
Bioinformatic Analysis Identified Hub Genes Associated with Heterocyclic Amines Induced Cytotoxicity of Peripheral Blood Mononuclear Cells

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. GEO Data Accession
2.2. WGCNA Analysis
2.3. LIMMA Analysis
2.4. Functional Analysis
2.5. Network Construction and Hub Gene Identification
2.6. Statistical Analysis and Plotting
3. Results
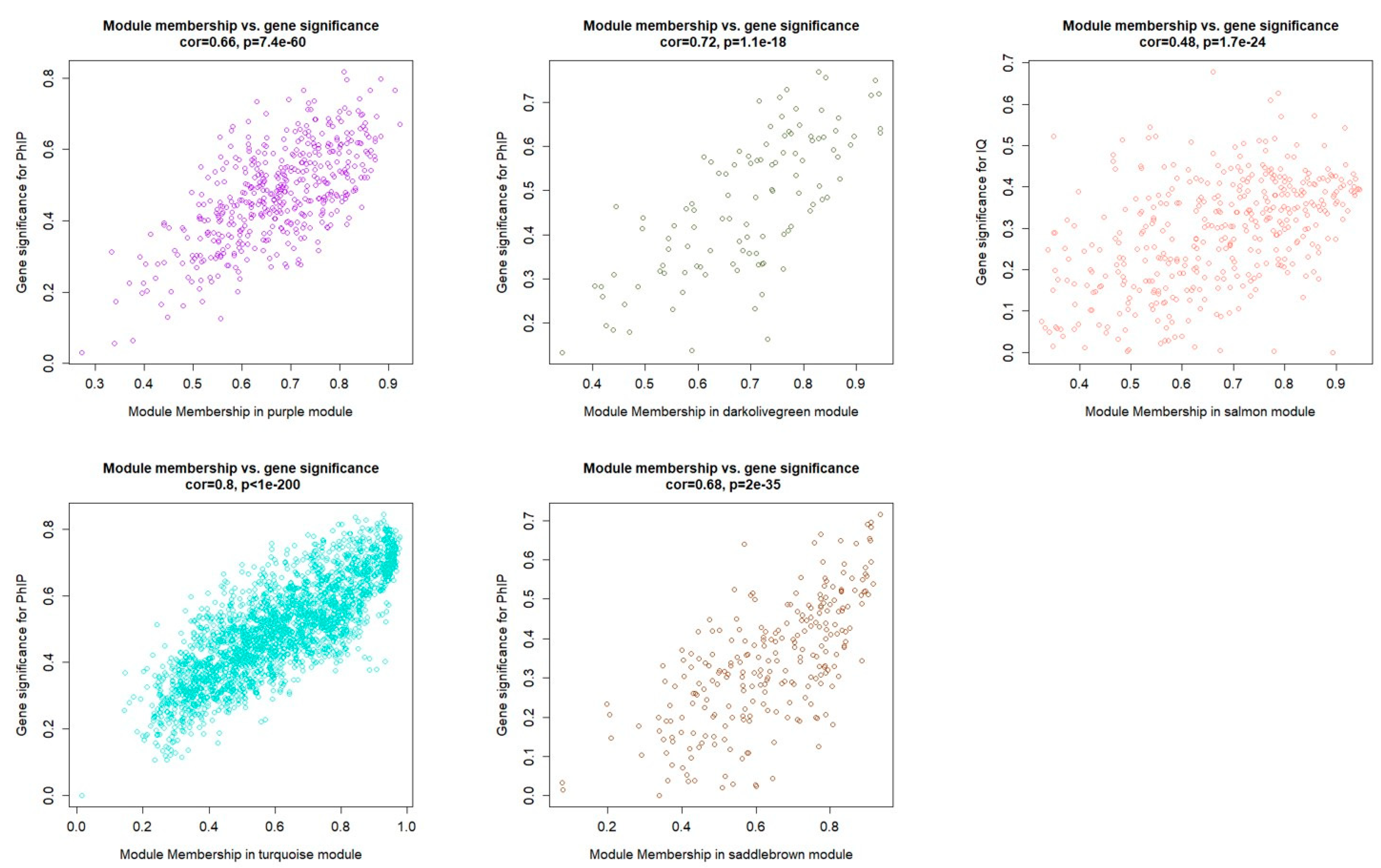
3.1. Weighted Gene Coexpression Network Analysis
3.2. Functional Analysis of the Correlated Gene Moudules
3.3. The Identification of Differentially Expressed Genes
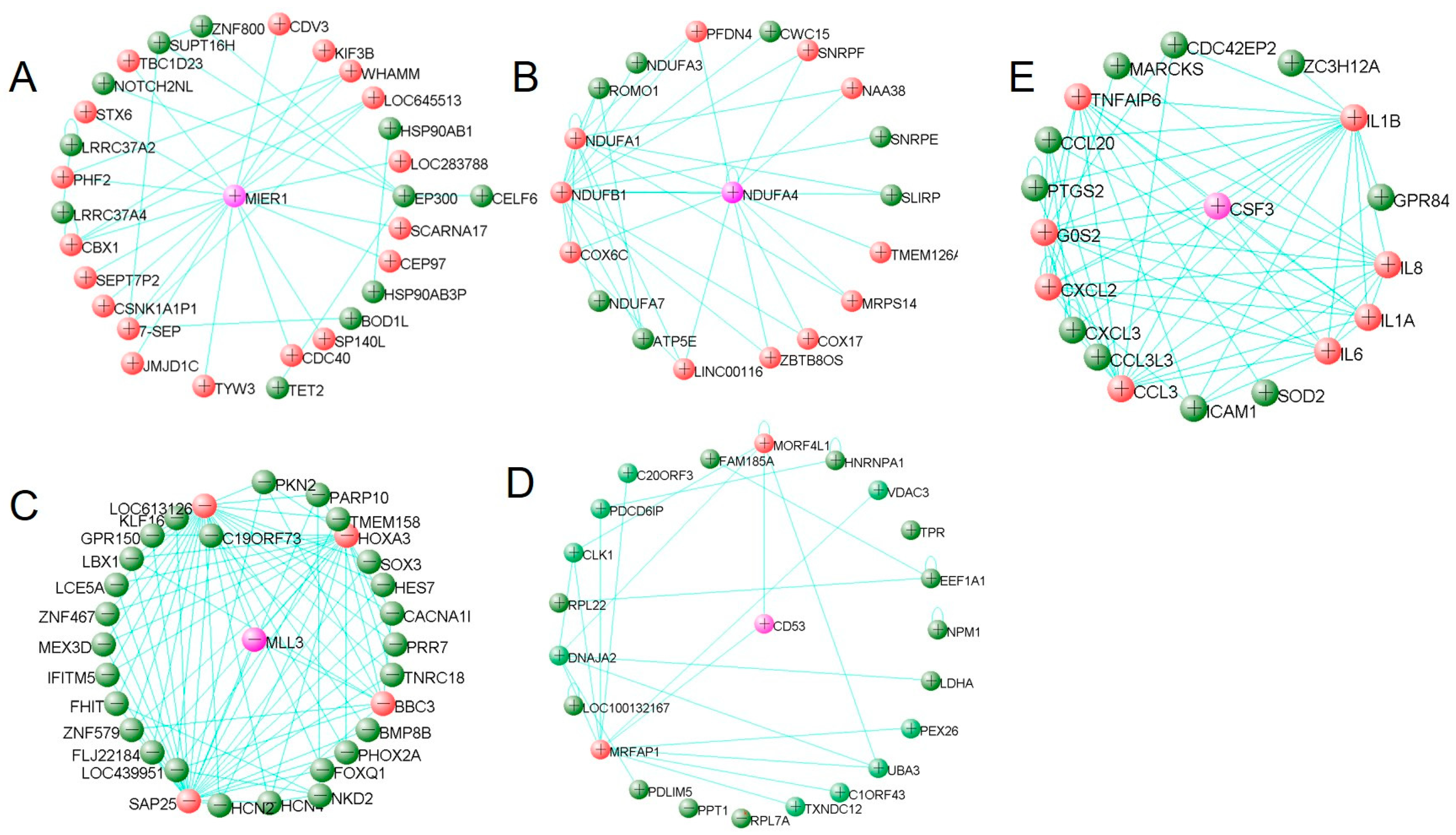
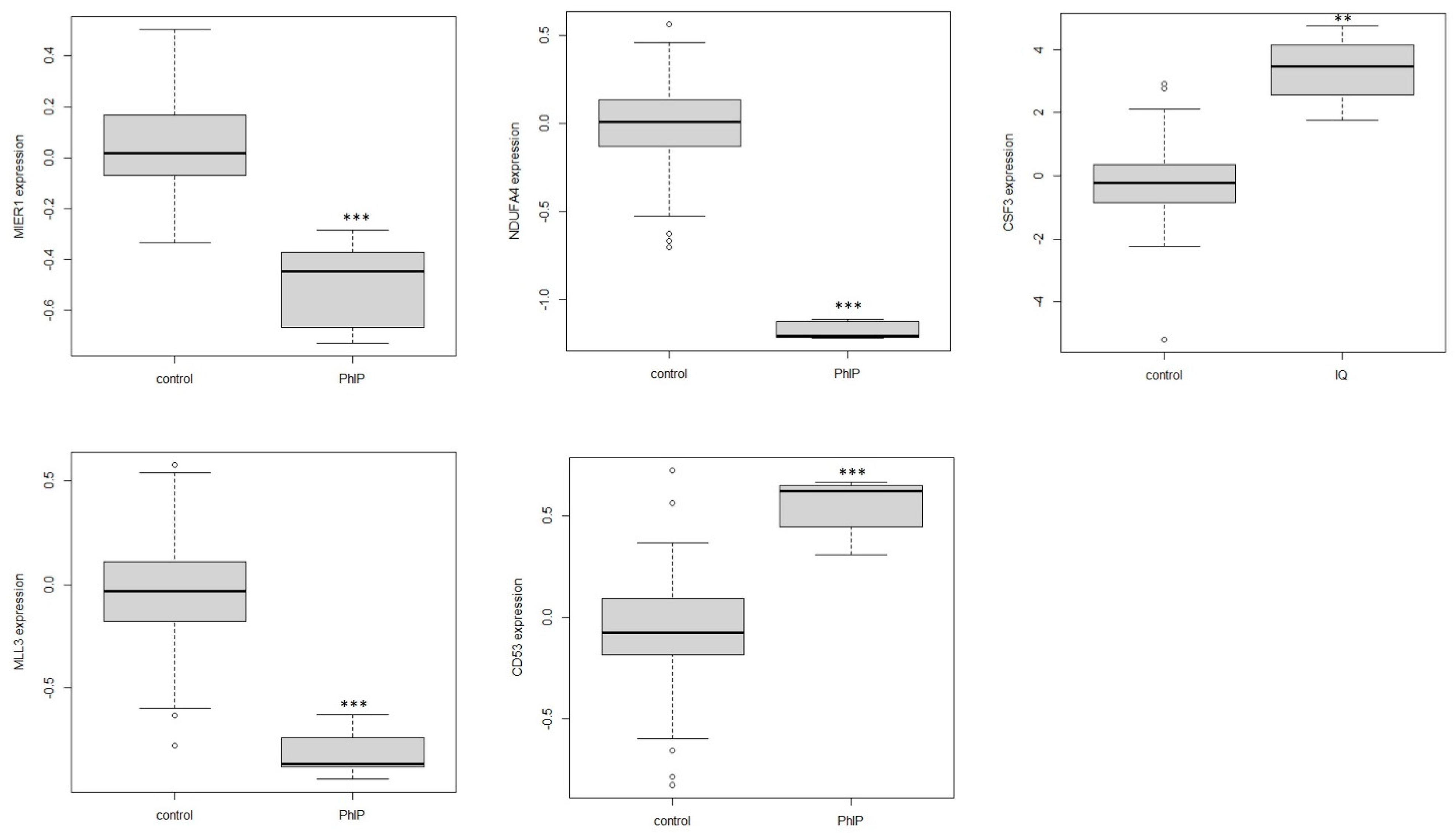
3.4. Hub Gene Identification
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Zamora, R.; Hidalgo, F.J. Formation of heterocyclic aromatic amines with the structure of aminoimidazoazarenes in food products. Food Chem. 2020, 313, 126128. [Google Scholar] [CrossRef]
- Bellamri, M.; Walmsley, S.J.; Turesky, R.J. Metabolism and biomarkers of heterocyclic aromatic amines in humans. Genes Environ. 2021, 43, 29. [Google Scholar] [CrossRef]
- Turesky, R.J. DNA adducts of heterocyclic aromatic amines, arylazides and 4-nitroquinoline 1-oxide. IARC Sci. Publ. 1994, 125, 217–228. [Google Scholar]
- Nakagama, H.; Ochiai, M.; Ubagai, T.; Tajima, R.; Fujiwara, K.; Sugimura, T.; Nagao, M. A rat colon cancer model induced by 2-amino-1-methyl-6-phenylimidazo[4,5-b]pyridine, PhIP. Mutat. Res. 2002, 506–507, 137–144. [Google Scholar] [CrossRef]
- Lo, J.J.; Park, Y.M.; Sinha, R.; Sandler, D.P. Association between meat consumption and risk of breast cancer: Findings from the Sister Study. Int. J. Cancer 2020, 146, 2156–2165. [Google Scholar] [CrossRef]
