
Genome-Wide Selective Signatures Reveal Candidate Genes Associated with Hair Follicle Development and Wool Shedding in Sheep

 , , ,
, , ,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Ethics Statement
2.2. Sample and Data Processing
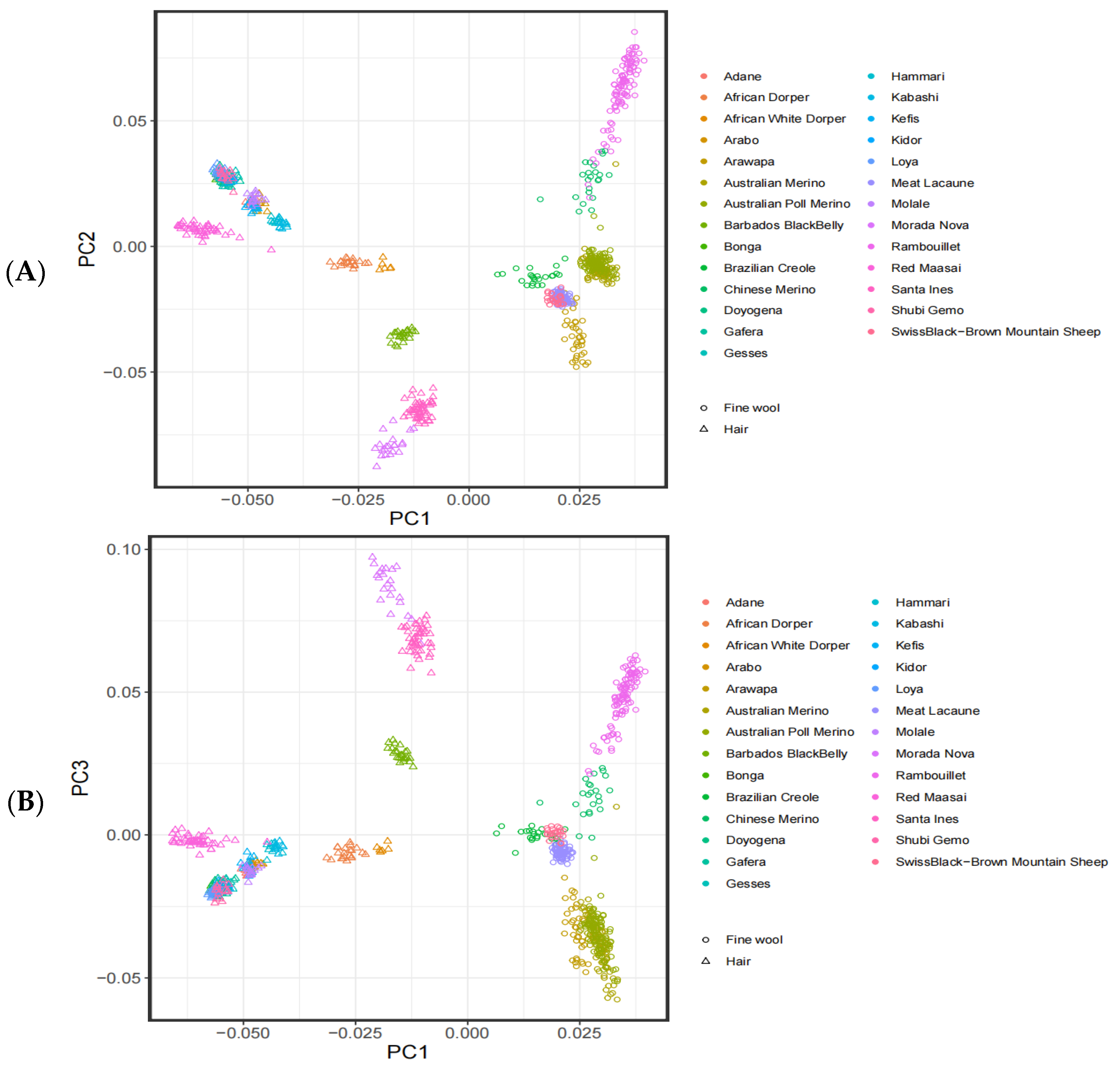
2.3. Population Structure
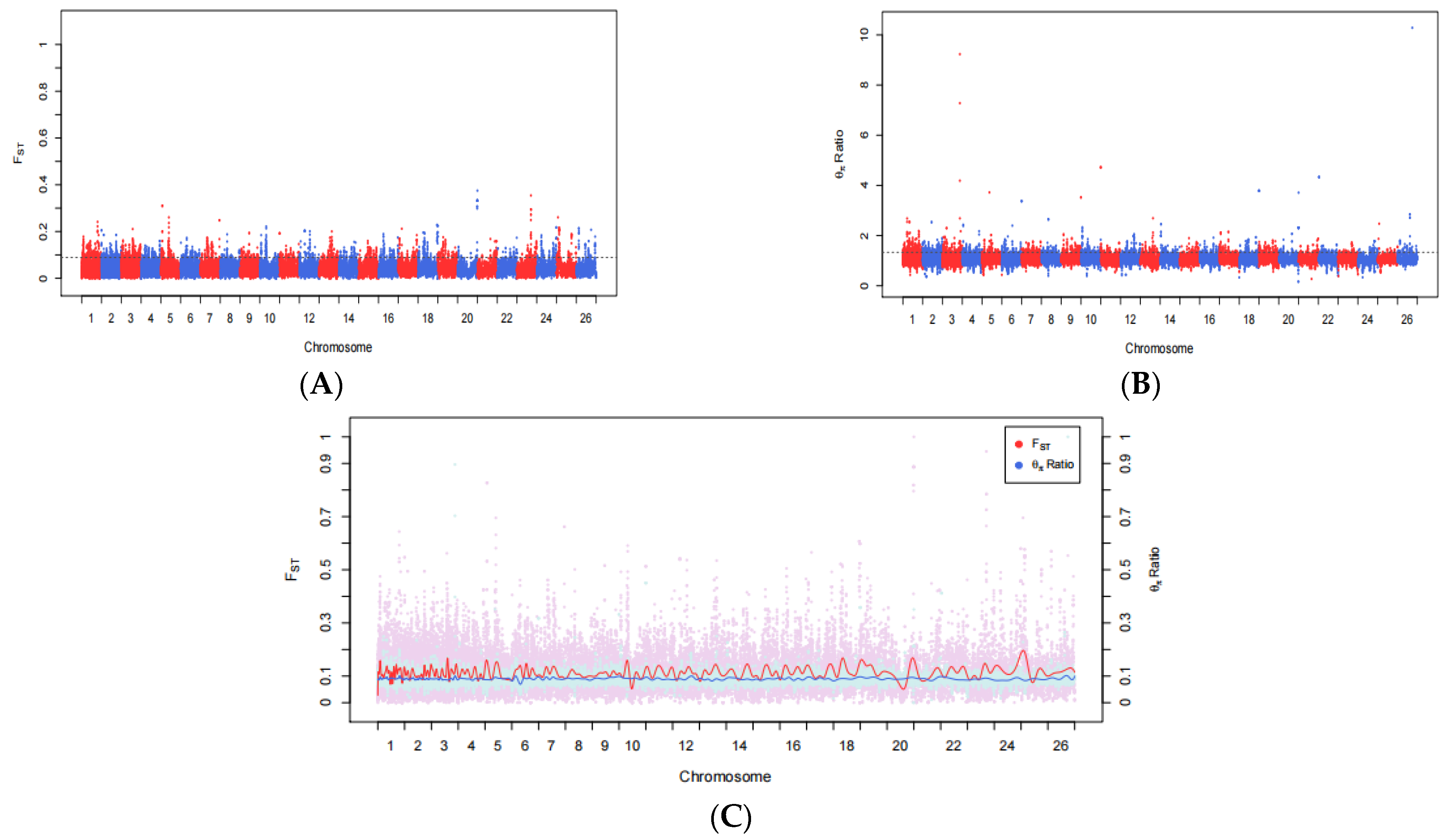
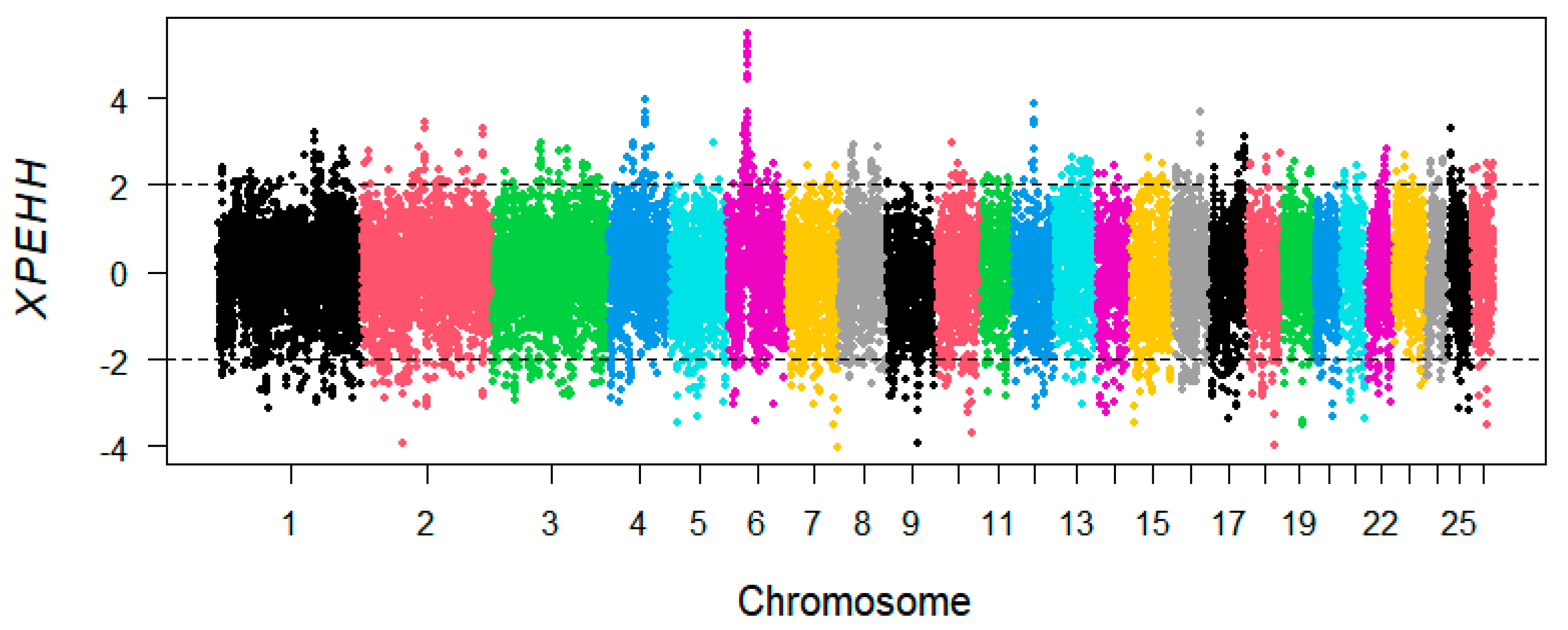
2.4. Selective Signatures
2.5. Candidate Gene Detection and Annotation
3. Results and Data Analyses
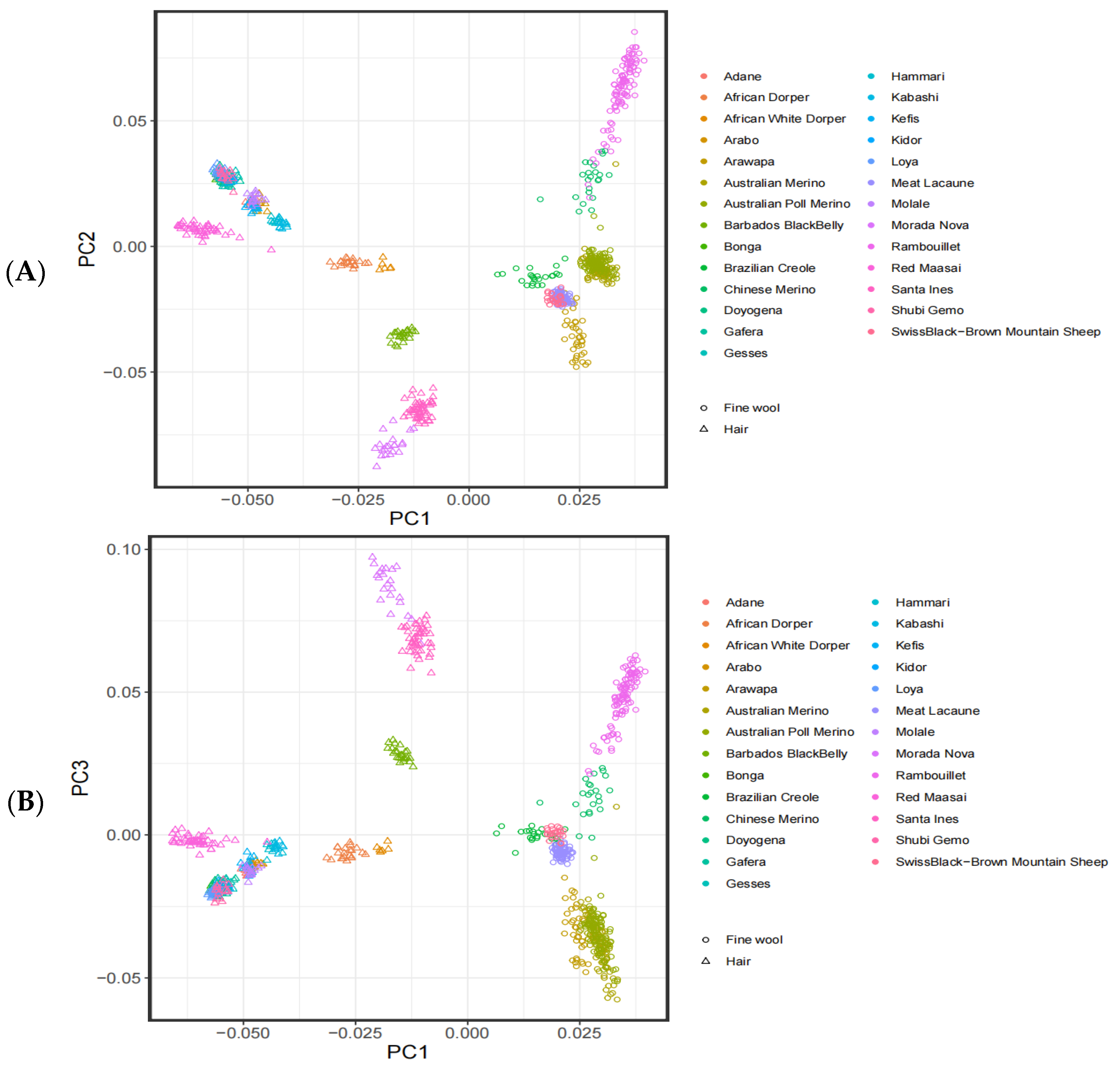
3.1. Analysis of the Population Structure
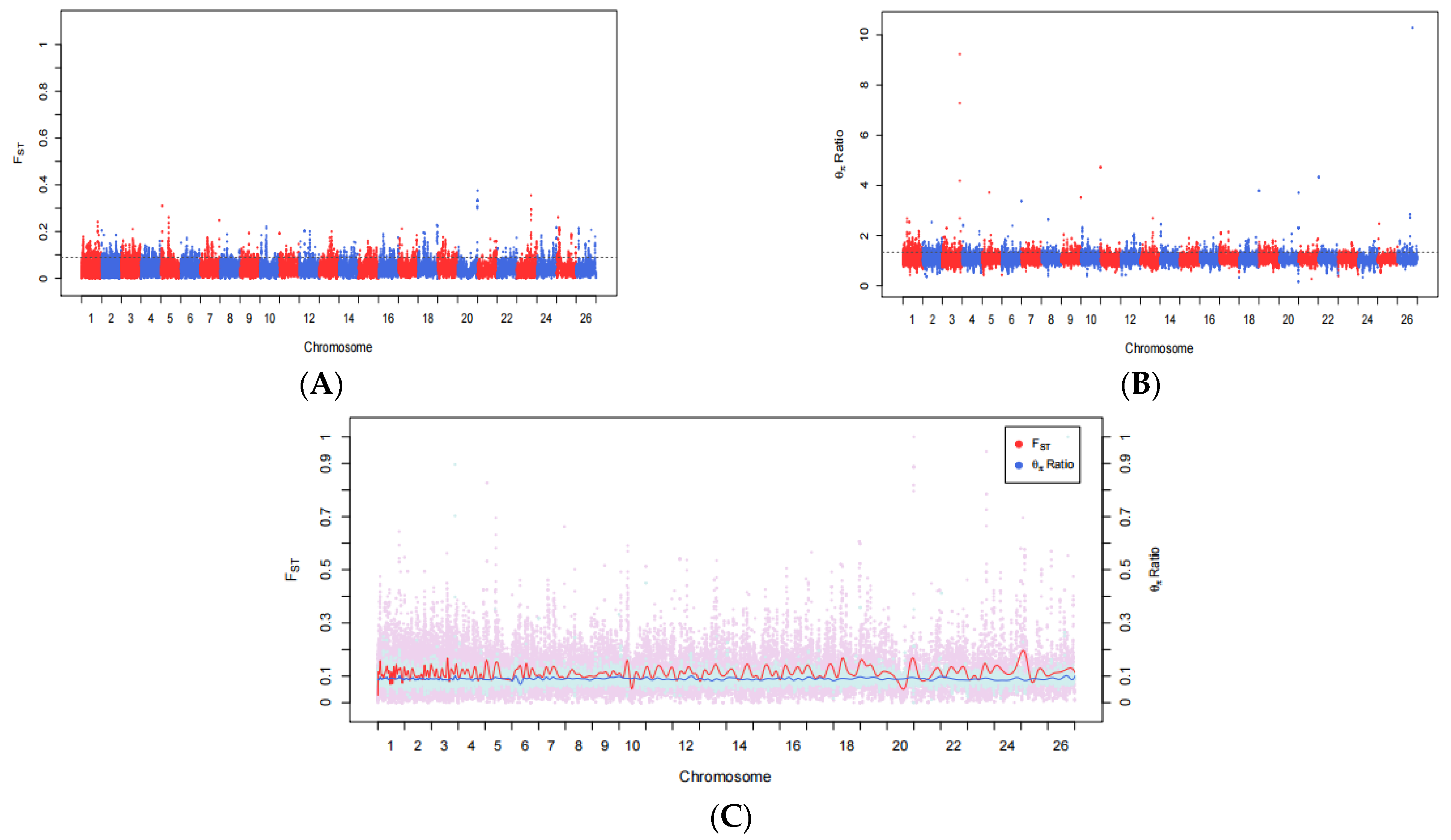
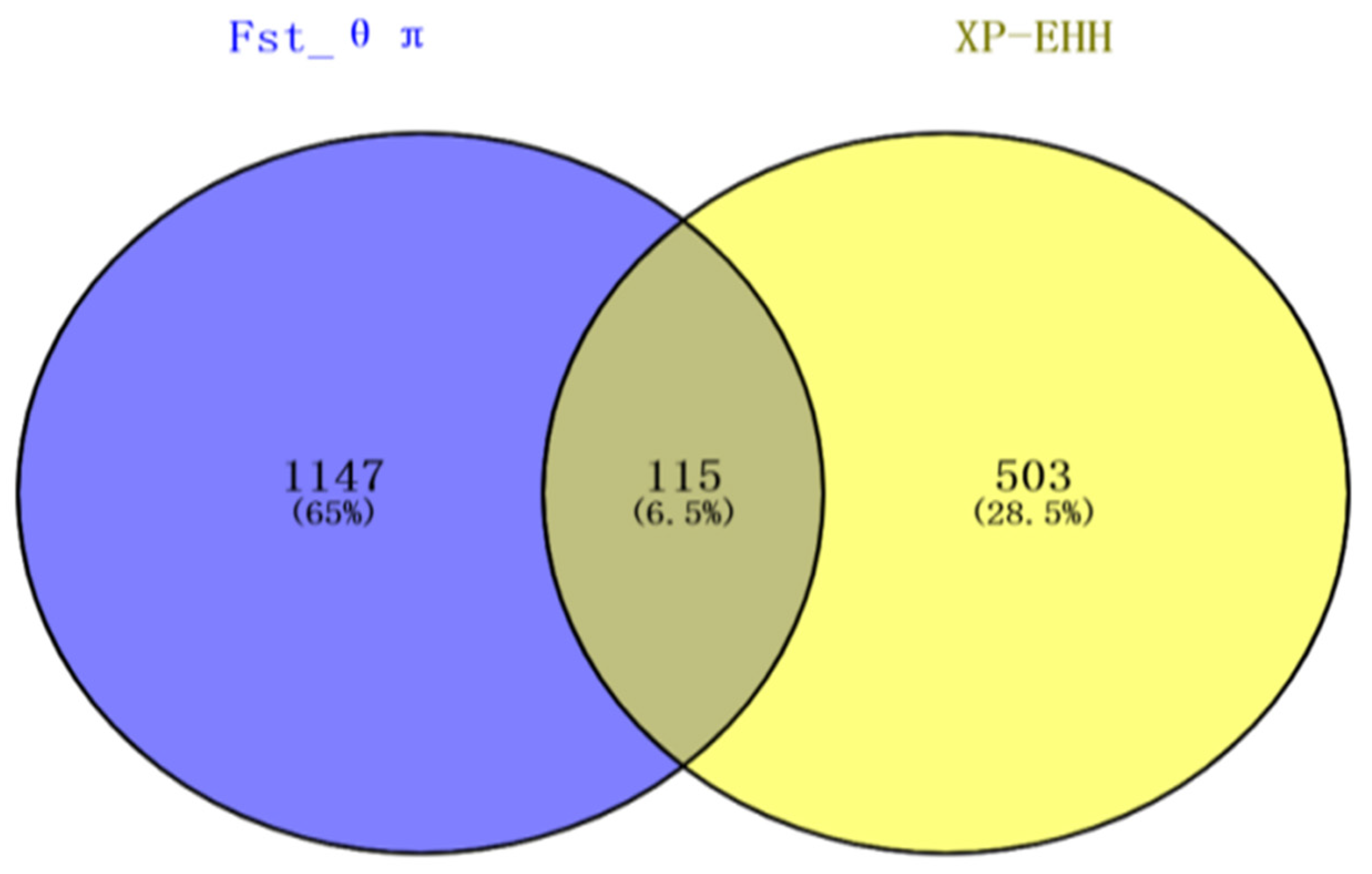
3.2. Fst and θπ Ratio
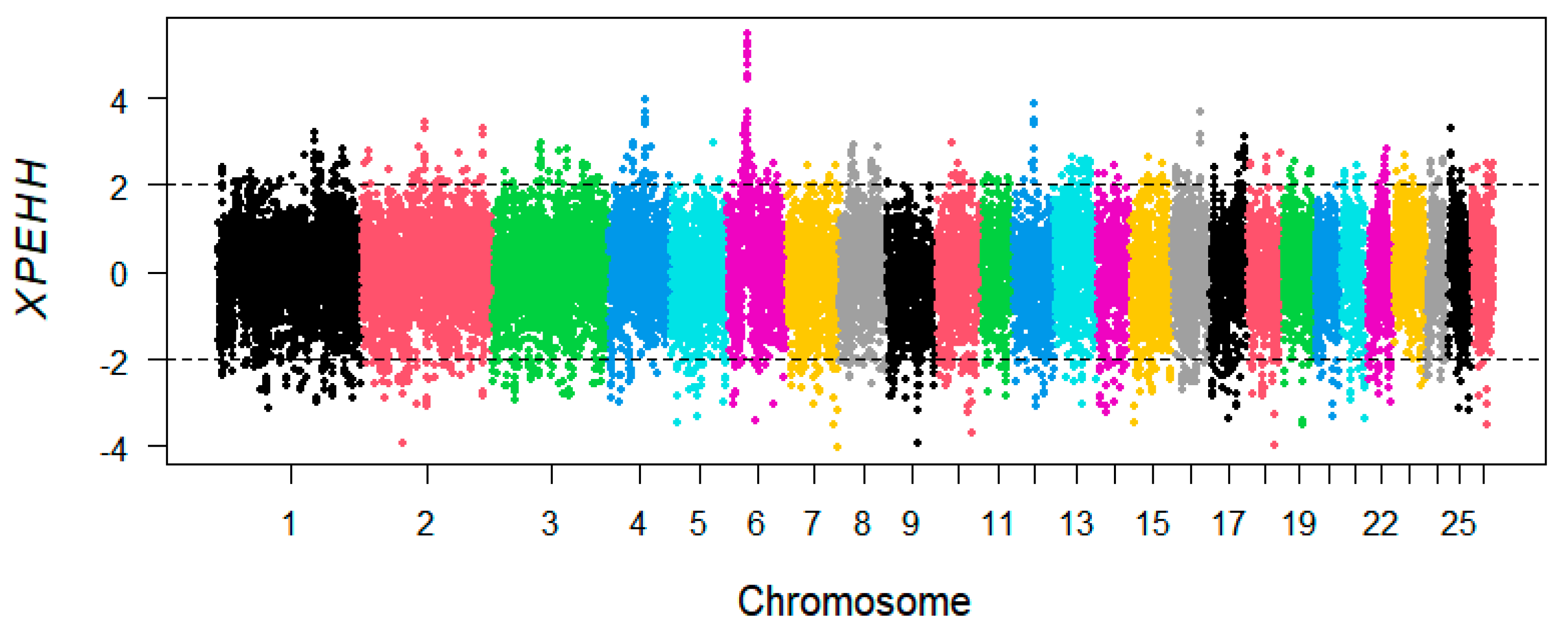
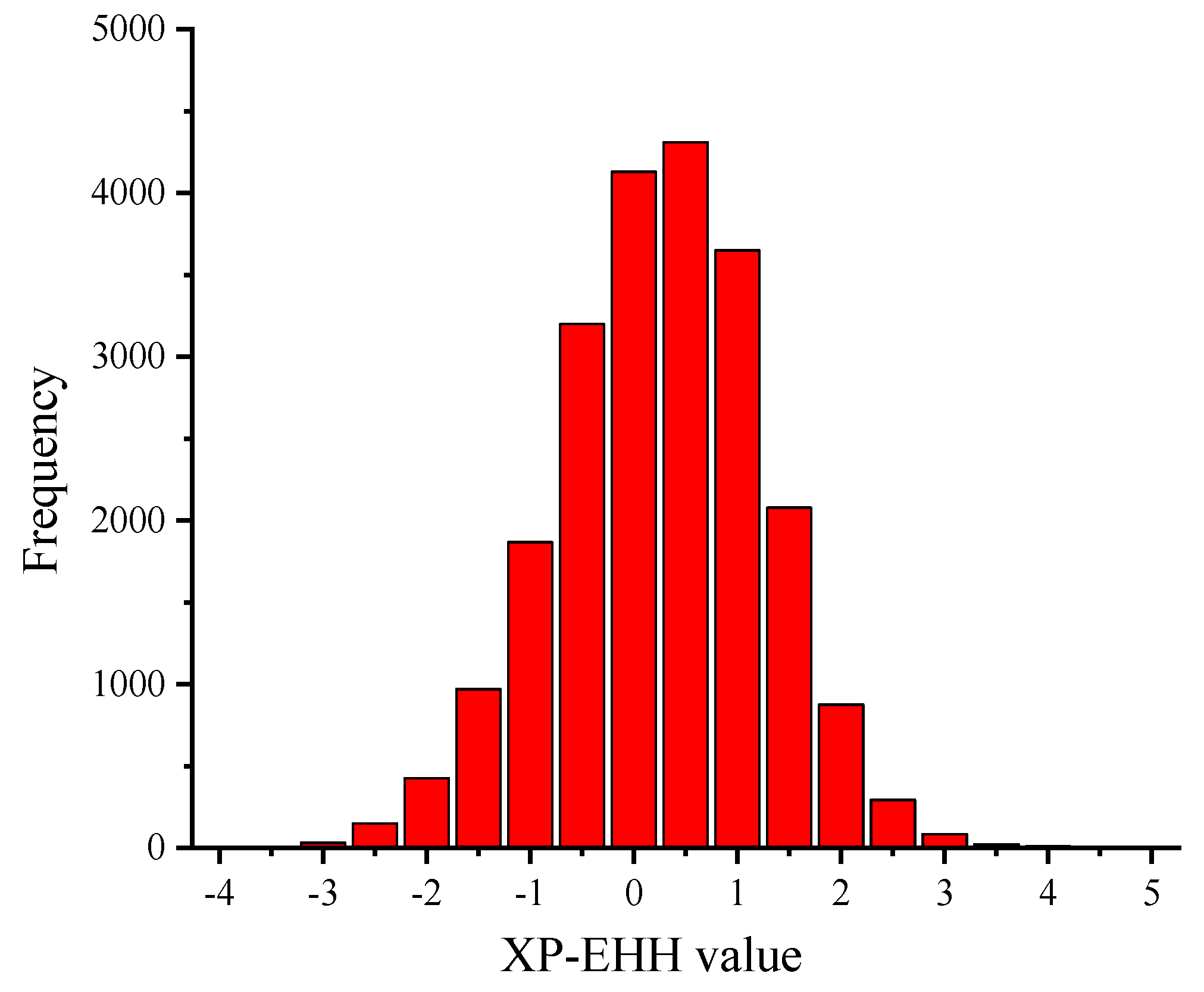
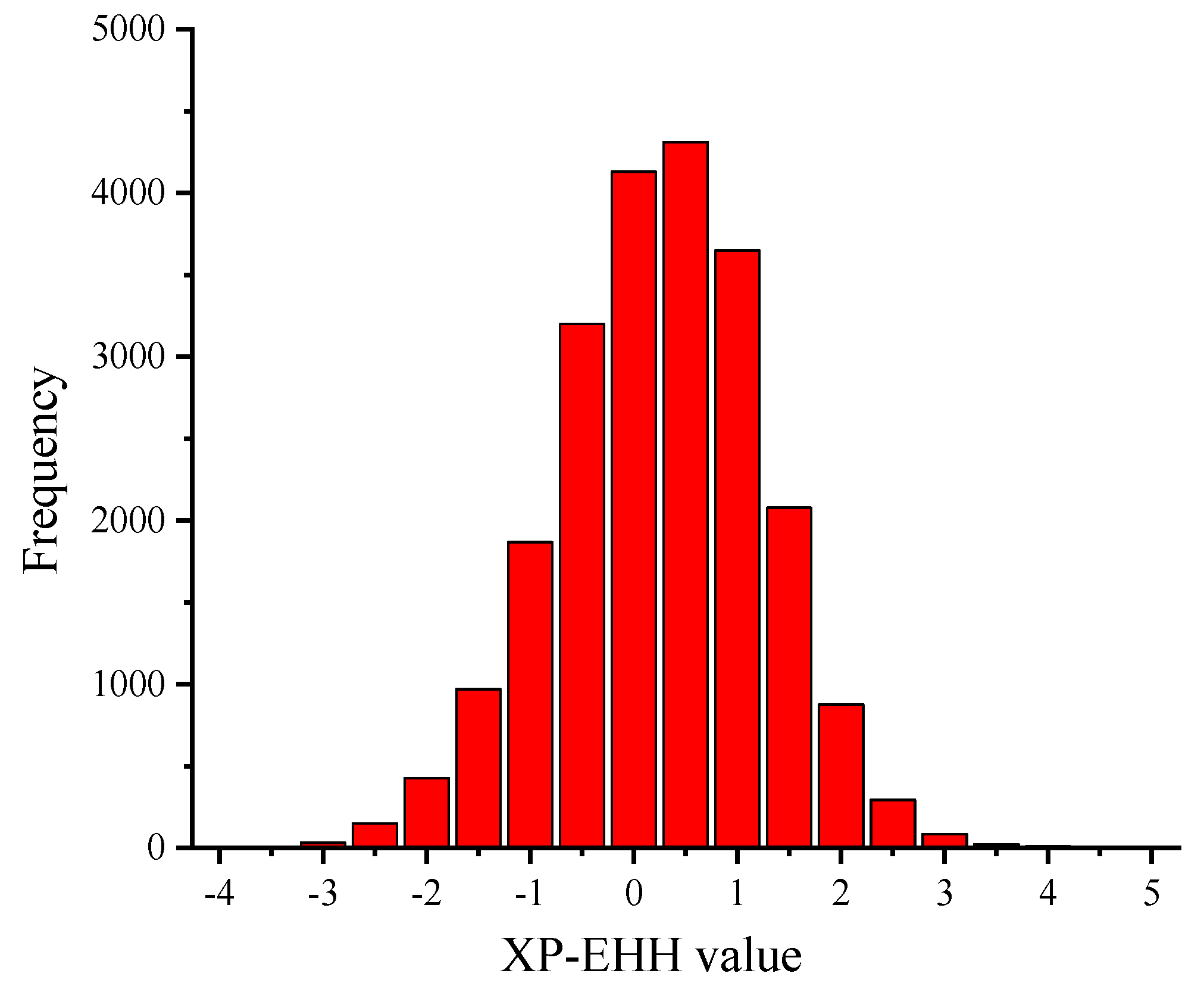
3.3. XP-EHH Analysis
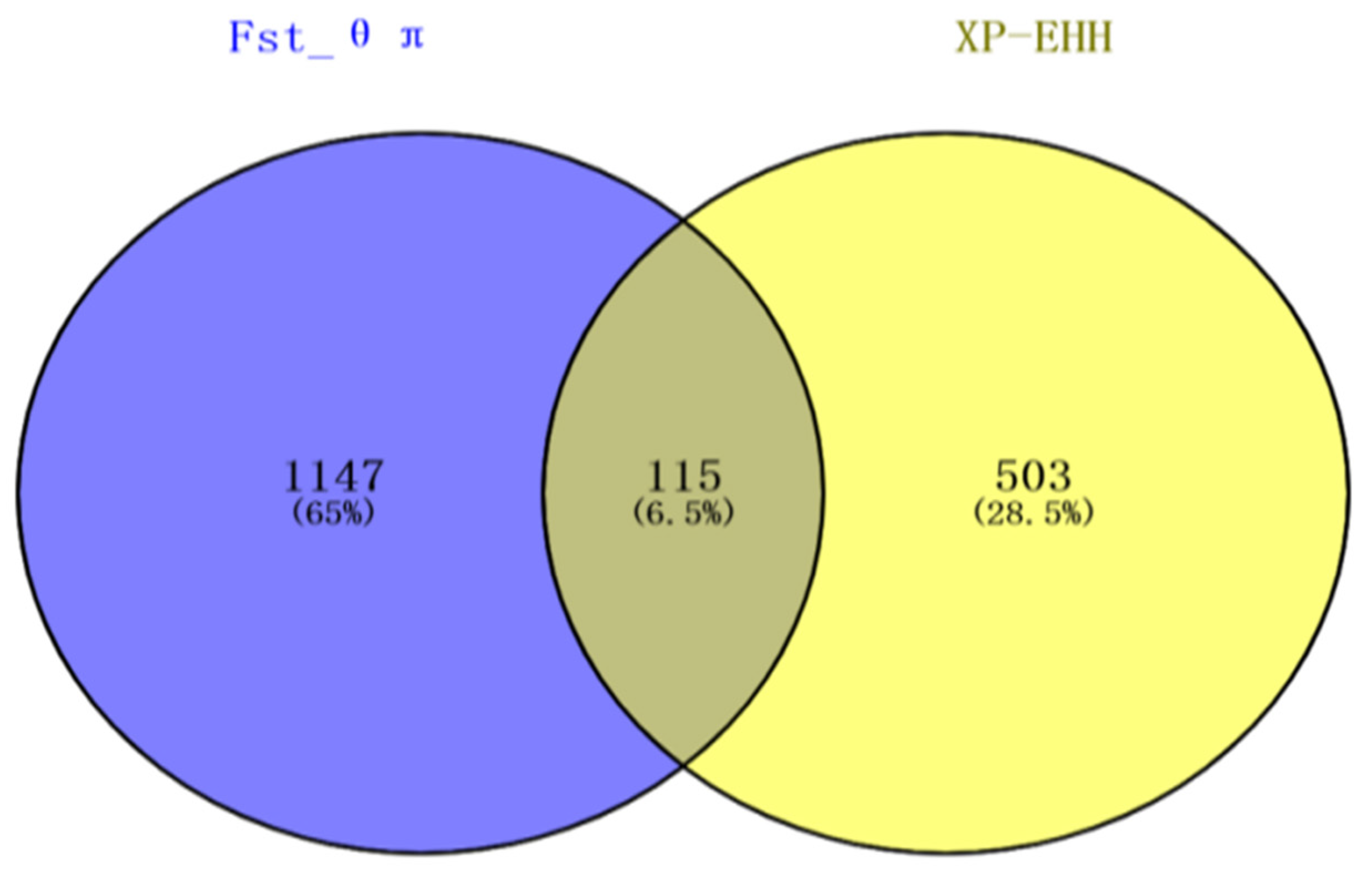
3.4. Gene Mapping and Functional Annotation
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Jurado, N.V.; Leymaster, K.A.; Kuehn, L.A. Genetic Analysis of Wool Shedding Scores of Ewes from a Composite Flock using a Threshold Model and Bayesian Methodologies. In Proceedings of the 10th World Congress on Genetics Applied to Livestock Production, Vancouver, BC, Canada, 17–22 August 2014. [Google Scholar]
- Pollott, G.E. A suggested mode of inheritance for wool shedding in sheep1. J. Anim. Sci. 2011, 89, 2316–2325. [Google Scholar] [CrossRef] [Green Version]
- Cloete, S.; Snyman, M.; Herselman, M. Productive performance of Dorper sheep. Small Rumin. Res. 2000, 36, 119–135. [Google Scholar] [CrossRef]
- Rose, G.; Mulder, H.; Thompson, A.; Van Der Werf, J.; Van Arendonk, J. Varying pasture growth and commodity prices change the value of traits in sheep breeding objectives. Agric. Syst. 2014, 131, 94–104. [Google Scholar] [CrossRef]
