
Rare Pathogenic Variants in Genes Implicated in Glutamatergic Neurotransmission Pathway Segregate with Schizophrenia in Pakistani Families

 , ,
, , {kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Subjects
2.2. Analysis for Copy Number Variations (CNVs)
2.2.1. Validation of CNV by Quantitative Real-Time PCR (qPCR)
2.2.2. Next-Generation Mate Pair Sequencing (MPS)
2.2.3. Validation of Breakpoints (BPs) with Sanger Sequencing
2.3. Whole-Exome Sequencing (WES)
In Silico Prediction of Structural Alternation in Mutated Protein
2.4. Rare Variants Screening through Amplified Fragment Length Polymorphism (AFLP)
3. Results
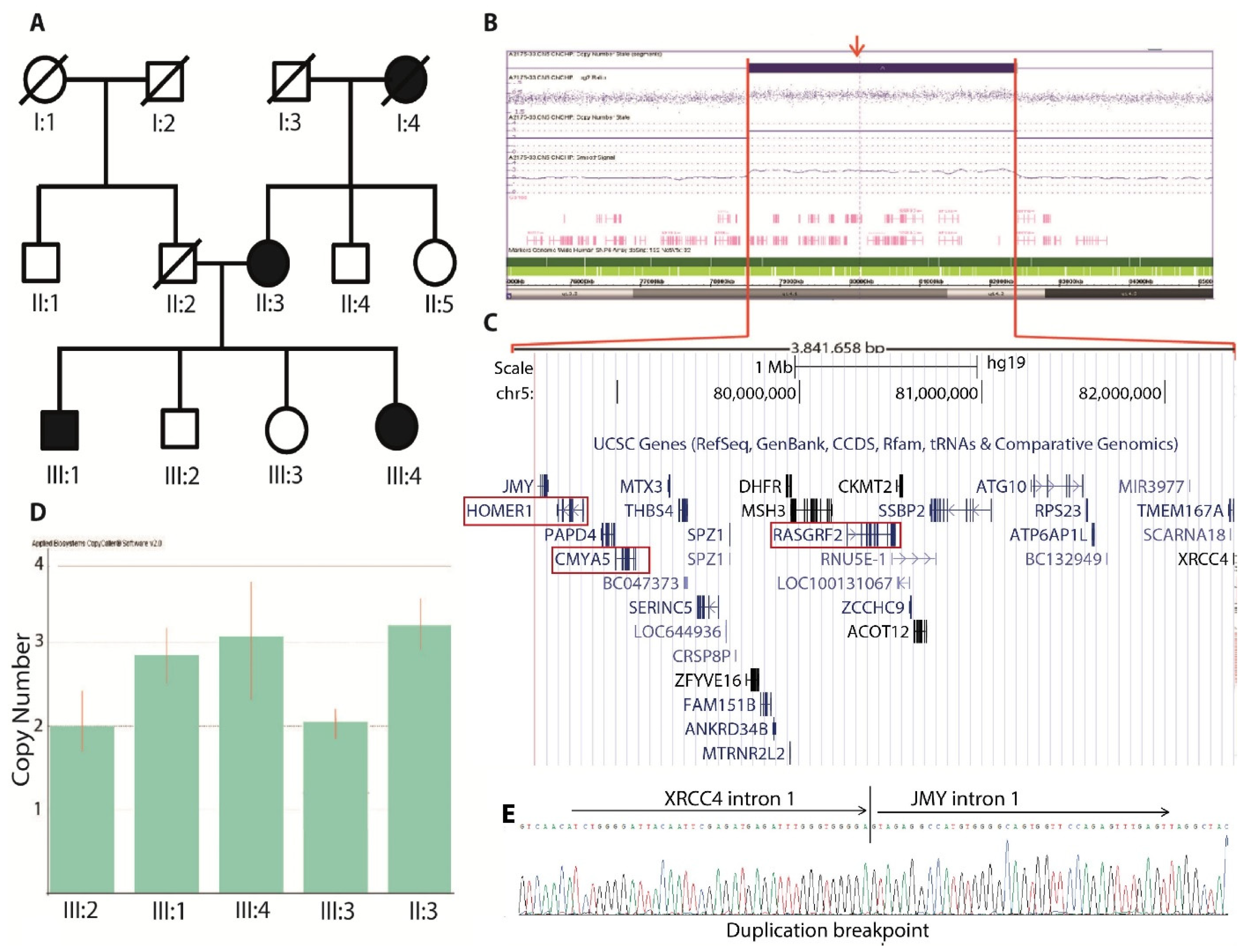
3.1. CNV Analysis
3.1.1. CNV Confirmation
3.1.2. MPS and Breakpoint Detection
3.1.3. Breakpoint Screening
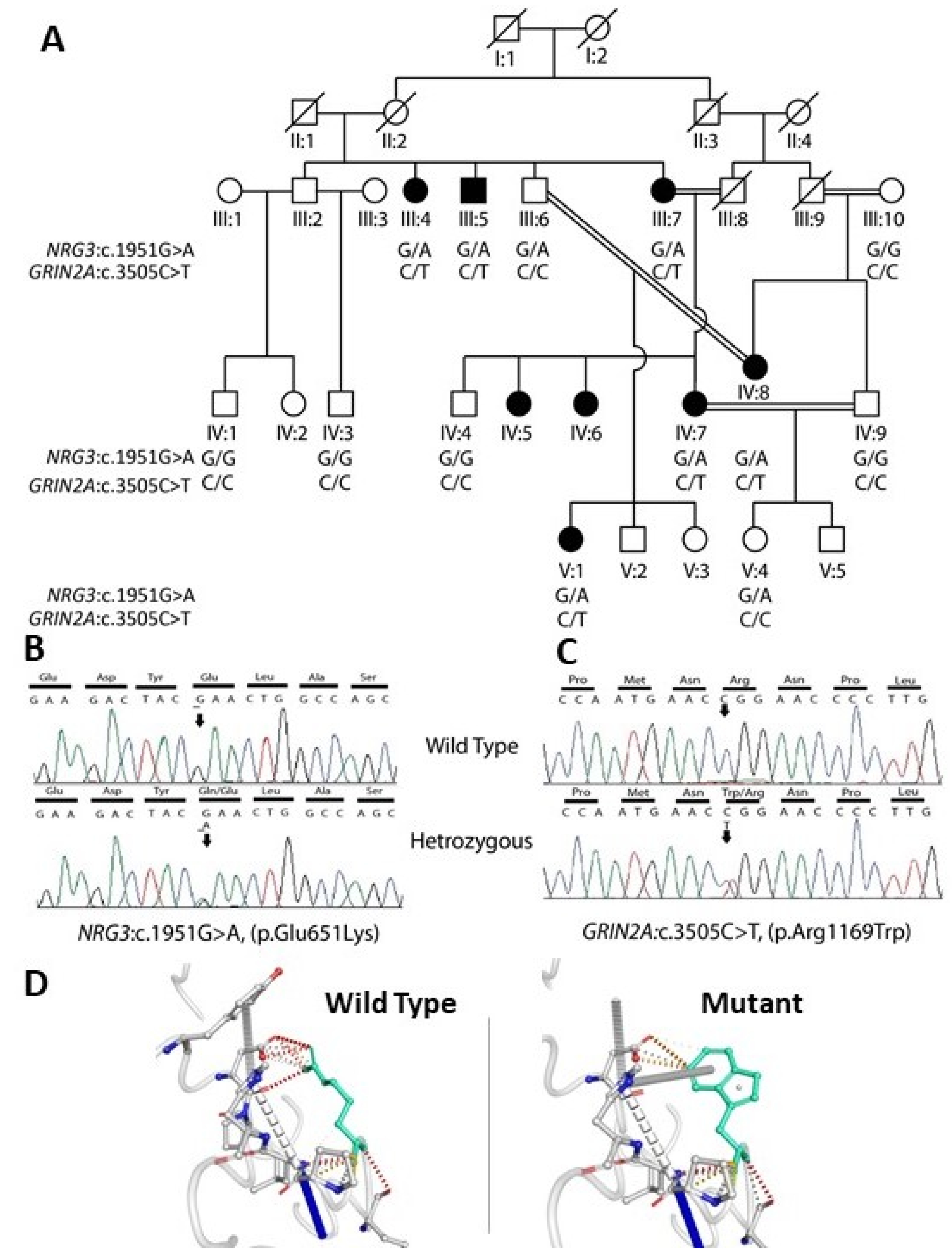
3.2. Whole-Exome Sequencing
3.3. In Silico Prediction
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Saha, S.; Chant, D.; Welham, J.; McGrath, J. A Systematic Review of the Prevalence of Schizophrenia. PLoS Med. 2005, 2, e141. [Google Scholar] [CrossRef] [PubMed]
- Sullivan, P.F. The genetics of schizophrenia. PLoS Med. 2005, 2, e212. [Google Scholar] [CrossRef] [Green Version]
- McCutcheon, R.A.; Marques, T.R.; Howes, O.D. Schizophrenia—An overview. JAMA Psychiatry 2020, 77, 201–210. [Google Scholar] [CrossRef] [PubMed]
- Hilker, R.; Helenius, D.; Fagerlund, B.; Skytthe, A.; Christensen, K.; Werge, T.M.; Nordentoft, M.; Glenthøj, B. Heritability of Schizophrenia and Schizophrenia Spectrum Based on the Nationwide Danish Twin Register. Biol. Psychiatry 2018, 83, 492–498. [Google Scholar] [CrossRef] [Green Version]
- Cardno, A.G.; Marshall, E.J.; Coid, B.; Macdonald, A.M.; Ribchester, T.R.; Davies, N.J.; Venturi, P.; Jones, L.A.; Lewis, S.W.; Sham, P.C. Heritability estimates for psychotic disorders: The Maudsley twin psychosis series. Arch. Gen. Psychiatry 1999, 56, 162–168. [Google Scholar] [CrossRef] [PubMed]
- Murray, R.M.; Vassos, E. Nature, Nurture, and the Polygenic Risk Score for Schizophrenia. Schizophr. Bull. 2020, 46, 1363–1365. [Google Scholar] [CrossRef] [PubMed]
- Corvin, A.; Ormond, C.; Cole, A. Genomics of schizophrenia. In Personalized Psychiatry; Elsevier BV: Amsterdam, The Netherlands, 2020; pp. 173–186. [Google Scholar]
- Nesic, M.J.; Stojkovic, B.; Maric, N.P. On the origin of schizophrenia: Testing evolutionary theories in the post-genomic era. Psychiatry Clin. Neurosci. 2019, 73, 723–730. [Google Scholar] [CrossRef] [PubMed]
- Ripke, S.; Neale, B.M.; Corvin, A.; Walters, J.T.; Farh, K.-H.; Holmans, P.A.; Lee, P.; Bulik-Sullivan, B.; Collier, D.A.; Huang, H. Biological insights from 108 schizophrenia-associated genetic loci. Nature 2014, 511, 421–427. [Google Scholar]
