
Epithelial–Mesenchymal Transition Signaling and Prostate Cancer Stem Cells: Emerging Biomarkers and Opportunities for Precision Therapeutics

 ,
,  , and
, and
Abstract
:1. Prostate Cancer Progression
2. Epithelial to Mesenchymal Transition (EMT) and Cancer Progression
3. Mouse Model and In Vitro Studies of EMT in PCa
4. Transcriptional Signatures and EMT Pathways of Progression
5. Epigenomic Regulation of EMT and Lineage Plasticity in Prostate Cancer
6. Noncoding RNA Regulation of EMT and Lineage Plasticity
7. Clinical Trials
8. Future Directions and Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Conflicts of Interest
References
- Bray, F.; Ferlay, J.; Soerjomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. Global Cancer Statistics 2018: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J. Clin. 2018, 68, 394–424. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer Statistics, 2020. CA Cancer J. Clin. 2020, 70, 7–30. [Google Scholar] [CrossRef] [PubMed]
- Rebello, R.J.; Oing, C.; Knudsen, K.E.; Loeb, S.; Johnson, D.C.; Reiter, R.E.; Gillessen, S.; Van der Kwast, T.; Bristow, R.G. Prostate Cancer. Nat. Rev. Dis. Primers 2021, 7, 9. [Google Scholar] [CrossRef]
- Mottet, N.; van den Bergh, R.C.N.; Briers, E.; Van den Broeck, T.; Cumberbatch, M.G.; De Santis, M.; Fanti, S.; Fossati, N.; Gandaglia, G.; Gillessen, S.; et al. EAU-EANM-ESTRO-ESUR-SIOG Guidelines on Prostate Cancer-2020 Update. Part 1: Screening, Diagnosis, and Local Treatment with Curative Intent. Eur. Urol. 2021, 79, 243–262. [Google Scholar] [CrossRef] [PubMed]
- Teo, M.Y.; Rathkopf, D.E.; Kantoff, P. Treatment of Advanced Prostate Cancer. Annu. Rev. Med. 2019, 70, 479–499. [Google Scholar] [CrossRef]
- Abeshouse, A.; Ahn, J.; Akbani, R.; Ally, A.; Amin, S.; Andry, C.D.; Annala, M.; Aprikian, A.; Armenia, J.; Arora, A.; et al. The Molecular Taxonomy of Primary Prostate Cancer. Cell 2015, 163, 1011–1025. [Google Scholar] [CrossRef] [Green Version]
- Armenia, J.; Wankowicz, S.A.M.; Liu, D.; Gao, J.; Kundra, R.; Reznik, E.; Chatila, W.K.; Chakravarty, D.; Han, G.C.; Coleman, I.; et al. The Long Tail of Oncogenic Drivers in Prostate Cancer. Nat. Genet. 2018, 50, 645–651. [Google Scholar] [CrossRef]
- Robinson, D.; Van Allen, E.M.; Wu, Y.-M.; Schultz, N.; Lonigro, R.J.; Mosquera, J.-M.; Montgomery, B.; Taplin, M.-E.; Pritchard, C.C.; Attard, G.; et al. Integrative Clinical Genomics of Advanced Prostate Cancer. Cell 2015, 161, 1215–1228. [Google Scholar] [CrossRef] [Green Version]
- Scaravilli, M.; Koivukoski, S.; Latonen, L. Androgen-Driven Fusion Genes and Chimeric Transcripts in Prostate Cancer. Front. Cell Dev. Biol. 2021, 9, 623809. [Google Scholar] [CrossRef]
- Angelergues, A.; Maillet, D.; Flechon, A.; Ozguroglu, M.; Mercier, F.; Guillot, A.; Le Moulec, S.; Gravis, G.; Beuzeboc, P.; Massard, C.; et al. Duration of Response to Androgen-Deprivation Therapy (ADT) and Efficacy of Secondary Hormone Therapy, Docetaxel (D), and Cabazitaxel (C) in Metastatic Castration-Resistant Prostate Cancer (MCRPC). JCO 2014, 32, 282. [Google Scholar] [CrossRef]
- Bluemn, E.G.; Coleman, I.M.; Lucas, J.M.; Coleman, R.T.; Hernandez-Lopez, S.; Tharakan, R.; Bianchi-Frias, D.; Dumpit, R.F.; Kaipainen, A.; Corella, A.N.; et al. Androgen Receptor Pathway-Independent Prostate Cancer Is Sustained through FGF Signaling. Cancer Cell 2017, 32, 474–489. [Google Scholar] [CrossRef] [Green Version]
- Hankey, W.; Chen, Z.; Wang, Q. Shaping Chromatin States in Prostate Cancer by Pioneer Transcription Factors. Cancer Res. 2020, 80, 2427–2436. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hanahan, D.; Weinberg, R.A. Hallmarks of Cancer: The Next Generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [Green Version]
- Melo, C.M.; Vidotto, T.; Chaves, L.P.; Lautert-Dutra, W.; Reis, R.B.d.; Squire, J.A. The Role of Somatic Mutations on the Immune Response of the Tumor Microenvironment in Prostate Cancer. Int. J. Mol. Sci. 2021, 22, 9550. [Google Scholar] [CrossRef] [PubMed]
- Kalluri, R.; Weinberg, R.A. The Basics of Epithelial-Mesenchymal Transition. J. Clin. Investig. 2009, 119, 1420–1428. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nieto, M.A.; Huang, R.Y.-J.; Jackson, R.A.; Thiery, J.P. EMT: 2016. Cell 2016, 166, 21–45. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Craene, B.D.; Berx, G. Regulatory Networks Defining EMT during Cancer Initiation and Progression. Nat. Rev. Cancer 2013, 13, 97–110. [Google Scholar] [CrossRef]