- Matika, O.; Bishop, S.C.; Pong-Wong, R.; Riggio, V.; Headon, D.J. Genetic factors controlling wool shedding in a composite Easycare sheep flock. Anim. Genet. 2013, 44, 742–749. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Combs, W. A History of the Barbados Blackbelly Sheep, 1st ed.; Westview Press: Boulder, CO, USA, 1983; pp. 179–197. [Google Scholar]
- Almeid, A. Barbados Blackbelly: The Caribbean ovine genetic resource. Trop. Anim. Health Prod. 2018, 50, 239–250. [Google Scholar] [CrossRef]
- Zhong, X.; Hao, S.; Zhe, Z.; Zhao, Q.B.; Olasege, B.S.; Li, Q.M.; Yue, Y. Genome-wide detection of selective signatures in a Jinhua pig population. J. Integr. Agric. 2020, 19, 1314–1322. [Google Scholar]
- Yang, F.-Y.; Guo, J.-J.; Liu, N.; Zhang, R.-Z. Genetic structure of the invasive Colorado potato beetle Leptinotarsa decemlineata populations in China. J. Integr. Agric. 2020, 19, 350–359. [Google Scholar] [CrossRef]
- Ji, Y.S.; Wang, C.Y.; Liu, R. Exploitation of freezing-tolerant genes in pea (Pisum sativum L.) based on selective sweeping analysis. China Veg. 2020, 3, 33–42. (In Chinese) [Google Scholar]
- Li, X.; Su, R.; Wan, W.; Zhang, W.; Jiang, H.; Qiao, X.; Fan, Y.; Zhang, Y.; Wang, R.; Liu, Z.; et al. Identification of selection signals by large-scale whole-genome resequencing of cashmere goats. Sci. Rep. 2017, 7, 15142. [Google Scholar] [CrossRef] [Green Version]
- Zhao, F.-P.; Wei, C.-H.; Zhang, L.; Liu, J.-S.; Wang, G.-K.; Zeng, T.; DU, L.-X. A genome scan of recent positive selection signatures in three sheep populations. J. Integr. Agric. 2016, 15, 162–174. [Google Scholar] [CrossRef]
- Yin, H.; Li, D.; Wang, Y.; Zhu, Q. Whole-genome resequencing analysis of Pengxian Yellow Chicken to identify genome-wide SNPs and signatures of selection. 3 Biotech 2019, 9, 383. [Google Scholar] [CrossRef]
- Zhang, Y.; Hu, Y.; Wang, X.; Jiang, Q.; Zhao, H.; Wang, J.; Ju, Z.; Yang, L.; Gao, Y.; Wei, X.; et al. Population Structure, and Selection Signatures Underlying High-Altitude Adaptation Inferred From Genome-Wide Copy Number Variations in Chinese Indigenous Cattle. Front. Genet. 2020, 10, 1404. [Google Scholar] [CrossRef] [PubMed]