- Pardiñas, A.F.; Holmans, P.; Pocklington, A.; Escott-Price, V.; Ripke, S.; Carrera, N.; Legge, S.E.; Bishop, S.; Cameron, D.; Hamshere, M.L.; et al. Common schizophrenia alleles are enriched in mutation-intolerant genes and in regions under strong background selection. Nat. Genet. 2018, 50, 381–389. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ripke, S.; Walters, J.T.; O’Donovan, M.C.; Schizophrenia Working Group of the Psychiatric Genomics Consortium. Mapping genomic loci prioritises genes and implicates synaptic biology in schizophrenia. MedRxiv 2020. [Google Scholar] [CrossRef]
- Consortium, I.S. Rare chromosomal deletions and duplications increase risk of schizophrenia. Nature 2008, 455, 237. [Google Scholar]
- Consortium, I.S. Common polygenic variation contributes to risk of schizophrenia and bipolar disorder. Nature 2009, 460, 748. [Google Scholar]
- Buizer-Voskamp, J.E.; Muntjewerff, J.-W.; Strengman, E.; Sabatti, C.; Stefansson, H.; Vorstman, J.A.; Ophoff, R.A. Genome-Wide Analysis Shows Increased Frequency of Copy Number Variation Deletions in Dutch Schizophrenia Patients. Biol. Psychiatry 2011, 70, 655–662. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ingason, A.; GROUP Investigators; Rujescu, D.; Cichon, S.; Sigurdsson, E.; Sigmundsson, T.; Pietiläinen, O.P.H.; Buizer-Voskamp, J.E.; Strengman, E.; Francks, C.; et al. Copy number variations of chromosome 16p13.1 region associated with schizophrenia. Mol. Psychiatry 2009, 16, 17–25. [Google Scholar] [CrossRef] [Green Version]
- Ferreira, M.A.R.; Wellcome Trust Case Control Wellcome Trust Case Control Consortium; O’Donovan, M.C.; Meng, Y.A.; Jones, I.R.; Ruderfer, D.M.; Jones, L.; Fan, J.; Kirov, G.; Perlis, R.H.; et al. Collaborative genome-wide association analysis supports a role for ANK3 and CACNA1C in bipolar disorder. Nat. Genet. 2008, 40, 1056–1058. [Google Scholar] [CrossRef] [Green Version]
- Marshall, C.R.; Psychosis Endophenotypes International Consortium; Howrigan, D.P.; Merico, D.; Thiruvahindrapuram, B.; Wu, W.; Greer, D.S.; Antaki, D.; Shetty, A.; Holmans, P.A.; et al. Contribution of copy number variants to schizophrenia from a genome-wide study of 41,321 subjects. Nat. Genet. 2017, 49, 27–35. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Singh, T.; Poterba, T.; Curtis, D.; Akil, H.; Eissa, M.A.; Barchas, J.D.; Bass, N.; Bigdeli, T.B.; Breen, G.; Bromet, E.J. Exome sequencing identifies rare coding variants in 10 genes which confer substantial risk for schizophrenia. MedRxiv 2020. [Google Scholar] [CrossRef]