- Thiery, J.P.; Acloque, H.; Huang, R.Y.J.; Nieto, M.A. Epithelial-Mesenchymal Transitions in Development and Disease. Cell 2009, 139, 871–890. [Google Scholar] [CrossRef]
- Stemmler, M.P.; Eccles, R.L.; Brabletz, S.; Brabletz, T. Non-Redundant Functions of EMT Transcription Factors. Nat. Cell Biol. 2019, 21, 102–112. [Google Scholar] [CrossRef]
- Armstrong, A.J.; Healy, P.; Halabi, S.; Vollmer, R.; Lark, A.; Kemeny, G.; Ware, K.; Freedland, S.J. Evaluation of an Epithelial Plasticity Biomarker Panel in Men with Localized Prostate Cancer. Prostate Cancer Prostatic Dis. 2016, 19, 40–45. [Google Scholar] [CrossRef]
- Gravdal, K.; Halvorsen, O.J.; Haukaas, S.A.; Akslen, L.A. A Switch from E-Cadherin to N-Cadherin Expression Indicates Epithelial to Mesenchymal Transition and Is of Strong and Independent Importance for the Progress of Prostate Cancer. Clin. Cancer Res. 2007, 13, 7003–7011. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kwon, J.T.W.; Bryant, R.J.; Parkes, E.E. The Tumor Microenvironment and Immune Responses in Prostate Cancer Patients. Endocr. Relat. Cancer 2021, 28, T95–T107. [Google Scholar] [CrossRef] [PubMed]
- Pastushenko, I.; Blanpain, C. EMT Transition States during Tumor Progression and Metastasis. Trends Cell Biol. 2019, 29, 212–226. [Google Scholar] [CrossRef] [Green Version]
- Lambert, A.W.; Weinberg, R.A. Linking EMT Programmes to Normal and Neoplastic Epithelial Stem Cells. Nat. Rev. Cancer 2021, 21, 325–338. [Google Scholar] [CrossRef]
- Williams, E.D.; Gao, D.; Redfern, A.; Thompson, E.W. Controversies around Epithelial–Mesenchymal Plasticity in Cancer Metastasis. Nat. Rev. Cancer 2019, 19, 716–732. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Batlle, E.; Clevers, H. Cancer Stem Cells Revisited. Nat. Med. 2017, 23, 1124–1134. [Google Scholar] [CrossRef]
- Shibue, T.; Weinberg, R.A. EMT, CSCs, and Drug Resistance: The Mechanistic Link and Clinical Implications. Nat. Rev. Clin. Oncol. 2017, 14, 611–629. [Google Scholar] [CrossRef] [Green Version]
- Kahounová, Z.; Remšík, J.; Fedr, R.; Bouchal, J.; Mičková, A.; Slabáková, E.; Binó, L.; Hampl, A.; Souček, K. Slug-Expressing Mouse Prostate Epithelial Cells Have Increased Stem Cell Potential. Stem Cell Res. 2020, 46, 101844. [Google Scholar] [CrossRef]
- Patrawala, L.; Calhoun, T.; Schneider-Broussard, R.; Li, H.; Bhatia, B.; Tang, S.; Reilly, J.G.; Chandra, D.; Zhou, J.; Claypool, K.; et al. Highly Purified CD44+ Prostate Cancer Cells from Xenograft Human Tumors Are Enriched in Tumorigenic and Metastatic Progenitor Cells. Oncogene 2006, 25, 1696–1708. [Google Scholar] [CrossRef] [Green Version]
- Maitland, N.J.; Frame, F.M.; Polson, E.S.; Lewis, J.L.; Collins, A.T. Prostate Cancer Stem Cells: Do They Have a Basal or Luminal Phenotype? Horm. Cancer 2011, 2, 47–61. [Google Scholar] [CrossRef] [Green Version]
- Drost, J.; Karthaus, W.R.; Gao, D.; Driehuis, E.; Sawyers, C.L.; Chen, Y.; Clevers, H. Organoid Culture Systems for Prostate Epithelial Tissue and Prostate Cancer Tissue. Nat. Protoc. 2016, 11, 347–358. [Google Scholar] [CrossRef] [Green Version]
- Mei, W.; Lin, X.; Kapoor, A.; Gu, Y.; Zhao, K.; Tang, D. The Contributions of Prostate Cancer Stem Cells in Prostate Cancer Initiation and Metastasis. Cancers 2019, 11, 434. [Google Scholar] [CrossRef] [Green Version]
- Robinton, D.A.; Daley, G.Q. The Promise of Induced Pluripotent Stem Cells in Research and Therapy. Nature 2012, 481, 295–305. [Google Scholar] [CrossRef] [PubMed]
- Kleiman, R.J.; Engle, S.J. Human Inducible Pluripotent Stem Cells: Realization of Initial Promise in Drug Discovery. Cell Stem Cell 2021, 28, 1507–1515. [Google Scholar] [CrossRef] [PubMed]
- Gómez-Cuadrado, L.; Tracey, N.; Ma, R.; Qian, B.; Brunton, V.G. Mouse Models of Metastasis: Progress and Prospects. Dis. Model Mech. 2017, 10, 1061–1074. [Google Scholar] [CrossRef] [Green Version]