- Bylsma, L.C.; Alexander, D.D. A review and meta-analysis of prospective studies of red and processed meat, meat cooking methods, heme iron, heterocyclic amines and prostate cancer. Nutr. J. 2015, 14, 125. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sugimura, T.; Wakabayashi, K.; Nakagama, H.; Nagao, M. Heterocyclic amines: Mutagens/carcinogens produced during cooking of meat and fish. Cancer Sci. 2004, 95, 290–299. [Google Scholar] [CrossRef]
- Im, J.; Choi, H.S.; Kim, S.K.; Woo, S.S.; Ryu, Y.H.; Kang, S.S.; Yun, C.H.; Han, S.H. A food-born heterocyclic amine, 2-amino-1-methyl-6-phenylimidazo[4,5-b]pyridine (PhIP), suppresses tumor necrosis factor-α expression in lipoteichoic acid-stimulated RAW 264.7 cells. Cancer Lett. 2009, 274, 109–117. [Google Scholar] [CrossRef]
- Fuccelli, R.; Rosignoli, P.; Servili, M.; Veneziani, G.; Taticchi, A.; Fabiani, R. Genotoxicity of heterocyclic amines (HCAs) on freshly isolated human peripheral blood mononuclear cells (PBMC) and prevention by phenolic extracts derived from olive, olive oil and olive leaves. Food Chem. Toxicol. 2018, 122, 234–241. [Google Scholar] [CrossRef]
- Angarica, V.E.; Del Sol, A. Bioinformatics Tools for Genome-Wide Epigenetic Research. Adv. Exp. Med. Biol. 2017, 978, 489–512. [Google Scholar] [CrossRef]
- Jean-Quartier, C.; Jeanquartier, F.; Jurisica, I.; Holzinger, A. In silico cancer research towards 3R. BMC Cancer 2018, 18, 408. [Google Scholar] [CrossRef]
- Zhao, X.; Shao, Z.; Zhou, X.; Lin, Y.; Guo, J.; Guo, J.; Zhang, Y.; Wang, S. Sub-chronic exposure to PhIP induces oxidative damage and DNA damage, and disrupts the amino acid metabolism in the colons of Wistar rats. Food Chem. Toxicol. 2021, 153, 112249. [Google Scholar] [CrossRef] [PubMed]
- Gu, S.; Sun, D.; Li, X.; Zhang, Z. Alterations of miRNAs and Their Potential Roles in Arsenite-Induced Transformation of Human Bronchial Epithelial Cells. Genes 2017, 8, 254. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, H.; Liu, J.; Li, J.; Zang, D.; Wang, X.; Chen, Y.; Gu, T.; Su, W.; Song, N. Identification of gene modules and hub genes in colon adenocarcinoma associated with pathological stage based on WGCNA analysis. Cancer Genet. 