- Timms, A.E.; Dorschner, M.O.; Wechsler, J.; Choi, K.Y.; Kirkwood, R.; Girirajan, S.; Baker, C.; Eichler, E.E.; Korvatska, O.; Roche, K.W.; et al. Support for the N -Methyl-D-Aspartate Receptor Hypofunction Hypothesis of Schizophrenia from Exome Sequencing in Multiplex Families. JAMA Psychiatry 2013, 70, 582–590. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kos, M.Z.; Carless, M.A.; Peralta, J.; Blackburn, A.; Almeida, M.; Roalf, D.; Pogue-Geile, M.F.; Prasad, K.; Gur, R.C.; Nimgaonkar, V.; et al. Exome Sequence Data from Multigenerational Families Implicate AMPA Receptor Trafficking in Neurocognitive Impairment and Schizophrenia Risk. Schizophr. Bull. 2015, 42, 288–300. [Google Scholar] [CrossRef] [Green Version]
- Xu, B.; Ionita-Laza, I.; Roos, J.; Boone, B.; Woodrick, S.; Sun, Y.; Levy, S.; Gogos, J.A.; Karayiorgou, M. De novo gene mutations highlight patterns of genetic and neural complexity in schizophrenia. Nat. Genet. 2012, 44, 1365–1369. [Google Scholar] [CrossRef] [Green Version]
- Howrigan, D.P.; Rose, S.A.; Samocha, K.E.; Fromer, M.; Cerrato, F.; Chen, W.J.; Churchhouse, C.; Chambert, K.; Chandler, S.D.; Daly, M.J.; et al. Exome sequencing in schizophrenia-affected parent–offspring trios reveals risk conferred by protein-coding de novo mutations. Nat. Neurosci. 2020, 23, 185–193. [Google Scholar] [CrossRef] [PubMed]
- Rees, E.; GROUP Investigators; Han, J.; Morgan, J.; Carrera, N.; Escott-Price, V.; Pocklington, A.J.; Duffield, M.; Hall, L.S.; Legge, S.E.; et al. De novo mutations identified by exome sequencing implicate rare missense variants in SLC6A1 in schizophrenia. Nat. Neurosci. 2020, 23, 179–184. [Google Scholar] [CrossRef]
- Purcell, S.M.; Moran, J.; Fromer, M.; Ruderfer, D.; Solovieff, N.; Roussos, P.; O’Dushlaine, C.; Chambert, K.; Bergen, S.; Kähler, A.; et al. A polygenic burden of rare disruptive mutations in schizophrenia. Nat. Cell Biol. 2014, 506, 185–190. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Singh, T.; INTERVAL Study; Walters, J.; Johnstone, M.; Curtis, D.; Suvisaari, J.; Torniainen, M.; Rees, E.; Iyegbe, C.; Blackwood, D.; et al. The contribution of rare variants to risk of schizophrenia in individuals with and without intellectual disability. Nat. Genet. 2017, 49, 1167–1173. [Google Scholar] [CrossRef] [PubMed]
- Genovese, G.; Fromer, M.; Stahl, E.A.; Ruderfer, D.M.; Chambert, K.; Landén, M.; Moran, J.; Purcell, S.M.; Sklar, P.; Sullivan, P.F.; et al. Increased burden of ultra-rare protein-altering variants among 4877 individuals with schizophrenia. Nat. Neurosci. 2016, 19, 1433–1441. [Google Scholar] [CrossRef] [PubMed]