- Ruscetti, M.; Quach, B.; Dadashian, E.L.; Mulholland, D.J.; Wu, H. Tracking and Functional Characterization of Epithelial–Mesenchymal Transition and Mesenchymal Tumor Cells during Prostate Cancer Metastasis. Cancer Res. 2015, 75, 2749–2759. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Salm, S.N.; Burger, P.E.; Coetzee, S.; Goto, K.; Moscatelli, D.; Wilson, E.L. TGF-{beta} Maintains Dormancy of Prostatic Stem Cells in the Proximal Region of Ducts. J. Cell Biol. 2005, 170, 81–90. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Giannoni, E.; Bianchini, F.; Masieri, L.; Serni, S.; Torre, E.; Calorini, L.; Chiarugi, P. Reciprocal Activation of Prostate Cancer Cells and Cancer-Associated Fibroblasts Stimulates Epithelial-Mesenchymal Transition and Cancer Stemness. Cancer Res. 2010, 70, 6945–6956. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Acevedo, V.D.; Gangula, R.D.; Freeman, K.W.; Li, R.; Zhang, Y.; Wang, F.; Ayala, G.E.; Peterson, L.E.; Ittmann, M.; Spencer, D.M. Inducible FGFR-1 Activation Leads to Irreversible Prostate Adenocarcinoma and an Epithelial-to-Mesenchymal Transition. Cancer Cell 2007, 12, 559–571. [Google Scholar] [CrossRef] [Green Version]
- Comito, G.; Giannoni, E.; Segura, C.P.; Barcellos-de-Souza, P.; Raspollini, M.R.; Baroni, G.; Lanciotti, M.; Serni, S.; Chiarugi, P. Cancer-Associated Fibroblasts and M2-Polarized Macrophages Synergize during Prostate Carcinoma Progression. Oncogene 2014, 33, 2423–2431. [Google Scholar] [CrossRef] [Green Version]
- Ku, S.Y.; Rosario, S.; Wang, Y.; Mu, P.; Seshadri, M.; Goodrich, Z.W.; Goodrich, M.M.; Labbé, D.P.; Gomez, E.C.; Wang, J.; et al. Rb1 and Trp53 Cooperate to Suppress Prostate Cancer Lineage Plasticity, Metastasis, and Antiandrogen Resistance. Science 2017, 355, 78–83. [Google Scholar] [CrossRef] [Green Version]
- Mu, P.; Zhang, Z.; Benelli, M.; Karthaus, W.R.; Hoover, E.; Chen, C.-C.; Wongvipat, J.; Ku, S.-Y.; Gao, D.; Cao, Z.; et al. SOX2 Promotes Lineage Plasticity and Antiandrogen Resistance in TP53- and RB1-Deficient Prostate Cancer. Science 2017, 355, 84–88. [Google Scholar] [CrossRef] [Green Version]
- Liang, Y.; Ma, C.; Li, F.; Nie, G.; Zhang, H. The Role of Contactin 1 in Cancers: What We Know So Far. Front. Oncol. 2020, 10, 2042. [Google Scholar] [CrossRef] [PubMed]
- Chen, B.; Zhang, Y.; Li, C.; Xu, P.; Gao, Y.; Xu, Y. CNTN-1 Promotes Docetaxel Resistance and Epithelial-to-Mesenchymal Transition via the PI3K/Akt Signaling Pathway in Prostate Cancer. Arch. Med. Sci. 2021, 17, 152–165. [Google Scholar] [CrossRef] [PubMed]
- Nath, D.; Li, X.; Mondragon, C.; Post, D.; Chen, M.; White, J.R.; Hryniewicz-Jankowska, A.; Caza, T.; Kuznetsov, V.A.; Hehnly, H.; et al. Abi1 Loss Drives Prostate Tumorigenesis through Activation of EMT and Non-Canonical WNT Signaling. Cell Commun. Signal. 2019, 17, 120. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, Q.; Gu, M.; Cai, Z.; Zhao, H.; Sun, S.; Liu, C.; Zhan, M.; Chen, Y.; Wang, Z. TGF-Β1 Promotes Epithelial-to-Mesenchymal Transition and Stemness of Prostate Cancer Cells by Inducing PCBP1 Degradation and Alternative Splicing of CD44. Cell. Mol. Life Sci. 2021, 78, 949–962. [Google Scholar] [CrossRef] [PubMed]
- Shen, Y.; Cao, J.; Liang, Z.; Lin, Q.; Wang, J.; Yang, X.; Zhang, R.; Zong, J.; Du, X.; Peng, Y.; et al. Estrogen Receptor α-NOTCH1 Axis Enhances Basal Stem-like Cells and Epithelial-Mesenchymal Transition Phenotypes in Prostate Cancer. Cell Commun. Signal. 2019, 17, 50. [Google Scholar] [CrossRef] [Green Version]
- Nolan, K.D.; Franco, O.E.; Hance, M.W.; Hayward, S.W.; Isaacs, J.S. Tumor-Secreted Hsp90 Subverts Polycomb Function to Drive Prostate Tumor Growth and Invasion *. J. Biol. Chem. 2015, 290, 8271–8282. [Google Scholar] [CrossRef] [Green Version]
- Yoo, Y.A.; Roh, M.; Naseem, A.F.; Lysy, B.; Desouki, M.M.; Unno, K.; Abdulkadir, S.A. Bmi1 Marks Distinct Castration-Resistant Luminal Progenitor Cells Competent for Prostate Regeneration and Tumor Initiation. Nat. Commun. 2016, 7, 12943. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, B.; Kwon, O.-J.; Henry, G.; Malewska, A.; Wei, X.; Zhang, L.; Brinkley, W.; Zhang, Y.; Castro, P.D.; Titus, M.; et al. Non-Cell-Autonomous Regulation of Prostate Epithelial Homeostasis by Androgen Receptor. Mol. Cell 2016, 63, 976–989. [Google Scholar] [CrossRef] [Green Version]
- Su, W.; Han, H.H.; Wang, Y.; Zhang, B.; Zhou, B.; Cheng, Y.; Rumandla, A.; Gurrapu, S.; Chakraborty, G.; Su, J.; et al. The Polycomb Repressor Complex 1 Drives Double-Negative Prostate Cancer Metastasis by Coordinating Stemness and Immune Suppression. Cancer Cell 2019, 36, 139–155.e10. [Google Scholar] [CrossRef]