2020, 242, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Zhang, B.; Horvath, S. A general framework for weighted gene co-expression network analysis. Stat. Appl. Genet. Mol. Biol. 2005, 4, 17. [Google Scholar] [CrossRef]
- Langfelder, P.; Horvath, S. WGCNA: An R package for weighted correlation network analysis. BMC Bioinform. 2008, 9, 559. [Google Scholar] [CrossRef] [Green Version]
- Ritchie, M.E.; Phipson, B.; Wu, D.; Hu, Y.; Law, C.W.; Shi, W.; Smyth, G.K. limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 2015, 43, e47. [Google Scholar] [CrossRef]
- Hochstenbach, K.; van Leeuwen, D.M.; Gmuender, H.; Stolevik, S.B.; Nygaard, U.C.; Lovik, M.; Granum, B.; Namork, E.; van Delft, J.H.; van Loveren, H. Transcriptomic profile indicative of immunotoxic exposure: In vitro studies in peripheral blood mononuclear cells. Toxicol. Sci. 2010, 118, 19–30. [Google Scholar] [CrossRef] [Green Version]
- Davis, S.; Meltzer, P.S. GEOquery: A bridge between the Gene Expression Omnibus (GEO) and BioConductor. Bioinformatics 2007, 23, 1846–1847. [Google Scholar] [CrossRef] [Green Version]
- Ashburner, M.; Ball, C.A.; Blake, J.A.; Botstein, D.; Butler, H.; Cherry, J.M.; Davis, A.P.; Dolinski, K.; Dwight, S.S.; Eppig, J.T.; et al. Gene ontology: Tool for the unification of biology. Gene Ontol. Consortium. Nat. Genet. 2000, 25, 25–29. [Google Scholar] [CrossRef] [Green Version]
- Gene Ontology Consortium. The Gene Ontology resource: Enriching a GOld mine. Nucleic Acids Res. 2021, 49, D325–D334. [Google Scholar] [CrossRef]
- Yu, G.; Wang, L.G.; Han, Y.; He, Q.Y. clusterProfiler: An R package for comparing biological themes among gene clusters. Omics 2012, 16, 284–287. [Google Scholar] [CrossRef] [PubMed]