- John, J.; Bhattacharyya, U.; Yadav, N.; Kukshal, P.; Bhatia, T.; Nimgaonkar, V.; Deshpande, S.N.; Thelma, B. Multiple rare inherited variants in a four generation schizophrenia family offer leads for complex mode of disease inheritance. Schizophr. Res. 2019, 216, 288–294. [Google Scholar] [CrossRef]
- Guermouche, A.D.; Guipponi, M.; Prados, J.; Antonarakis, S. Wes of a Consanguineous Family with Schizophrenia and Mental Retardation in North Algeria. Eur. Psychiatry 2015, 30, 1421. [Google Scholar] [CrossRef]
- Amin A., G.; Ahsan A., V. Mental Health Morbidity Pattern in Pakistan. JCPSP 1999, 9, 362–365. [Google Scholar]
- Gadit, A.A.M. Psychiatry in Pakistan: 1947–2006: A new balance sheet. J. Pak. Med. Assoc. 2007, 57, 453–463. [Google Scholar] [PubMed]
- Nazaryan, L.; Stefanou, E.G.; Hansen, C.; Kosyakova, N.; Bak, M.; Sharkey, F.H.; Mantziou, T.; Papanastasiou, A.D.; Velissariou, V.; Liehr, T.; et al. The strength of combined cytogenetic and mate-pair sequencing techniques illustrated by a germline chromothripsis rearrangement involving FOXP. Eur. J. Hum. Genet. 2013, 22, 338–343. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, X.; Lee, G.; Maher, B.S.; Fanous, A.H.; Chen, J.; Zhao, Z.; Guo, A.; Oord, E.V.D.; Sullivan, P.F.; Shi, J.; et al. GWA study data mining and independent replication identify cardiomyopathy-associated 5 (CMYA5) as a risk gene for schizophrenia. Mol. Psychiatry 2010, 16, 1117–1129. [Google Scholar] [CrossRef]
- Rietschel, M.; Mattheisen, M.; Frank, J.; Treutlein, J.; Degenhardt, F.; Breuer, R.; Steffens, M.; Mier, D.; Esslinger, C.; Walter, H.; et al. Genome-Wide Association-, Replication-, and Neuroimaging Study Implicates HOMER1 in the Etiology of Major Depression. Biol. Psychiatry 2010, 68, 578–585. [Google Scholar] [CrossRef]
- Xiang, B.; Wu, J.-Y.; Ma, X.-H.; Wang, Y.-C.; Deng, W.; Chen, Z.-F.; Li, M.-L.; Wang, Q.; He, Z.-L.; Jiang, L.-J.; et al. Genome-wide association study with memory measures as a quantitative trait locus for schizophrenia. Chin. J. Med Genet. 2012, 29, 255–259. [Google Scholar] [CrossRef]
- Luo, P.; Li, X.; Fei, Z.; Poon, W. Scaffold protein Homer 1: Implications for neurological diseases. Neurochem. Int. 2012, 61, 731–738. [Google Scholar] [CrossRef]
- Fagni, L.; Worley, P.F.; Ango, F. Homer as Both a Scaffold and Transduction Molecule. Sci. Signal. 2002, 2002, re8. [Google Scholar] [CrossRef] [PubMed]
- Lominac, K.; Oleson, E.B.; Pava, M.; Klugmann, M.; Schwarz, M.K.; Seeburg, P.H.; During, M.J.; Worley, P.F.; Kalivas, P.W.; Szumlinski, K.K. Distinct Roles for Different Homer1 Isoforms in Behaviors and Associated Prefrontal Cortex Function. J. Neurosci. 2005, 25, 11586–11594. [Google Scholar] [CrossRef] [PubMed]