- Bezzi, M.; Seitzer, N.; Ishikawa, T.; Reschke, M.; Chen, M.; Wang, G.; Mitchell, C.; Ng, C.; Katon, J.; Lunardi, A.; et al. Diverse Genetic-Driven Immune Landscapes Dictate Tumor Progression through Distinct Mechanisms. Nat. Med. 2018, 24, 165–175. [Google Scholar] [CrossRef]
- Isaacs, J. Control of cell proliferation and cell death in the normal and neoplastic prostate: A stem cell model. In Benign Prostatic Hyperplasia; Rodgers, C.H., Coffey, D.S., Cunha, G., Grayhack, J.T., Hinman, F., Jr., Horton, R., Eds.; US Department of Health and Human Services: Bethesda, MD, USA, 1985; pp. 85–94. [Google Scholar]
- Karthaus, W.R.; Hofree, M.; Choi, D.; Linton, E.L.; Turkekul, M.; Bejnood, A.; Carver, B.; Gopalan, A.; Abida, W.; Laudone, V.; et al. Regenerative Potential of Prostate Luminal Cells Revealed by Single-Cell Analysis. Science 2020, 368, 497–505. [Google Scholar] [CrossRef]
- Stoyanova, T.; Cooper, A.R.; Drake, J.M.; Liu, X.; Armstrong, A.J.; Pienta, K.J.; Zhang, H.; Kohn, D.B.; Huang, J.; Witte, O.N.; et al. Prostate Cancer Originating in Basal Cells Progresses to Adenocarcinoma Propagated by Luminal-like Cells. Proc. Natl. Acad. Sci. USA 2013, 110, 20111–20116. [Google Scholar] [CrossRef] [Green Version]
- Taylor, R.A.; Toivanen, R.; Frydenberg, M.; Pedersen, J.; Harewood, L.; Bioresource, A.P.C.; Collins, A.T.; Maitland, N.J.; Risbridger, G.P. Human Epithelial Basal Cells Are Cells of Origin of Prostate Cancer, Independent of CD133 Status. Stem Cells 2012, 30, 1087–1096. [Google Scholar] [CrossRef]
- Xin, L.; Lawson, D.A.; Witte, O.N. The Sca-1 Cell Surface Marker Enriches for a Prostate-Regenerating Cell Subpopulation That Can Initiate Prostate Tumorigenesis. Proc. Natl. Acad. Sci. USA 2005, 102, 6942–6947. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.; Xu, H.; Cheng, C.; Ji, Z.; Zhao, H.; Sheng, Y.; Li, X.; Wang, J.; Shu, Y.; He, Y.; et al. Identification of a Zeb1 Expressing Basal Stem Cell Subpopulation in the Prostate. Nat. Commun. 2020, 11, 706. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.; Xu, L.; Li, A.; Han, X. The Roles of ZEB1 in Tumorigenic Progression and Epigenetic Modifications. Biomed. Pharmacother. 2019, 110, 400–408. [Google Scholar] [CrossRef]
- Zhang, D.; Park, D.; Zhong, Y.; Lu, Y.; Rycaj, K.; Gong, S.; Chen, X.; Liu, X.; Chao, H.-P.; Whitney, P.; et al. Stem Cell and Neurogenic Gene-Expression Profiles Link Prostate Basal Cells to Aggressive Prostate Cancer. Nat. Commun. 2016, 7, 10798. [Google Scholar] [CrossRef] [Green Version]
- Alumkal, J.J.; Sun, D.; Lu, E.; Beer, T.M.; Thomas, G.V.; Latour, E.; Aggarwal, R.; Cetnar, J.; Ryan, C.J.; Tabatabaei, S.; et al. Transcriptional Profiling Identifies an Androgen Receptor Activity-Low, Stemness Program Associated with Enzalutamide Resistance. Proc. Natl. Acad. Sci. USA 2020, 117, 12315–12323. [Google Scholar] [CrossRef]
- Banyard, J.; Bielenberg, D.R. The Role of EMT and MET in Cancer Dissemination. Connect. Tissue Res. 2015, 56, 403–413. [Google Scholar] [CrossRef] [Green Version]
- Byers, L.A.; Diao, L.; Wang, J.; Saintigny, P.; Girard, L.; Peyton, M.; Shen, L.; Fan, Y.; Giri, U.; Tumula, P.K.; et al. An Epithelial–Mesenchymal Transition Gene Signature Predicts Resistance to EGFR and PI3K Inhibitors and Identifies Axl as a Therapeutic Target for Overcoming EGFR Inhibitor Resistance. Clin. Cancer Res. 2013, 19, 279–290. [Google Scholar] [CrossRef] [Green Version]
- Bocci, F.; Jolly, M.K.; George, J.T.; Levine, H.; Onuchic, J.N. A Mechanism-Based Computational Model to Capture the Interconnections among Epithelial-Mesenchymal Transition, Cancer Stem Cells and Notch-Jagged Signaling. Oncotarget 2018, 9, 29906–29920. [Google Scholar] [CrossRef] [Green Version]
- George, J.T.; Jolly, M.K.; Xu, S.; Somarelli, J.A.; Levine, H. Survival Outcomes in Cancer Patients Predicted by a Partial EMT Gene Expression Scoring Metric. Cancer Res. 2017, 77, 6415–6428. [Google Scholar] [CrossRef] [Green Version]
- Chae, Y.K.; Chang, S.; Ko, T.; Anker, J.; Agte, S.; Iams, W.; Choi, W.M.; Lee, K.; Cruz, M. Epithelial-Mesenchymal Transition (EMT) Signature Is Inversely Associated with T-Cell Infiltration in Non-Small Cell Lung Cancer (NSCLC). Sci. Rep. 2018, 8, 2918. [Google Scholar] [CrossRef] [Green Version]