- Purcell, S.; Neale, B.; Todd-Brown, K.; Thomas, L.; Ferreira, M.A.R.; Bender, D.; Maller, J.; Sklar, P.; de Bakker, P.I.W.; Daly, M.J.; et al. PLINK: A Tool Set for Whole-Genome Association and Population-Based Linkage Analyses. Am. J. Hum. Genet. 2007, 81, 559–575. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yuan, Z.H. Analysis of Selection Signatures Identify Genes Associated with Tail Type in Sheep. Anim. Genet. 2016, 48, 55–56. [Google Scholar] [CrossRef] [PubMed]
- Sankhyan, V.; Thakur, Y.P.; Katoch, S.; Dogra, P.K.; Thakur, R. Morphological structuring using principal component analysis of Rampur-Bushair sheep under transhumance production in western Himalayan region, India. Indian J. Anim. Res. 2017. [Google Scholar] [CrossRef] [Green Version]
- Wickham, H. Ggplot2: Elegant Graphics for Data Analysis; Springer Publishing Company, Incorporated: Berlin/Heidelberg, Germany, 2009. [Google Scholar]
- Danecek, P.; Auton, A.; Abecasis, G.; Albers, C.A.; Banks, E.; DePristo, M.A.; Handsaker, R.E.; Lunter, G.; Marth, G.T.; Sherry, S.T.; et al. The variant call format and VCFtools. Bioinformatics 2011, 27, 2156–2158. [Google Scholar] [CrossRef]
- Zeng, L.; Liu, H.-Q.; Tu, X.-L.; Ji, C.-M.; Gou, X.; Esmailizadeh, A.; Wang, S.; Wang, M.-S.; Wang, M.-C.; Li, X.-L.; et al. Genomes reveal selective sweeps in kiang and donkey for high-altitude adaptation. Zool. Res. 2021, 42, 450–460. [Google Scholar] [CrossRef]
- Wu, D.-D.; Yang, C.-P.; Wang, M.-S.; Dong, K.-Z.; Yan, D.-W.; Hao, Z.-Q.; Fan, S.-Q.; Chu, S.-Z.; Shen, Q.-S.; Jiang, L.-P.; et al. Convergent genomic signatures of high-altitude adaptation among domestic mammals. Natl. Sci. Rev. 2019, 7, 952–963. [Google Scholar] [CrossRef] [Green Version]
- Abied, A. Genome-wide Characterization of the Sudanese and Chinese Indigenous sheep populations by High Density SNP arrays. Ph.D. Thesis, Chinese Academy of Agricultural Sciences, Beijing, China, 2020. [Google Scholar]