- Oliveros, J.C.; VENNY. An Interactive Tool for Comparing Lists with Venn Diagrams. 2007. Available online: https://bioinfogp.cnb.csic.es/tools/venny/index.html (accessed on 18 November 2021).
- Clements, J.A.; Mercer, F.C.; Paterno, G.D.; Gillespie, L.L. Differential splicing alters subcellular localization of the α but not β isoform of the MIER1 transcriptional regulator in breast cancer cells. PLoS ONE 2012, 7, e32499. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McCarthy, P.L.; Paterno, G.D.; Gillespie, L.L. Protein expression pattern of human MIER1 α, a novel estrogen receptor binding protein. J. Mol. Histol. 2013, 44, 469–479. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Malik, D.E.; David, R.M.; Gooderham, N.J. Ethanol potentiates the genotoxicity of the food-derived mammary carcinogen PhIP in human estrogen receptor-positive mammary cells: Mechanistic support for lifestyle factors (cooked red meat and ethanol) associated with mammary cancer. Arch. Toxicol. 2018, 92, 1639–1655. [Google Scholar] [CrossRef] [Green Version]
- Bennion, B.J.; Cosman, M.; Lightstone, F.C.; Knize, M.G.; Montgomery, J.L.; Bennett, L.M.; Felton, J.S.; Kulp, K.S. PhIP carcinogenicity in breast cancer: Computational and experimental evidence for competitive interactions with human estrogen receptor. Chem. Res. Toxicol. 2005, 18, 1528–1536. [Google Scholar] [CrossRef]
- Pitceathly, R.D.S.; Taanman, J.W. NDUFA4 (Renamed COXFA4) Is a Cytochrome-c Oxidase Subunit. Trends Endocrinol. Metab. 2018, 29, 452–454. [Google Scholar] [CrossRef]
- Zhao, L.; Zhou, N.; Zhang, H.; Pan, F.; Ai, X.; Wang, Y.; Hao, S.; Wang, C. Cyanidin-3-O-glucoside and its metabolite protocatechuic acid ameliorate 2-amino-1-methyl-6-phenylimidazo[4,5-b]pyridine (PhIP) induced cytotoxicity in HepG2 cells by regulating apoptotic and Nrf2/p62 pathways. Food Chem. Toxicol. 2021, 157, 112582. [Google Scholar] [CrossRef]