- Norton, N.; Williams, H.; Williams, N.; Spurlock, G.; Zammit, S.; Jones, G.; Jones, S.; Owen, R.; O’Donovan, M.; Owen, M. Mutation screening of theHomer gene family and association analysis in schizophrenia. Am. J. Med Genet. 2003, 120B, 18–21. [Google Scholar] [CrossRef] [PubMed]
- Wagner, K.V.; Häusl, A.S.; Pöhlmann, M.L.; Hartmann, J.; Labermaier, C.; Müller, M.B.; Schmidt, M.V. Hippocampal Homer1 Levels Influence Motivational Behavior in an Operant Conditioning Task. PLoS ONE 2014, 9, e85975. [Google Scholar] [CrossRef] [PubMed]
- Yoon, S.; Piguel, N.H.; Khalatyan, N.; Dionisio, L.E.; Savas, J.N.; Penzes, P. Homer1 promotes dendritic spine growth through ankyrin-G and its loss reshapes the synaptic proteome. Mol. Psychiatry 2021, 26, 1775–1789. [Google Scholar] [CrossRef]
- Wagner, K.V.; Hartmann, J.; Labermaier, C.; Häusl, A.S.; Zhao, G.; Harbich, D.; Schmid, B.; Wang, X.-D.; Santarelli, S.; Kohl, C. Homer1/mGluR5 activity moderates vulnerability to chronic social stress. Neuropsychopharmacology 2015, 40, 1222–1233. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Verpelli, C.; Schmeisser, M.J.; Sala, C.; Boeckers, T.M. Scaffold Proteins at the Postsynaptic Density. In Advances in Experimental Medicine and Biology; Springer: Singapore, 2012; Volume 970, pp. 29–61. [Google Scholar]
- De Bartolomeis, A.; Latte, G.; Tomasetti, C.; Iasevoli, F. Glutamatergic postsynaptic density protein dysfunctions in synaptic plasticity and dendritic spines morphology: Relevance to schizophrenia and other behavioral disorders pathophysiology, and implications for novel therapeutic approaches. Mol. Neurobiol. 2014, 49, 484–511. [Google Scholar] [CrossRef]
- Matosin, N.; Frank, E.; Deng, C.; Huang, X.-F.; Newell, K.A. Metabotropic glutamate receptor 5 binding and protein expression in schizophrenia and following antipsychotic drug treatment. Schizophr. Res. 2013, 146, 170–176. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Newell, K.A. Metabotropic glutamate receptor 5 in schizophrenia: Emerging evidence for the development of antipsychotic drugs. Futur. Med. Chem. 2013, 5, 1471–1474. [Google Scholar] [CrossRef] [Green Version]
- Brambilla, C.R.; Veselinović, T.; Rajkumar, R.; Mauler, J.; Orth, L.; Ruch, A.; Ramkiran, S.; Heekeren, K.; Kawohl, W.; Wyss, C.; et al. mGluR5 receptor availability is associated with lower levels of negative symptoms and better cognition in male patients with chronic schizophrenia. Hum. Brain Mapp. 2020, 41, 2762–2781. [Google Scholar] [CrossRef] [Green Version]