- Gautier, M.; Klassmann, A.; Vitalis, R. REHH 2.0: A reimplementation of the R package REHH to detect positive selection from haplotype structure. Mol. Ecol. Resour. 2017, 17, 78–90. [Google Scholar] [CrossRef]
- Ye, R.; Tian, Y.; Huang, Y.; Zhang, Y.; Wang, J.; Sun, X.; Zhou, H.; Zhang, D.; Pan, W. Genome-wide Population Genetics Analysis of Plasmodium fallciparum Isolates from China-Myanmar border. Frontiers 2019. [Google Scholar] [CrossRef]
- Yan, J.; Zou, D.; Li, C.; Zhang, Z.; Song, S.; Wang, X. SR4R: An Integrative SNP Resource for Genomic Breeding and Population Research in Rice. Genom. Proteom. Bioinform. 2020, 18, 173–185. [Google Scholar] [CrossRef] [PubMed]
- Kiptoo, C.E.; Cheruiyot, B.R.; Oluoch, A.J. Signatures of Selection in Admixed Dairy Cattle in Tanzania. Front. Genet. 2018, 9, 607. [Google Scholar]
- Jolliffe, I.T.; Cadima, J. Principal component analysis: A review and recent developments. Philos. Trans. R. Soc. A Math. Phys. Eng. Sci. 2016, 374, 20150202. [Google Scholar] [CrossRef]
- Manel, S.; Gaggiotti, O.; Waples, R.S. Assignment methods: Matching biological questions with appropriate techniques. Trends Ecol. Evol. 2005, 20, 136–142. [Google Scholar] [CrossRef] [Green Version]
- Moradi, M.H.; Khaltabadi-Farahani, A.H.; Khodaei-Motlagh, M.; Kazemi-Bonchenari, M.; McEwan, J. Genome-wide selection of discriminant SNP markers for breed assignment in indigenous sheep breeds. Ann. Anim. Sci. 2021, 21. [Google Scholar] [CrossRef]
- Wright, S. The Genetical Structure of Populations. Ann. Eugenic. 1950, 15, 323–354. [Google Scholar] [CrossRef]
- Manzari, Z.; Mehrabani, H.; Nejati, A.; Moradi, H.; Gholizadeh, M. Detecting selection signatures in three Iranian sheep breeds. Anim. Genet. 2019, 50, 298–302. [Google Scholar] [CrossRef]
- Ma, Y.; Ding, X.; Qanbari, S.; Weigend, S.; Zhang, Q.; Simianer, H. Properties of different selection signature statistics and a new strategy for combining them. Heredity 2015, 115, 426–436. [Google Scholar] [CrossRef] [Green Version]
