized haplotype clustering. Am. J. Hum. Genet. 2007, 81, 1084–1097. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Purcell, S.; Neale, B.; Todd-Brown, K.; Thomas, L.; Ferreira, M.A.; Bender, D.; Maller, J.; Sklar, P.; de Bakker, P.I.; Daly, M.J.; et al. PLINK: A tool set for whole-genome association and population-based linkage analyses. Am. J. Hum. Genet. 2007, 81, 559–575. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, J.; Lee, S.H.; Goddard, M.E.; Visscher, P.M. GCTA: A tool for genome-wide complex trait analysis. Am. J. Hum. Genet. 2011, 88, 76–82. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Speed, D.; Hemani, G.; Johnson, M.R.; Balding, D.J. Improved heritability estimation from genome-wide SNPs. Am. J. Hum. Genet. 2012, 91, 1011–1021. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Erbe, M.; Hayes, B.J.; Matukumalli, L.K.; Goswami, S.; Bowman, P.J.; Reich, C.M.; Mason, B.A.; Goddard, M.E. Improving accuracy of genomic predictions within and between dairy cattle breeds with imputed high-density single nucleotide polymorphism panels. J. Dairy Sci. 2012, 95, 4114–4129. [Google Scholar] [CrossRef] [Green Version]
- Mollandin, F.; Rau, A.; Croseau, P. An evaluation of the predictive performance and mapping power of the BayesR model for genomic prediction. G3 Genes Genomes Genet. 2021, 11, 225. [Google Scholar] [CrossRef]
- Li, M.X.; Yeung, J.M.; Cherny, S.S.; Sham, P.C. Evaluating the effective numbers of independent tests and significant p-value thresholds in commercial genotyping arrays and public imputation reference datasets. Human Genet. 2011, 131, 747–756. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, X.; Stephens, M. Genome-wide efficient mixed-model analysis for association studies. Nat. Genet. 2012, 44, 821–824. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Norton, E.M.; Schultz, N.; Geor, R.; McFarlane, D.; Mickelson, J.R.; McCue, M.E. Genome-wide association analyses of equine metabolic syndrome phenotypes in Welsh pones and Morgan horses. Genes 2019, 10, 893. [Google Scholar] [CrossRef]
- Durinck, S.; Spellman, P.T.; Birney, E.; Huber, W. Mapping identifiers for the integration of genomic datasets with the R/Bioconductor package biomaRt. Nat. Protoc. 2009, 4, 1184–1191. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ács, V.; Bokor, Á.; Nagy, I. Population Structure Analysis of the Border Collie Dog Breed in Hungary. Animals 2019, 9, 250. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carvalho, B.S.; Louis, T.A.; Irizarry, R.A. Quantifying uncertainty in genotype calls. Bioinformatics 2010, 26, 242–249. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, S.H.; Wray, N.R.; Goddard, M.E.; Visscher, P.M. Estimating missing heritability for disease from genome-wide association studies. Am. J. Hum. Genet. 2011, 88, 294–305. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moser, G.; Lee, S.H.; Hayes, B.J.; Goddard, M.E.; Wray, N.R.; Visscher, P.M. Simultaneous discovery, estimation and prediction analysis of complex traits using a bayesian mixture model. PLoS Genet. 2015, 11, e1004969. [Google Scholar] [CrossRef]
- Parker, H.G.; Dreger, D.L.; Rimbault, M.; Davis, B.W.; Mullen, A.B.; Carpintero-Ramirez, G.; Ostrander, E.A. Genomic Analyses Reveal the Influence of Geographic Origin, Migration, and Hybridization on Modern Dog Breed Development. Cell Rep. 2017, 19, 697–708. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
{kind=link}
| Prevalence | h2SNP | SE | p-Value |
|---|---|---|---|
| 0.05 | 0.49 | 0.15 | 3.19 × 10−5 |
| 0.08 | 0.57 | 0.17 | 3.19 × 10−5 |
| 0.10 | 0.61 | 0.18 | 3.19 × 10−5 |
| CHR | Total SNPs | Significant SNPs | Min LD Region | Max LD Region | Total % h2SNP | Positional Coding Genes | OtherPositional Genes |
|---|---|---|---|---|---|---|---|
| 1 | 10 | 1 | 17039965 | 20022599 | 0.36% | 13 | 13 |
| 3 | 2 | 54214259 | 54884985 | 0.15% | 7 | 2 | |
| 6 | 21 | 8 | 75980913 | 77466517 | 0.66% | 5 | 10 |
| 11 | 15 | 13 | 23858342 | 25374425 | 0.64% | 8 | 6 |
| 12 | 6 | 0 | 28213995 | 29672357 | 0.03% | 3 | 3 |
| 13 | 2 | 0 | 11870489 | 12736069 | 0.01% | 2 | 0 |
| 15 | 2 | 0 | 57796133 | 58553452 | 0.05% | 3 | 1 |
| 17 | 17 | 0 | 39495267 | 42135498 | 0.45% | 16 | 10 |
| 20 | 10 | 6 | 6702324 | 8206842 | 0.28% | 13 | 2 |
| 26 | 10 | 39976489 | 41516852 | 0.20% | 45 | 7 | |
| 28 | 3 | 2 | 34428475 | 34905524 | 0.05% | 2 | 5 |
| 23 | 8 | 35367805 | 37688937 | 1.85% | 9 | 10 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Norton, E.M.; Minor, K.M.; Taylor, S.M.; McCue, M.E.; Mickelson, J.R. Heritability and Genomic Architecture of Episodic Exercise-Induced Collapse in Border Collies. Genes 2021, 12, 1927. https://doi.org/10.3390/genes12121927
Norton EM, Minor KM, Taylor SM, McCue ME, Mickelson JR. Heritability and Genomic Architecture of Episodic Exercise-Induced Collapse in Border Collies. Genes. 2021; 12(12):1927. https://doi.org/10.3390/genes12121927
Chicago/Turabian StyleNorton, Elaine M., Katie M. Minor, Susan M. Taylor, Molly E. McCue, and James R. Mickelson. 2021. "Heritability and Genomic Architecture of Episodic Exercise-Induced Collapse in Border Collies" Genes 12, no. 12: 1927. https://doi.org/10.3390/genes12121927
APA StyleNorton, E. M., Minor, K. M., Taylor, S. M., McCue, M. E., & Mickelson, J. R. (2021). Heritability and Genomic Architecture of Episodic Exercise-Induced Collapse in Border Collies. Genes, 12(12), 1927. https://doi.org/10.3390/genes12121927