- Tomasetti, C.; Iasevoli, F.; Buonaguro, E.F.; De Berardis, D.; Fornaro, M.; Fiengo, A.L.C.; Martinotti, G.; Orsolini, L.; Valchera, A.; Di Giannantonio, M.; et al. Treating the Synapse in Major Psychiatric Disorders: The Role of Postsynaptic Density Network in Dopamine-Glutamate Interplay and Psychopharmacologic Drugs Molecular Actions. Int. J. Mol. Sci. 2017, 18, 135. [Google Scholar] [CrossRef]
- Zimmerman, A.J.; Hafez, A.K.; Amoah, S.K.; Rodriguez, B.A.; Dell’Orco, M.; Lozano, E.; Hartley, B.J.; Alural, B.; Lalonde, J.; Chander, P.; et al. A psychiatric disease-related circular RNA controls synaptic gene expression and cognition. Mol. Psychiatry 2020, 25, 2712–2727. [Google Scholar] [CrossRef] [Green Version]
- Schwechter, B.; Rosenmund, C.; Tolias, K.F. RasGRF2 Rac-GEF activity couples NMDA receptor calcium flux to enhanced synaptic transmission. Proc. Natl. Acad. Sci. USA 2013, 110, 14462–14467. [Google Scholar] [CrossRef] [Green Version]
- Darcy, M.J.; Trouche, S.; Jin, S.-X.; Feig, L.A. Ras-GRF2 mediates long-term potentiation, survival, and response to an enriched environment of newborn neurons in the hippocampus. Hippocampus 2014, 24, 1317–1329. [Google Scholar] [CrossRef] [Green Version]
- Minor, S.S.I. A common variant of the cardiomyopathy associated 5 gene (CMYA5) is associated with schizophrenia in Chinese population. Schizophr. Res. 2011, 129, 217–219. [Google Scholar]
- Zhang, R.; Zhang, H.; Li, M.; Li, H.; Li, Y.; Valenzuela, R.K.; Su, B.; Ma, J. Genetic analysis of common variants in the CMYA5 (cardiomyopathy-associated 5) gene with schizophrenia. Prog. Neuro-Psychopharmacol. Biol. Psychiatry 2013, 46, 64–69. [Google Scholar] [CrossRef]
- Durham, J.T.; Brand, O.M.; Arnold, M.; Reynolds, J.G.; Muthukumar, L.; Weiler, H.; Richardson, J.A.; Naya, F.J. Myospryn Is a Direct Transcriptional Target for MEF2A That Encodes a Striated Muscle, α-Actinin-interacting, Costamere-localized Protein. J. Biol. Chem. 2006, 281, 6841–6849. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reynolds, J.G.; McCalmon, S.A.; Donaghey, J.A.; Naya, F.J. Deregulated Protein Kinase A Signaling and Myospryn Expression in Muscular Dystrophy. J. Biol. Chem. 2008, 283, 8070–8074. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sarparanta, J.; Blandin, G.; Charton, K.; Vihola, A.; Marchand, S.; Milic, A.; Hackman, P.; Ehler, E.; Richard, I.; Udd, B. Interactions with M-band Titin and Calpain 3 Link Myospryn (CMYA5) to Tibial and Limb-girdle Muscular Dystrophies. J. Biol. Chem. 2010, 285, 30304–30315. [Google Scholar] [CrossRef] [Green Version]
- Hsiung, A.; Naya, F.J.; Chen, X.; Shiang, R. A schizophrenia associated CMYA5 allele displays differential binding with desmin. J. Psychiatr. Res. 2019, 111, 8–15. [Google Scholar] [CrossRef]