- Lee, S.-C.; Na, Y.-P.; Lee, J.-B. Expression of peroxiredoxin II in vascular tumors of the skin: A novel vascular marker of endothelial cells. J. Am. Acad. Dermatol. 2003, 49, 487–491. [Google Scholar] [CrossRef]
- Lee, J.; Yun, S.; Chae, H.; Won, Y.; Kim, Y.; Lee, S.-C. Expression of peroxiredoxin and thioredoxin in dermatological disorders. Br. J. Dermatol. 2002, 146, 710–712. [Google Scholar] [CrossRef]
- Lee, J.E.; Kwon, B.D.; Lee, J.-B.; Won, Y.-H.; Kim, Y.P.; Lee, S.-C.; Chae, H.Z.; Ahn, K.Y. Peroxiredoxin is Ubiquitously Expressed in Rat Skin: Isotype-Specific Expression in the Epidermis and Hair Follicle. J. Investig. Dermatol. 2000, 115, 1108–1114. [Google Scholar] [CrossRef] [Green Version]
- DeRouen, M.C.; Zhen, H.; Tan, S.H.; Williams, S.; Marinkovich, M.P.; Oro, A.E. Laminin-511 and integrin beta-1 in hair follicle development and basal cell carcinoma formation. BMC Dev. Biol. 2010, 10, 112. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Valenzuela, I.; Fernadez, P.; Plaja, A. Further delineation of the SOX18-related Hypotrichosis, Lymphedema, Telangiectasia syndrome (HTLS). Eur. J. Med. Genet. 2018, 3, 269–272. [Google Scholar] [CrossRef] [PubMed]
- Duong, T.; Koltowska, K.; Pichol-Thievend, C.; Le Guen, L.; Fontaine, F.; Smith, K.A.; Truong, V.; Skoczylas, R.; Stacker, S.A.; Achen, M.G.; et al. VEGFD regulates blood vascular development by modulating SOX18 activity. Blood 2014, 123, 1102–1112. [Google Scholar] [CrossRef] [Green Version]
- Francois, M.; Caprini, A.; Hosking, B.; Orsenigo, F.; Wilhelm, D.; Browne, C.; Paavonen, K.; Karnezis, T.; Shayan, R.; Downes, M.; et al. Sox18 induces development of the lymphatic vasculature in mice. Nature 2008, 456, 643–647. [Google Scholar] [CrossRef] [PubMed]
- Pennisi, D.; Gardner, J.; Chambers, D.; Hosking, B.; Peters, J.; Muscat, G.; Abbott, C.; Koopman, P. Mutations in Sox18 underlie cardiovascular and hair follicle defects in ragged mice. Nat. Genet. 2000, 24, 434–437. [Google Scholar] [CrossRef] [PubMed]