- Dorighi, K.M.; Swigut, T.; Henriques, T.; Bhanu, N.V.; Scruggs, B.S.; Nady, N.; Still, C.D., 2nd; Garcia, B.A.; Adelman, K.; Wysocka, J. Mll3 and Mll4 Facilitate Enhancer RNA Synthesis and Transcription from Promoters Independently of H3K4 Monomethylation. Mol. Cell 2017, 66, 568–576.e564. [Google Scholar] [CrossRef] [Green Version]
- Im, J.; Kang, S.S.; Yang, J.S.; Yun, C.H.; Yang, Y.; Han, S.H. 3-Amino-1,4-dimethyl-5H-pyrido[4,3-b]indole (Trp-P-1) attenuates LPS-induced IL-8 expression by decreasing mRNA stability in THP-1 cells. Toxicol. Lett. 2008, 177, 108–115. [Google Scholar] [CrossRef]
- Dunlock, V.E. Tetraspanin CD53: An overlooked regulator of immune cell function. Med. Microbiol. Immunol. 2020, 209, 545–552. [Google Scholar] [CrossRef] [PubMed]
- Shi, Y.J.; Hu, S.J.; Zhao, Q.Q.; Liu, X.S.; Liu, C.; Wang, H. Toll-like receptor 4 (TLR4) deficiency aggravates dextran sulfate sodium (DSS)-induced intestinal injury by down-regulating IL6, CCL2 and CSF3. Ann. Transl. Med. 2019, 7, 713. [Google Scholar] [CrossRef] [PubMed]
- Hirt, N.; Body-Malapel, M. Immunotoxicity and intestinal effects of nano- and microplastics: A review of the literature. Part. Fibre Toxicol. 2020, 17, 57. [Google Scholar] [CrossRef] [PubMed]
- Mose, T.; Mathiesen, L.; Karttunen, V.; Nielsen, J.K.; Sieppi, E.; Kummu, M.; Mørck, T.A.; Myöhänen, K.; Partanen, H.; Vähäkangas, K.; et al. Meta-analysis of data from human ex vivo placental perfusion studies on genotoxic and immunotoxic agents within the integrated European project NewGeneris. Placenta 2012, 33, 433–439. [Google Scholar] [CrossRef] [PubMed]
- Davis, D.A.; Archuleta, M.M.; Born, J.L.; Knize, M.G.; Felton, J.S.; Burchiel, S.W. Inhibition of humoral immunity and mitogen responsiveness of lymphoid cells following oral administration of the heterocyclic food mutagen 2-amino-1-methyl-6-phenylimidazo[4,5-b]pyridine (PhIP) to B6C3F1 mice. Fundam. Appl. Toxicol. 1994, 23, 81–86. [Google Scholar] [CrossRef]