- Burdick, K.E.; Goldberg, T.E.; Funke, B.; Bates, J.A.; Lencz, T.; Kucherlapati, R.; Malhotra, A.K. DTNBP1 genotype influences cognitive decline in schizophrenia. Schizophr. Res. 2007, 89, 169–172. [Google Scholar] [CrossRef] [Green Version]
- Wang, H.; Xu, J.; Lazarovici, P.; Zheng, W. Dysbindin-1 Involvement in the Etiology of Schizophrenia. Int. J. Mol. Sci. 2017, 18, 2044. [Google Scholar] [CrossRef] [Green Version]
- Numakawa, T.; Yagasaki, Y.; Ishimoto, T.; Okada, T.; Suzuki, T.; Iwata, N.; Ozaki, N.; Taguchi, T.; Tatsumi, M.; Kamijima, K.; et al. Evidence of novel neuronal functions of dysbindin, a susceptibility gene for schizophrenia. Hum. Mol. Genet. 2004, 13, 2699–2708. [Google Scholar] [CrossRef] [Green Version]
- Adams, J.; Crosbie, J.; Wigg, K.; Ickowicz, A.; Pathare, T.; Roberts, W.; Malone, M.; Schachar, R.; Tannock, R.; Kennedy, J.L.; et al. Glutamate receptor, ionotropic, N-methyl D-aspartate 2A (GRIN2A) gene as a positional candidate for attention-deficit/hyperactivity disorder in the 16p13 region. Mol. Psychiatry 2004, 9, 494–499. [Google Scholar] [CrossRef] [Green Version]
- Tarabeux, J.; Kebir, O.; Gauthier, J.; Hamdan, F.F.; Xiong, L.; Piton, A.; Spiegelman, D.; Henrion, É.; Millet, B.; Fathalli, F.; et al. Rare mutations in N-methyl-D-aspartate glutamate receptors in autism spectrum disorders and schizophrenia. Transl. Psychiatry 2011, 1, e55. [Google Scholar] [CrossRef] [Green Version]
- Guzmán-Jiménez, D.E.; Flores-Ramírez, E.L.; Velasco-Monroy, A.L. Genomic studies in epilepsy. Rev. Med. Hosp. Gen. Mex. 2019, 82, 33–38. [Google Scholar] [CrossRef]
- Endele, S.; Rosenberger, G.; Geider, K.; Popp, B.; Tamer, C.; Stefanova, I.; Milh, M.; Kortüm, F.; Fritsch, A.; Pientka, F.K.; et al. Mutations in GRIN2A and GRIN2B encoding regulatory subunits of NMDA receptors cause variable neurodevelopmental phenotypes. Nat. Genet. 2010, 42, 1021–1026. [Google Scholar] [CrossRef]
- Hornig, T.; Grüning, B.; Kundu, K.; Houwaart, T.; Backofen, R.; Biber, K.; Normann, C. GRIN3B missense mutation as an inherited risk factor for schizophrenia: Whole-exome sequencing in a family with a familiar history of psychotic disorders. Genet. Res. 2017, 99, e1. [Google Scholar] [CrossRef] [Green Version]
- John, J.; Kukshal, P.; Sharma, A.; Bhatia, T.; Nimgaonkar, V.; Deshpande, S.; Thelma, B. Rare variants in Protein tyrosine phosphatase, receptor type A (PTPRA) in schizophrenia: Evidence from a family based study. Schizophr. Res. 2019, 206, 75–81. [Google Scholar] [CrossRef] [PubMed]
- Nakazawa, K.; Sapkota, K. The origin of NMDA receptor hypofunction in schizophrenia. Pharmacol. Ther. 2020, 205, 107426. [Google Scholar] [CrossRef] [PubMed]