- Jung, A.R.; Jung, C.-H.; Noh, J.K.; Lee, Y.C.; Eun, Y.-G. Epithelial-Mesenchymal Transition Gene Signature Is Associated with Prognosis and Tumor Microenvironment in Head and Neck Squamous Cell Carcinoma. Sci. Rep. 2020, 10, 3652. [Google Scholar] [CrossRef]
- Thorsson, V.; Gibbs, D.L.; Brown, S.D.; Wolf, D.; Bortone, D.S.; Ou Yang, T.-H.; Porta-Pardo, E.; Gao, G.F.; Plaisier, C.L.; Eddy, J.A.; et al. The Immune Landscape of Cancer. Immunity 2018, 48, 812–830. [Google Scholar] [CrossRef] [Green Version]
- Chakraborty, P.; Chen, E.L.; McMullen, I.; Armstrong, A.J.; Kumar Jolly, M.; Somarelli, J.A. Analysis of Immune Subtypes across the Epithelial-Mesenchymal Plasticity Spectrum. Comput. Struct. Biotechnol. J. 2021, 19, 3842–3851. [Google Scholar] [CrossRef]
- Stark, T.W.; Hensley, P.J.; Spear, A.; Pu, H.; Strup, S.S.; Kyprianou, N. Predictive Value of Epithelial-Mesenchymal-Transition (EMT) Signature and PARP-1 in Prostate Cancer Radioresistance. Prostate 2017, 77, 1583–1591. [Google Scholar] [CrossRef]
- Jiménez, N.; Reig, Ò.; Montalbo, R.; Milà-Guasch, M.; Nadal-Dieste, L.; Castellano, G.; Lozano, J.J.; Victoria, I.; Font, A.; Rodriguez-Vida, A.; et al. Cell Plasticity-Related Phenotypes and Taxanes Resistance in Castration-Resistant Prostate Cancer. Front. Oncol. 2020, 10, 2358. [Google Scholar] [CrossRef]
- Stylianou, N.; Lehman, M.L.; Wang, C.; Fard, A.T.; Rockstroh, A.; Fazli, L.; Jovanovic, L.; Ward, M.; Sadowski, M.C.; Kashyap, A.S.; et al. A Molecular Portrait of Epithelial–Mesenchymal Plasticity in Prostate Cancer Associated with Clinical Outcome. Oncogene 2019, 38, 913–934. [Google Scholar] [CrossRef]
- Cmero, M.; Kurganovs, N.J.; Stuchbery, R.; McCoy, P.; Grima, C.; Ngyuen, A.; Chow, K.; Mangiola, S.; Macintyre, G.; Howard, N.; et al. Loss of SNAI2 in Prostate Cancer Correlates with Clinical Response to Androgen Deprivation Therapy. JCO Precis. Oncol. 2021, 5, 1048–1059. [Google Scholar] [CrossRef]
- Shafran, J.S.; Jafari, N.; Casey, A.N.; Győrffy, B.; Denis, G.V. BRD4 Regulates Key Transcription Factors That Drive Epithelial–Mesenchymal Transition in Castration-Resistant Prostate Cancer. Prostate Cancer Prostatic Dis. 2021, 24, 268–277. [Google Scholar] [CrossRef]
- Sandsmark, E.; Hansen, A.F.; Selnæs, K.M.; Bertilsson, H.; Bofin, A.M.; Wright, A.J.; Viset, T.; Richardsen, E.; Drabløs, F.; Bathen, T.F.; et al. A Novel Non-Canonical Wnt Signature for Prostate Cancer Aggressiveness. Oncotarget 2016, 8, 9572–9586. [Google Scholar] [CrossRef] [Green Version]
- Flavahan, W.A.; Gaskell, E.; Bernstein, B.E. Epigenetic Plasticity and the Hallmarks of Cancer. Science 2017, 357, eaal2380. [Google Scholar] [CrossRef] [Green Version]
- Ceder, Y.; Bjartell, A.; Culig, Z.; Rubin, M.A.; Tomlins, S.; Visakorpi, T. The Molecular Evolution of Castration-Resistant Prostate Cancer. Eur. Urol. Focus 2016, 2, 506–513. [Google Scholar] [CrossRef]
- Pomerantz, M.M.; Qiu, X.; Zhu, Y.; Takeda, D.Y.; Pan, W.; Baca, S.C.; Gusev, A.; Korthauer, K.D.; Severson, T.M.; Ha, G.; et al. Prostate Cancer Reactivates Developmental Epigenomic Programs during Metastatic Progression. Nat. Genet. 2020, 52, 790–799. [Google Scholar] [CrossRef]
- Labbé, D.P.; Brown, M. Transcriptional Regulation in Prostate Cancer. Cold Spring Harb. Perspect. Med. 2018, 8, a030437. [Google Scholar] [CrossRef]
- Figiel, S.; Vasseur, C.; Bruyere, F.; Rozet, F.; Maheo, K.; Fromont, G. Clinical Significance of Epithelial-Mesenchymal Transition Markers in Prostate Cancer. Hum. Pathol. 2017, 61, 26–32. [Google Scholar] [CrossRef]
- Skrypek, N.; Goossens, S.; De Smedt, E.; Vandamme, N.; Berx, G. Epithelial-to-Mesenchymal Transition: Epigenetic Reprogramming Driving Cellular Plasticity. Trends Genet. 2017, 33, 943–959. [Google Scholar] [CrossRef]
- Pistore, C.; Giannoni, E.; Colangelo, T.; Rizzo, F.; Magnani, E.; Muccillo, L.; Giurato, G.; Mancini, M.; Rizzo, S.; Riccardi, M.; et al. DNA Methylation Variations Are Required for Epithelial-to-Mesenchymal Transition Induced by Cancer-Associated Fibroblasts in Prostate Cancer Cells. Oncogene 2017, 36, 5551–5566. [Google Scholar] [CrossRef]
- He, M.X.; Cuoco, M.S.; Crowdis, J.; Bosma-Moody, A.; Zhang, Z.; Bi, K.; Kanodia, A.; Su, M.-J.; Ku, S.-Y.; Garcia, M.M.; et al. Transcriptional Mediators of Treatment Resistance in Lethal Prostate Cancer. Nat. Med. 2021, 27, 426–433. [Google Scholar] [CrossRef]