- Zhu, F.H.; Shi, J.Q. Research progress of transcription factor SOX18. J. Jining Med. Univ. 2016, 39, 280–282. (In Chinese) [Google Scholar]
- Olsson, J.; Kamachi, Y.; Penning, S.; Muscat, G.; Kondoh, H.; Koopman, P. Sox18 expression in blood vessels and feather buds during chicken embryogenesis. Gene 2001, 271, 151–158. [Google Scholar] [CrossRef]
- Hosking, B.M.; Wang, S.-C.; Chen, S.L.; Penning, S.; Koopman, P.; Muscat, G.E. SOX18 Directly Interacts with MEF2C in Endothelial Cells. Biochem. Biophys. Res. Commun. 2001, 287, 493–500. [Google Scholar] [CrossRef]
- Legrand, J.M.; Roy, E.; Ellis, J.J.; Francois, M.; Brooks, A.J.; Khosrotehrani, K. STAT5 Activation in the Dermal Papilla Is Important for Hair Follicle Growth Phase Induction. J. Investig. Dermatol. 2016, 136, 1781–1791. [Google Scholar] [CrossRef] [Green Version]
- Yamane, A.; Fukui, M.; Sugimura, Y.; Itoh, M.; Alea, M.P.; Thomas, V.; El Alaoui, S.; Akiyama, M.; Hitomi, K. Identification of a preferred substrate peptide for transglutaminase 3 and detection of in situ activity in skin and hair follicles. FEBS J. 2010, 277, 3564–3574. [Google Scholar] [CrossRef] [PubMed]
- Schmuth, M.; Gruber, R.; Elias, P.M.; Williams, M.L. Ichthyosis Update: Towards a Function-Driven Model of Pathogenesis of the Disorders of Cornification and the Role of Corneocyte Proteins in These Disorders. Adv. Dermatol. 2007, 23, 231–256. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- John, S.; Thiebach, L.; Frie, C. Epidermal transglutaminase (TGase 3) is required for proper hair development, but not the formation of the epidermal barrier. PLoS ONE 2017, 7, e34252. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zheng, X.; Ni, Y.M.; Xie, S.J. The characteristics of hair follicle stem cells and their application in skin repair. Tradit. Chin. Med. 2015, 27, 73–75. [Google Scholar]