- Balu, D.; Li, Y.; Puhl, M.D.; Benneyworth, M.A.; Basu, A.C.; Takagi, S.; Bolshakov, V.Y.; Coyle, J.T. Multiple risk pathways for schizophrenia converge in serine racemase knockout mice, a mouse model of NMDA receptor hypofunction. Proc. Natl. Acad. Sci. USA 2013, 110, E2400–E2409. [Google Scholar] [CrossRef] [Green Version]
- Stefansson, H.; Petursson, H.; Sigurdsson, E.; Steinthorsdottir, V.; Bjornsdottir, S.; Sigmundsson, T.; Ghosh, S.; Brynjolfsson, J.; Gunnarsdottir, S.; Ivarsson, O.; et al. Neuregulin 1 and Susceptibility to Schizophrenia. Am. J. Hum. Genet. 2002, 71, 877–892. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ananloo, E.; Yoosefee, S.; Karimipour, M. Neuregulin1 gene variants as a biomarker for cognitive impairments in patients with schizophrenia. Eur. J. Psychiatry 2020, 34, 11–19. [Google Scholar] [CrossRef]
- Kao, W.-T.; Wang, Y.; Kleinman, J.E.; Lipska, B.K.; Hyde, T.M.; Weinberger, D.R.; Law, A.J. Common genetic variation in Neuregulin 3 (NRG3) influences risk for schizophrenia and impacts NRG3 expression in human brain. Proc. Natl. Acad. Sci. USA 2010, 107, 15619–15624. [Google Scholar] [CrossRef] [Green Version]
- Li, Z.; Liu, L.; Lin, W.; Zhou, Y.; Zhang, G.; Du, X.; Li, Y.; Tang, W.; Zhang, X. NRG3 contributes to cognitive deficits in chronic patients with schizophrenia. Schizophr. Res. 2020, 215, 134–139. [Google Scholar] [CrossRef]
- Hayes, L.N.; Shevelkin, A.; Zeledon, M.; Steel, G.; Chen, P.-L.; Obie, C.; Pulver, A.; Avramopoulos, D.; Valle, D.; Sawa, A.; et al. Neuregulin 3 Knockout Mice Exhibit Behaviors Consistent with Psychotic Disorders. Mol. Neuropsychiatry 2016, 2, 79–87. [Google Scholar] [CrossRef] [PubMed] [Green Version]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fatima, A.; Abdullah, U.; Farooq, M.; Mang, Y.; Mehrjouy, M.M.; Asif, M.; Ali, Z.; Tommerup, N.; Baig, S.M. Rare Pathogenic Variants in Genes Implicated in Glutamatergic Neurotransmission Pathway Segregate with Schizophrenia in Pakistani Families. Genes 2021, 12, 1899. https://doi.org/10.3390/genes12121899
Fatima A, Abdullah U, Farooq M, Mang Y, Mehrjouy MM, Asif M, Ali Z, Tommerup N, Baig SM. Rare Pathogenic Variants in Genes Implicated in Glutamatergic Neurotransmission Pathway Segregate with Schizophrenia in Pakistani Families. Genes. 2021; 12(12):1899. https://doi.org/10.3390/genes12121899
Chicago/Turabian StyleFatima, Ambrin, Uzma Abdullah, Muhammad Farooq, Yuan Mang, Mana M. Mehrjouy, Maria Asif, Zafar Ali, Niels Tommerup, and Shahid M. Baig. 2021. "Rare Pathogenic Variants in Genes Implicated in Glutamatergic Neurotransmission Pathway Segregate with Schizophrenia in Pakistani Families" Genes 12, no. 12: 1899. https://doi.org/10.3390/genes12121899