- O’Brien, J.; Hayder, H.; Zayed, Y.; Peng, C. Overview of MicroRNA Biogenesis, Mechanisms of Actions, and Circulation. Front. Endocrinol. 2018, 9, 402. [Google Scholar] [CrossRef] [Green Version]
- Yao, Q.; Chen, Y.; Zhou, X. The Roles of MicroRNAs in Epigenetic Regulation. Curr. Opin. Chem. Biol. 2019, 51, 11–17. [Google Scholar] [CrossRef]
- Babaei, G.; Raei, N.; Toofani Milani, A.; Gholizadeh-Ghaleh Aziz, S.; Pourjabbar, N.; Geravand, F. The Emerging Role of MiR-200 Family in Metastasis: Focus on EMT, CSCs, Angiogenesis, and Anoikis. Mol. Biol. Rep. 2021, 48, 6935–6947. [Google Scholar] [CrossRef] [PubMed]
- Zaravinos, A. The Regulatory Role of MicroRNAs in EMT and Cancer. J. Oncol. 2015, 2015, 865816. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abisoye-Ogunniyan, A.; Lin, H.; Ghebremedhin, A.; Salam, A.B.; Karanam, B.; Theodore, S.; Jones-Trich, J.; Davis, M.; Grizzle, W.; Wang, H.; et al. Transcriptional Repressor Kaiso Promotes Epithelial to Mesenchymal Transition and Metastasis in Prostate Cancer through Direct Regulation of MiR-200c. Cancer Lett. 2018, 431, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Bucay, N.; Bhagirath, D.; Sekhon, K.; Yang, T.; Fukuhara, S.; Majid, S.; Shahryari, V.; Tabatabai, Z.; Greene, K.L.; Hashimoto, Y.; et al. A Novel MicroRNA Regulator of Prostate Cancer Epithelial–Mesenchymal Transition. Cell Death Differ. 2017, 24, 1263–1274. [Google Scholar] [CrossRef] [PubMed]
- Mozdarani, H.; Ezzatizadeh, V.; Rahbar Parvaneh, R. The Emerging Role of the Long Non-Coding RNA HOTAIR in Breast Cancer Development and Treatment. J. Transl. Med. 2020, 18, 152. [Google Scholar] [CrossRef] [Green Version]
- Chang, Z.; Cui, J.; Song, Y. Long Noncoding RNA PVT1 Promotes EMT via Mediating MicroRNA-186 Targeting of Twist1 in Prostate Cancer. Gene 2018, 654, 36–42. [Google Scholar] [CrossRef]
- Zhang, Y.; Li, Y. Long Non-Coding RNA NORAD Contributes to the Proliferation, Invasion and EMT Progression of Prostate Cancer via the MiR-30a-5p/RAB11A/WNT/β-Catenin Pathway. Cancer Cell Int. 2020, 20, 571. [Google Scholar] [CrossRef] [PubMed]
- Dong, B.; Qiu, Z.; Wu, Y. Tackle Epithelial-Mesenchymal Transition with Epigenetic Drugs in Cancer. Front. Pharmacol. 2020, 11, 1889. [Google Scholar] [CrossRef] [PubMed]
- De Las Rivas, J.; Brozovic, A.; Izraely, S.; Casas-Pais, A.; Witz, I.P.; Figueroa, A. Cancer Drug Resistance Induced by EMT: Novel Therapeutic Strategies. Arch. Toxicol. 2021, 95, 2279–2297. [Google Scholar] [CrossRef]
- Malek, R.; Wang, H.; Taparra, K.; Tran, P.T. Therapeutic Targeting of Epithelial Plasticity Programs: Focus on the Epithelial-Mesenchymal Transition. Cells Tissues Organs 2017, 203, 114–127. [Google Scholar] [CrossRef]
- Mallik, R.; Chowdhury, T.A. Metformin in Cancer. Diabetes Res. Clin. Pract. 2018, 143, 409–419. [Google Scholar] [CrossRef]
- Zaidi, S.; Gandhi, J.; Joshi, G.; Smith, N.L.; Khan, S.A. The Anticancer Potential of Metformin on Prostate Cancer. Prostate Cancer Prostatic Dis. 2019, 22, 351–361. [Google Scholar] [CrossRef] [PubMed]
- Chen, M.; Zhang, J.; Hu, F.; Liu, S.; Zhou, Z. Metformin Affects the Features of a Human Hepatocellular Cell Line (HepG2) by Regulating Macrophage Polarization in a Co-Culture Microenviroment. Diabetes/Metab. Res. Rev. 2015, 31, 781–789. [Google Scholar] [CrossRef] [PubMed]
- Rossini, M.; Martini, F.; Torreggiani, E.; Fortini, F.; Aquila, G.; Sega, F.V.D.; Patergnani, S.; Pinton, P.; Maniscalco, P.; Cavallesco, G.; et al. Metformin Induces Apoptosis and Inhibits Notch1 in Malignant Pleural Mesothelioma Cells. Front Cell Dev. Biol. 2021, 8, 534499. [Google Scholar] [CrossRef]
- Hamilton, D.H.; Huang, B.; Fernando, R.I.; Tsang, K.-Y.; Palena, C. WEE1 Inhibition Alleviates Resistance to Immune Attack of Tumor Cells Undergoing Epithelial-Mesenchymal Transition. Cancer Res. 2014, 74, 2510–2519. [Google Scholar] [CrossRef] [Green Version]
- Cheng, Y.; He, C.; Wang, M.; Ma, X.; Mo, F.; Yang, S.; Han, J.; Wei, X. Targeting Epigenetic Regulators for Cancer Therapy: Mechanisms and Advances in Clinical Trials. Signal Transduct. Target Ther. 2019, 4, 62. [Google Scholar] [CrossRef] [Green Version]