- Nguyen, H.; Rendl, M.; Fuchs, E. Tcf3 Governs Stem Cell Features and Represses Cell Fate Determination in Skin. Cell 2006, 127, 171–183. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dasgupta, R.; Fuchs, E. Multiple roles for activated LEF/TCF transcription complexes during hair follicle development and differentiation. Development 1999, 126, 4557–4568. [Google Scholar] [CrossRef]
- Lien, W.-H.; Polak, L.; Lin, M.; Lay, K.; Zheng, D.; Fuchs, E. In Vivo transcriptional governance of hair follicle stem cells by canonical Wnt regulators. Nature 2014, 16, 179–190. [Google Scholar] [CrossRef] [Green Version]
- Nguyen, H.; Merrill, B.J.; Polak, L.; Nikolova, M.; Rendl, M.; Shaver, T.M.; Pasolli, H.A.; Fuchs, E. Tcf3 and Tcf4 are essential for long-term homeostasis of skin epithelia. Nat. Genet. 2009, 41, 1068–1075. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, C.I.; Hoffman, J.A.; Shy, B.R. Function of Wnt/β-catenin in counteracting Tcf3 repression through the Tcf3–β-catenin interaction. Development 2012, 139, 2118–2129. [Google Scholar] [CrossRef] [Green Version]
- Luo, M.; Li, J.-F.; Yang, Q.; Zhang, K.; Wang, Z.-W.; Zheng, S.; Zhou, J.-J. Stem cell quiescence and its clinical relevance. World J. Stem Cells 2020, 12, 1307–1326. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Breed | Abbreviation | Samples Size | Wool Type |
|---|---|---|---|
| Arawapa | APA | 37 | fine-wool sheep |
| Australian Poll Merino | APM | 98 | fine-wool sheep |
| Brazilian Creole | BCS | 23 | fine-wool sheep |
| Chinese Merino | CME | 23 | fine-wool sheep |
| Meat Lacaune | LAC | 75 | fine-wool sheep |
| Australian Merino | MER | 88 | fine-wool sheep |
| Rambouillet | RMB | 102 | fine-wool sheep |
| SwissBlack-Brown Mountain Sheep | SBS | 24 | fine-wool sheep |
| African Dorper | ADP | 21 | hair sheep |
| Adane | AKD | 12 | hair sheep |
| Arabo | AKR | 10 | hair sheep |
| African White Dorper | AWD | 6 | hair sheep |
| Barbados Black Belly | BBB | 24 | hair sheep |
| Morada Nova | BMN | 22 | hair sheep |
| Bonga | BQ | 9 | hair sheep |
| Santa Ines | BSI | 47 | hair sheep |