- Toropova, A.P.; Toropov, A.A.; Benfenati, E. Semi-correlations as a tool to model for skin sensitization. Food Chem. Toxicol. 2021, 157, 112580. [Google Scholar] [CrossRef]
- Wang, X.Q.; Li, F.; Chen, J.W.; Ji, C.L.; Wu, H.F. Integration of Computational Toxicology, Toxicogenomics Data Mining, and Omics Techniques to Unveil Toxicity Pathways. ACS Sustain. Chem. Eng. 2021, 9, 4130–4138. [Google Scholar] [CrossRef]
- Wang, J.; Wang, C.Y. Integrated miRNA and mRNA omics reveal the anti-cancerous mechanism of Licochalcone B on Human Hepatoma Cell HepG2. Food Chem. Toxicol. 2021, 150, 112096. [Google Scholar] [CrossRef]
- Kleinstreuer, N.C.; Tong, W.; Tetko, I.V. Computational Toxicology. Chem. Res. Toxicol. 2020, 33, 687–688. [Google Scholar] [CrossRef] [Green Version]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, X.; Dong, L.; Yu, H.; Zhang, Y.; Wang, S. Bioinformatic Analysis Identified Hub Genes Associated with Heterocyclic Amines Induced Cytotoxicity of Peripheral Blood Mononuclear Cells. Genes 2021, 12, 1888. https://doi.org/10.3390/genes12121888
Li X, Dong L, Yu H, Zhang Y, Wang S. Bioinformatic Analysis Identified Hub Genes Associated with Heterocyclic Amines Induced Cytotoxicity of Peripheral Blood Mononuclear Cells. Genes. 2021; 12(12):1888. https://doi.org/10.3390/genes12121888
Chicago/Turabian StyleLi, Xinyang, Lu Dong, Huaning Yu, Yan Zhang, and Shuo Wang. 2021. "Bioinformatic Analysis Identified Hub Genes Associated with Heterocyclic Amines Induced Cytotoxicity of Peripheral Blood Mononuclear Cells" Genes 12, no. 12: 1888. https://doi.org/10.3390/genes12121888
APA StyleLi, X., Dong, L., Yu, H., Zhang, Y., & Wang, S. (2021). Bioinformatic Analysis Identified Hub Genes Associated with Heterocyclic Amines Induced Cytotoxicity of Peripheral Blood Mononuclear Cells. Genes, 12(12), 1888. https://doi.org/10.3390/genes12121888