- Feng, S.; De Carvalho, D.D. Clinical Advances in Targeting Epigenetics for Cancer Therapy. FEBS J. 2021, 29, 375–381. [Google Scholar] [CrossRef] [PubMed]
- Dzobo, K.; Senthebane, D.A.; Ganz, C.; Thomford, N.E.; Wonkam, A.; Dandara, C. Advances in Therapeutic Targeting of Cancer Stem Cells within the Tumor Microenvironment: An Updated Review. Cells 2020, 9, 1896. [Google Scholar] [CrossRef] [PubMed]
- Du, B.; Shim, J.S. Targeting Epithelial–Mesenchymal Transition (EMT) to Overcome Drug Resistance in Cancer. Molecules 2016, 21, 965. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beck, J.D.; Reidenbach, D.; Salomon, N.; Sahin, U.; Türeci, Ö.; Vormehr, M.; Kranz, L.M. MRNA Therapeutics in Cancer Immunotherapy. Mol. Cancer 2021, 20, 69. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
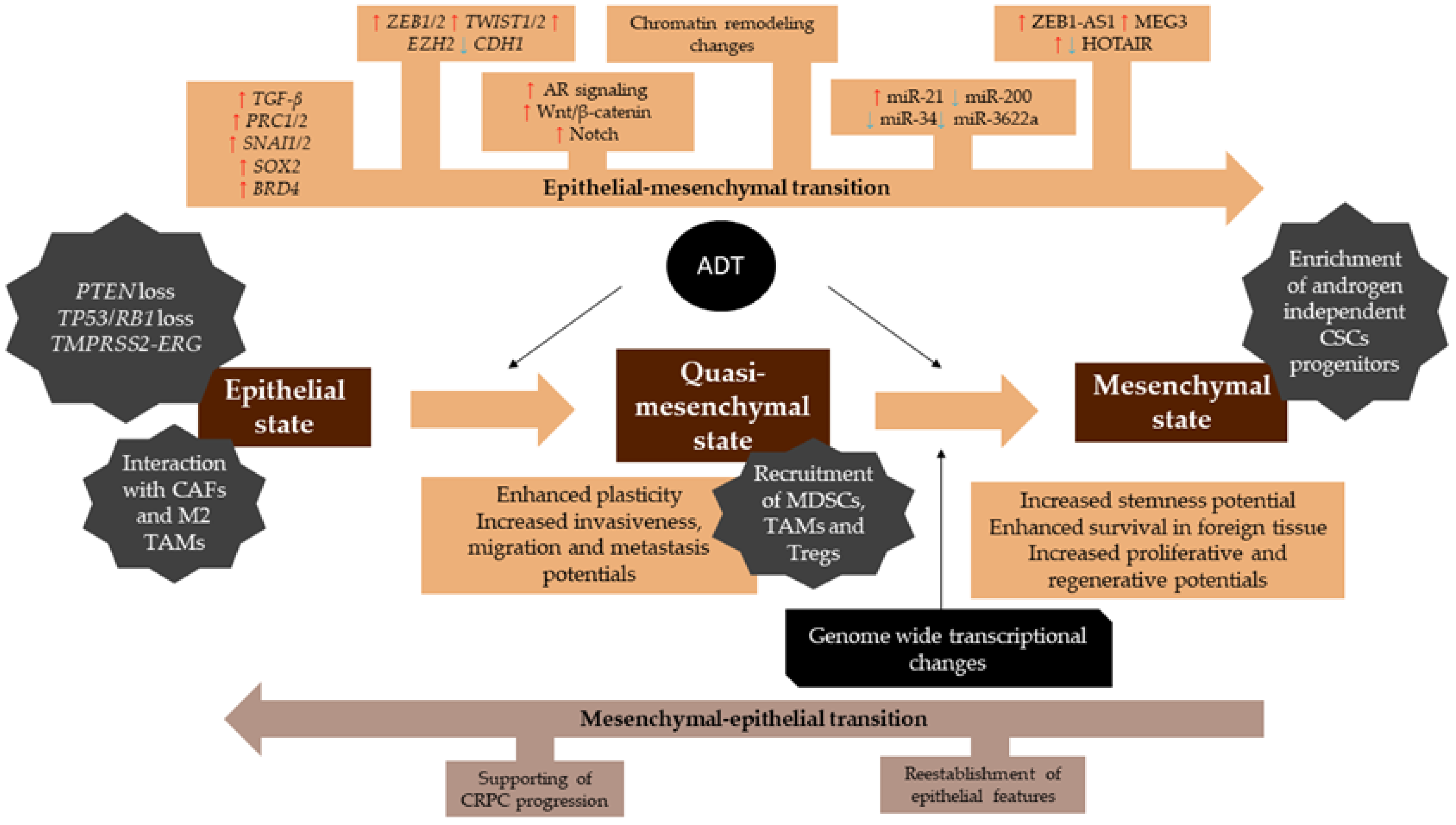
| Castration-resistant prostate cancer (CRPC) |
| CRPC is an incurable advanced prostate cancer that underwent genomic and phenotypical changes that promoted resistance to androgen deprivation therapy. CRPC overcomes the standard of care therapy and, therefore, its progression is independent of androgen signaling. It is believed that CRPC became resistant to hormonal therapy by reactivation of prostate developmental stem-cell-like gene expression. |
| Neuroendocrine differentiation |
| Neuroendocrine differentiation in prostate cancer is a well-recognized phenotypic change by which prostate cancer cells transdifferentiate into neuroendocrine-like cells. NE-like cells lack the expression of AR and prostate-specific antigen (PSA), and are resistant to androgen deprivation therapy. |
| Double-negative prostate cancer (DNPC) |
| DNPC are tumors that lack both AR expression and neuroendocrine differentiation. |
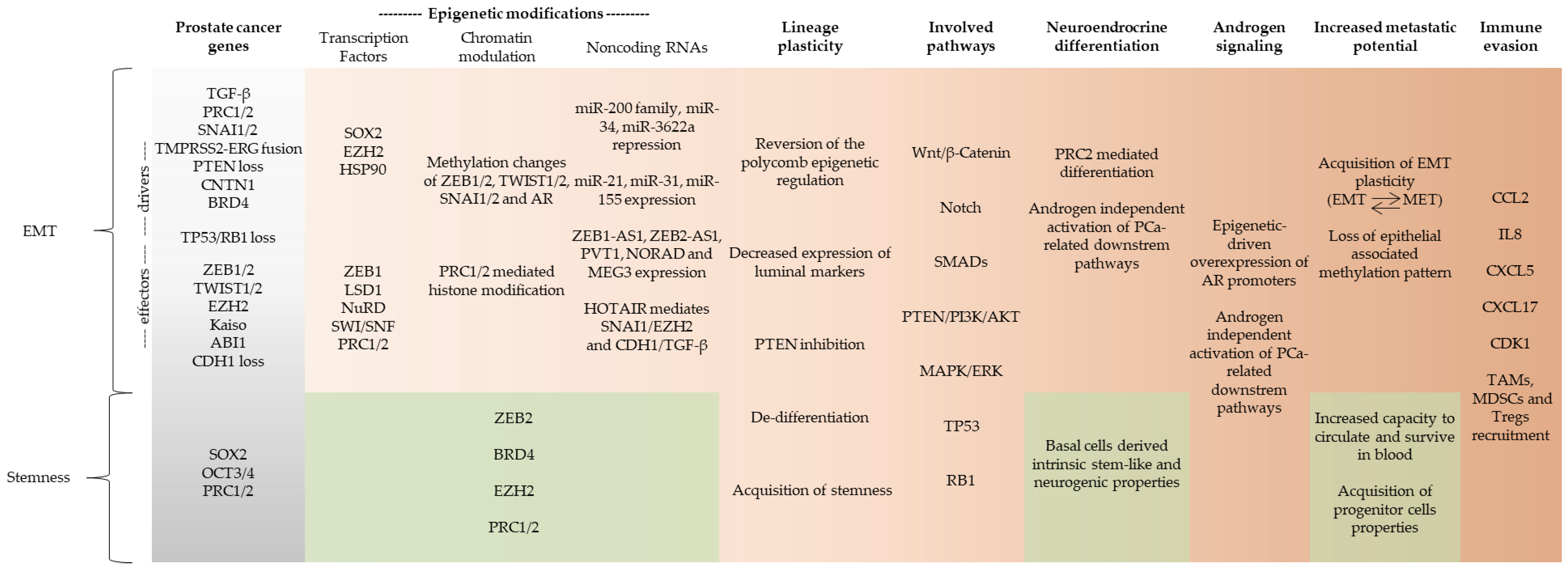
| Epithelial–mesenchymal transition (EMT) |
| EMT is a latent embryonic programming whereby epithelial cells change from cuboidal- to spindle-shaped morphology, losing their epithelial phenotype to acquire mesenchymal features. It is a pivotal process during homeostasis, but in the context of cancer, EMT programming can be reactivated to promote tumor initiation, invasion, migration, metastasis, and stemness. In this context, EMT drivers are responsible to start the EMT process that promotes expression of downstream EMT effectors. |
| Mesenchymal–epithelial transition (MET) |
| MET is the reverse of EMT programming, by which the cells undergo changes to lose the mesenchymal phenotype and acquire epithelial features. MET and EMT work together to promote lineage plasticity. |
| Lineage plasticity |
| Plasticity endows cancer cells with the capacity to shift dynamically between a differentiated state, with limited tumorigenic potential, and an undifferentiated or cancer stem-cell-like (CSC) state, which is responsible for long-term tumor growth. During lineage plasticity, cells constantly shift between EMT and MET to acquire therapy resistance and enhance stemness and metastatic potential. |
| Stemness |
| Stemness is a state of pluripotency that combines the ability of a cell to perpetuate its lineage, to give rise to differentiated cells, and to interact with its environment to maintain a balance between quiescence, proliferation, and regeneration. |
| Cancer stem cells (CSCs) |
| The cells in human tumors, such as PCa, are organized hierarchically, and CSCs comprise a tiny subset of cancer cells that are endowed with tumor-initiating and long-term tumor-propagating capabilities. These tumor-initiating cells display phenotypic and functional features characteristic of normal prostate stem cells and are involved in tumor initiation, metastasis, and drug resistance. |
| Chromatin remodeling |
| Chromatin remodeling is the main form of epigenetic control of expression. Through processes such as DNA methylation and histone modification, the chromatin can be opened or closed. A more opened euchromatin is less condensed and favors gene transcription, while a closer and more condensed heterochromatin suppresses gene expression. |
| Histone modification |
| Histone modification is a complex epigenetic process by which histone tails are acetylated, methylated, phosphorylated, ubiquitinated, or sumoylated to directly or indirectly alter their affinity to DNA. Histones structure the chromatin, and therefore, the more affinity they have to DNA, the more condensed the chromatin is. |