| Doyogena | DA/DH | 15 | hair sheep |
| Kefis | FKD | 13 | hair sheep |
| Gesses | GGD | 11 | hair sheep |
| Hammari | H | 11 | hair sheep |
| Kabashi | K | 9 | hair sheep |
| Kidor | KO | 10 | hair sheep |
| Loya | LA | 15 | hair sheep |
| Molale | MZ | 15 | hair sheep |
| Red Maasai | RMA | 45 | hair sheep |
| Shubi Gemo | SHG | 15 | hair sheep |
| Gafera | WA | 15 | hair sheep |
| Chromosome 1 | Position (bp) 2 | REF 3 | ALT 4 | Gene Name 5 | Distance(bp) 6 |
|---|---|---|---|---|---|
| 5 | 41339449 | G | T | TCF3 | 2996 |
| 13 | 52044015 | C | T | TGM3 | 825 |
| 13 | 52055150 | G | A | TGM3 | within |
| 13 | 53089456 | C | T | SOX18 | 26,609 |
| 14 | 48786396 | G | A | PRX | within |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lei, Z.; Sun, W.; Guo, T.; Li, J.; Zhu, S.; Lu, Z.; Qiao, G.; Han, M.; Zhao, H.; Yang, B.; et al. Genome-Wide Selective Signatures Reveal Candidate Genes Associated with Hair Follicle Development and Wool Shedding in Sheep. Genes 2021, 12, 1924. https://doi.org/10.3390/genes12121924
Lei Z, Sun W, Guo T, Li J, Zhu S, Lu Z, Qiao G, Han M, Zhao H, Yang B, et al. Genome-Wide Selective Signatures Reveal Candidate Genes Associated with Hair Follicle Development and Wool Shedding in Sheep. Genes. 2021; 12(12):1924. https://doi.org/10.3390/genes12121924
Chicago/Turabian StyleLei, Zhihui, Weibo Sun, Tingting Guo, Jianye Li, Shaohua Zhu, Zengkui Lu, Guoyan Qiao, Mei Han, Hongchang Zhao, Bohui Yang, and et al. 2021. "Genome-Wide Selective Signatures Reveal Candidate Genes Associated with Hair Follicle Development and Wool Shedding in Sheep" Genes 12, no. 12: 1924. https://doi.org/10.3390/genes12121924
APA StyleLei, Z., Sun, W., Guo, T., Li, J., Zhu, S., Lu, Z., Qiao, G., Han, M., Zhao, H., Yang, B., Zhang, L., Liu, J., Yuan, C., & Yue, Y. (2021). Genome-Wide Selective Signatures Reveal Candidate Genes Associated with Hair Follicle Development and Wool Shedding in Sheep. Genes, 12(12), 1924. https://doi.org/10.3390/genes12121924