| DNA methylation |
| DNA methylation is a reversible but stable epigenetic process by which a methyl group is attached to the 5-carbon of a cytosine in a CpG dinucleotide, catalyzed by DNA methyltransferase (DNMT). This process allows ligation of methyl-CpG binding-domain proteins (MBDs) that recruit histone-modifying enzymes to promote heterochromatinization. CpG islands, regions rich in CpG dinucleotides, are mostly found in regulatory gene loci, especially promoters and enhancers, and are essential to normal development and cell differentiation. |
| Noncoding RNA |
| Noncoding RNAs are single-stranded molecules of RNA that do not encode proteins. The main noncoding RNAs involved in epigenetic processes are micro-RNAs and long noncoding RNAs. Micro-RNAs are small molecules of RNA (19–25 nucleotides) that bind to complementary regions of messenger RNAs (mRNAs) to post-transcriptionally regulate gene expression. Long noncoding RNAs (lncRNAs) comprise RNA species with more than 200 nucleotides that regulate genes expression by controlling nuclear architecture, nuclear transcription, and mRNA stability. |
| Biomarkers Investigation | |||
| Target | Rationale | Clinical Trial Identifier | |
| Circulating biomarkers of epithelial plasticity | Biomarkers of epithelial plasticity and microtubule interacting protein variants may be related to docetaxel resistance and be enriched in patients failing abiraterone. | NCT02269982 | |
| Circulating tumor cells (CTCs) | Determine whether circulating tumor cells in patients with metastatic progressive castration-resistant prostate cancer or metastatic progressive breast cancer can be captured using a novel mesenchymal marker-based ferrofluid (N-cadherin- or O cadherin-based). | NCT02025413 | |
| CTCs, free DNA, and EMT antigens | Identification of biomarkers that may be predictive of outcome of activity of cabazitaxel treatment in castration-resistant prostate cancer. | NCT03381326 | |
| Therapeutical Agents | |||
| Target | Rationale | Drug | Clinical trials identifier |
| AMP-Kinase | Recent evidence shows that the drug may circumvent tumor growth and resistance to castration therapy. | Metformin | NCT01620593 NCT02176161 |
| WEE 1 Inhibitor | Specifically inhibiting WEE1 may restore CDK1 expression and re-establish immuno-mediated attack of mesenchymal-like cells. | Adavosertib | NCT03385655 NCT02465060 |
| Deacethylase inhibitors | Specifically blocking HDACs may prevent histone modifications implicated in cancer. | Romidepsin | NCT00106418 NCT00106301 |
| Panobinostat | NCT00667862 NCT00878436 | ||
| Pracinostat | NCT01075308 | ||
| Vorinostat | NCT00330161 NCT00589472 | ||
| Phenylbutyrate | NCT00006019 | ||
| EZH2 | Restraining EZH2 may repress PRC2-mediated EMT. | Tazemetostat | NCT04179864 |
| CPI-1205 | NCT03480646 | ||
| DNMTs | Specifically argeting DNMTs may avoid hypermetilation of CDH1 and tumor suppressors. | Azacitidine | NCT03572387 NCT00384839 |
| Decitabine | NCT02649790 NCT03572387 | ||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chaves, L.P.; Melo, C.M.; Saggioro, F.P.; Reis, R.B.d.; Squire, J.A. Epithelial–Mesenchymal Transition Signaling and Prostate Cancer Stem Cells: Emerging Biomarkers and Opportunities for Precision Therapeutics. Genes 2021, 12, 1900. https://doi.org/10.3390/genes12121900
Chaves LP, Melo CM, Saggioro FP, Reis RBd, Squire JA. Epithelial–Mesenchymal Transition Signaling and Prostate Cancer Stem Cells: Emerging Biomarkers and Opportunities for Precision Therapeutics. Genes. 2021; 12(12):1900. https://doi.org/10.3390/genes12121900
Chicago/Turabian StyleChaves, Luiz Paulo, Camila Morais Melo, Fabiano Pinto Saggioro, Rodolfo Borges dos Reis, and Jeremy Andrew Squire. 2021. "Epithelial–Mesenchymal Transition Signaling and Prostate Cancer Stem Cells: Emerging Biomarkers and Opportunities for Precision Therapeutics" Genes 12, no. 12: 1900. https://doi.org/10.3390/genes12121900
APA StyleChaves, L. P., Melo, C. M., Saggioro, F. P., Reis, R. B. d., & Squire, J. A. (2021). Epithelial–Mesenchymal Transition Signaling and Prostate Cancer Stem Cells: Emerging Biomarkers and Opportunities for Precision Therapeutics. Genes, 12(12), 1900. https://doi.org/10.3390/genes12121900