
Variant Selection and Interpretation: An Example of Modified VarSome Classifier of ACMG Guidelines in the Diagnostic Setting

 , , ,
, , ,  ,
,
Abstract
:1. Introduction
2. Materials and Methods
Filtering Benign/Likely Benign Variants through VarSome Stable-API
3. Results
3.1. Varsome Algorithm Modifications
3.2. Exceptions to BA1: Hypomorphic Variants
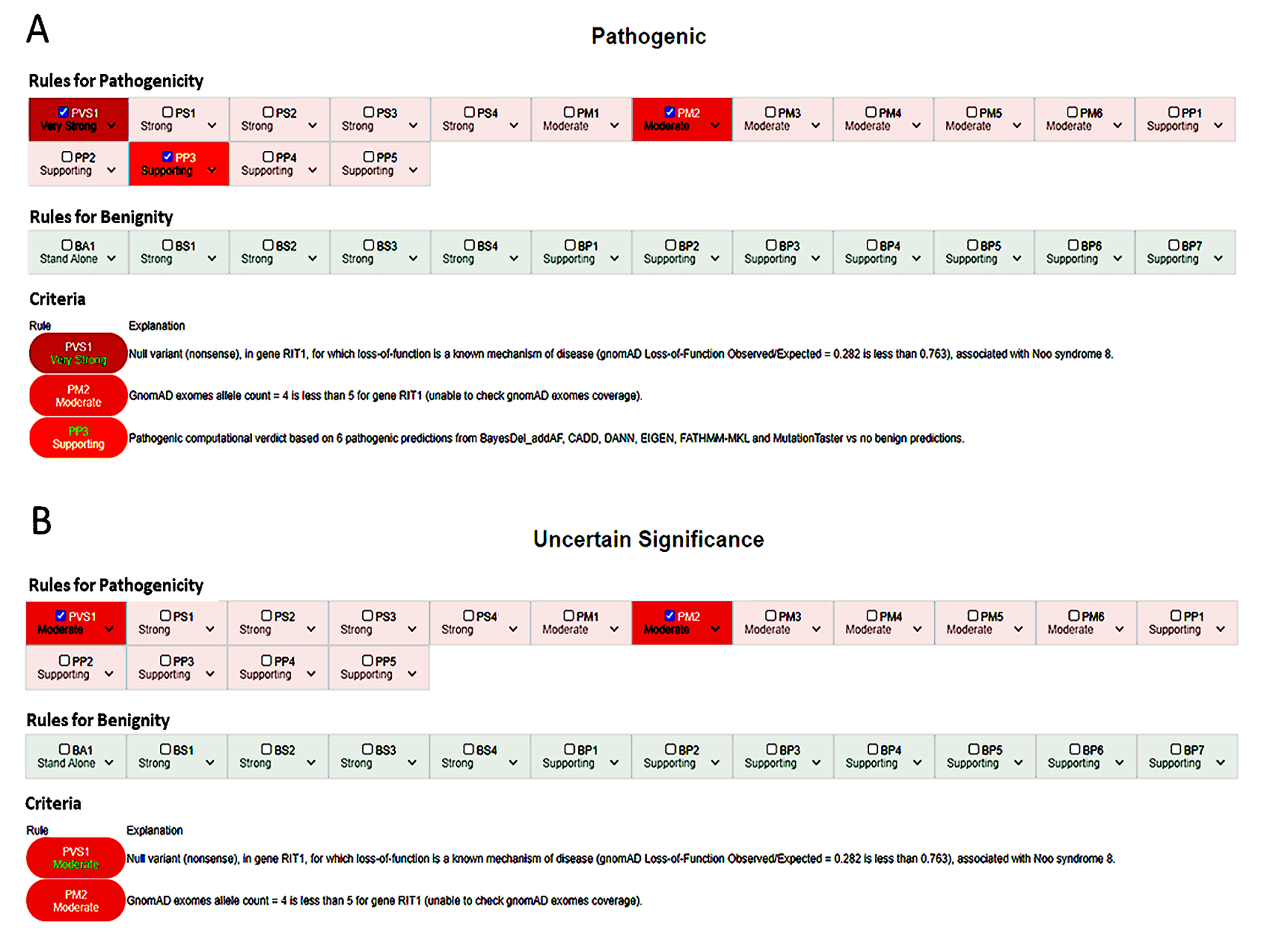
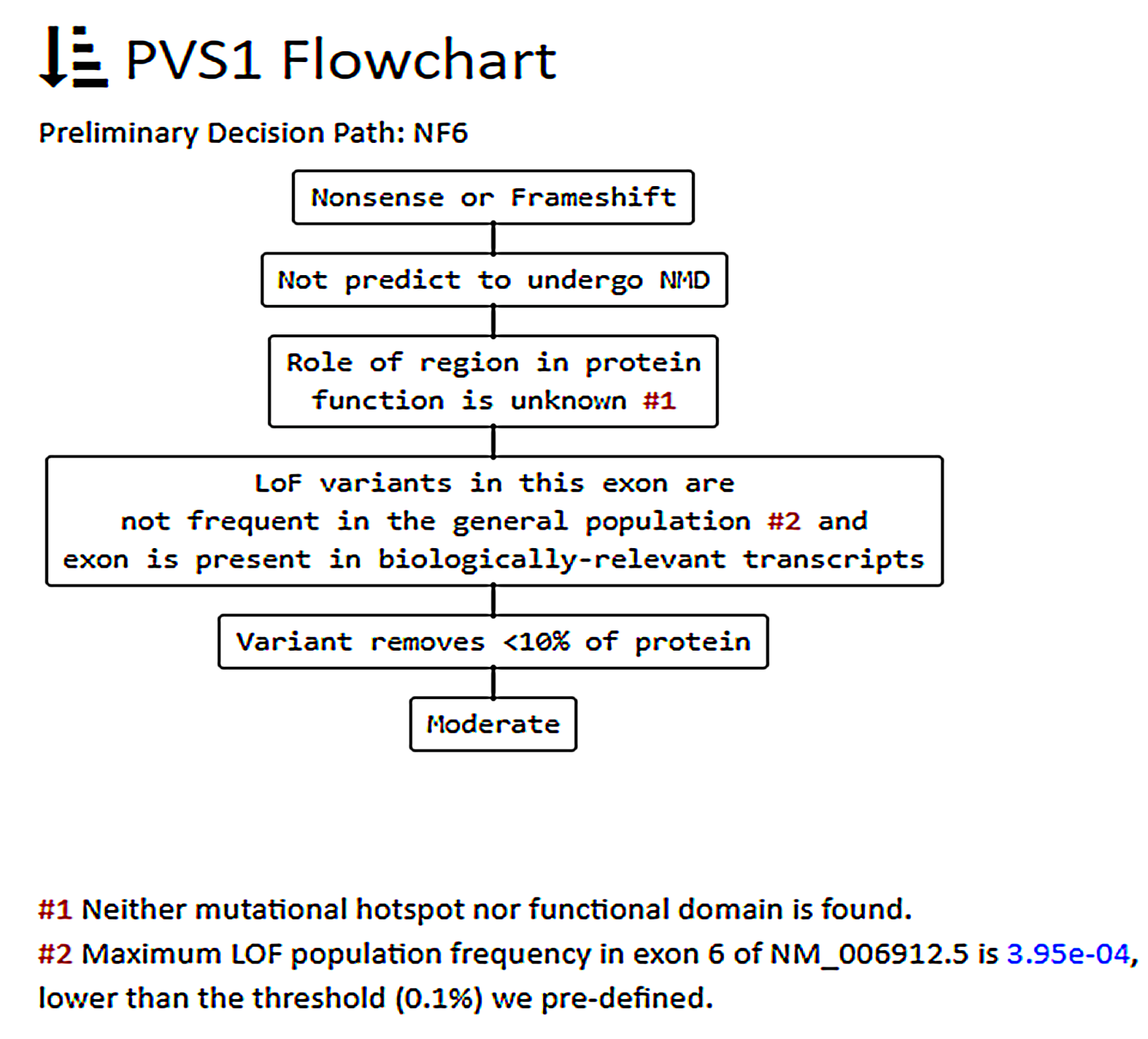
3.3. In-Depth Evaluation of the PVS1 Criterion
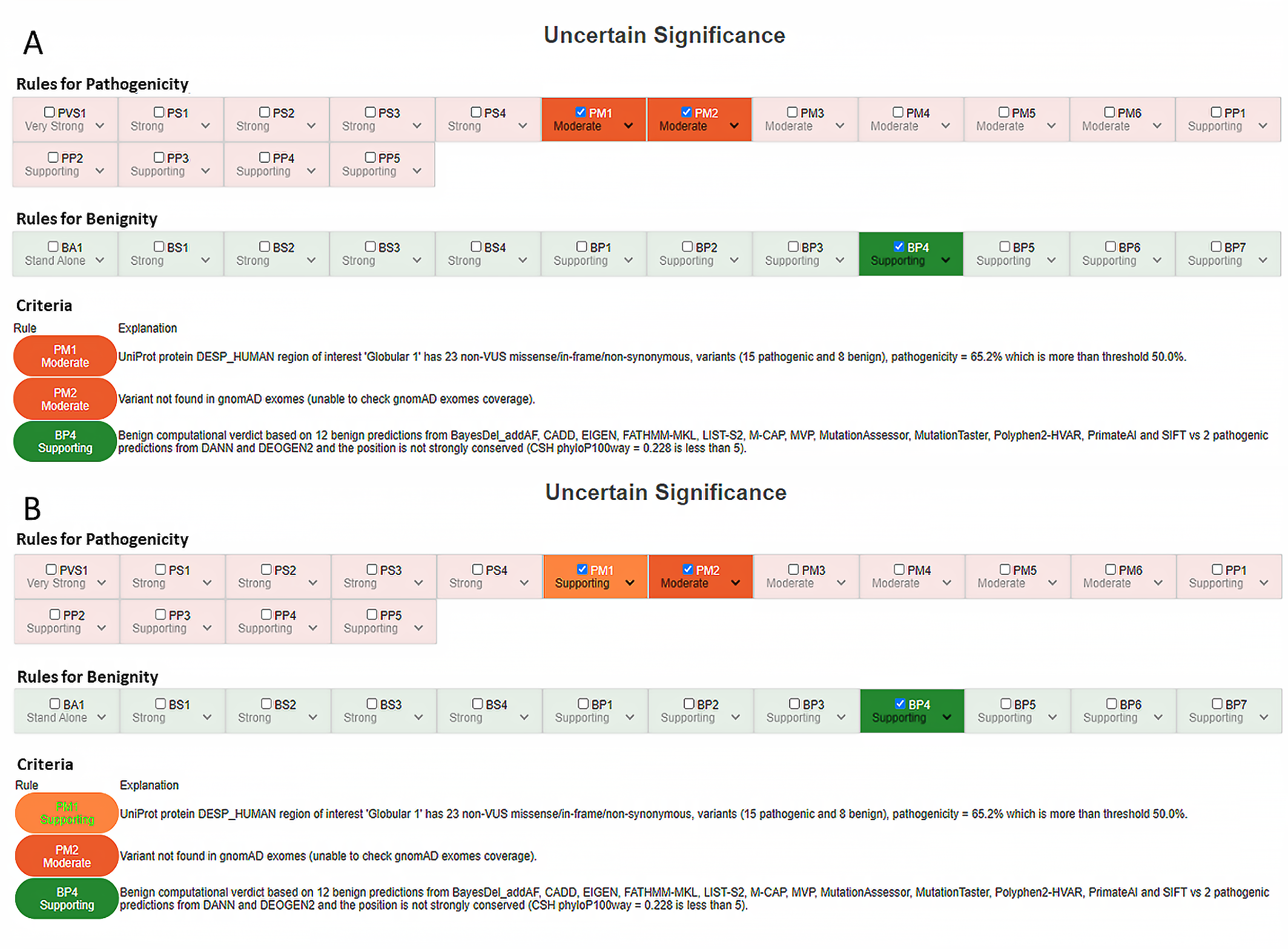
3.4. PM1 Criterion (Mutational Hotspot and/or Critical and Well-Established Functional Domain, without Benign Variation)
3.5. PP3/BP4 Criteria (Functional Predictors)
3.6. Observations on Genes and Associated Inheritance Patterns
3.7. Late-Onset Disorders
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Kazazian, H.; Boehm, C.; Seltzer, W. ACMG recommendations for standards for interpretation of sequence variations. Genet. Med. 2000, 2, 302–303. [Google Scholar] [CrossRef] [Green Version]
- Richards, C.S.; Bale, S.; Bellissimo, D.B.; Das, S.; Grody, W.W.; Hegde, M.R.; Lyon, E.; Ward, B.E. ACMG recommendations for standards for interpretation and reporting of sequence variations: Revisions 2007. Genet. Med. 2008, 10, 294–300. [Google Scholar] [CrossRef] [Green Version]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.; Hegde, M.; Lyon, E.; Spector, E.; et al. Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 2015, 17, 405–424. [Google Scholar] [CrossRef] [PubMed]
- Amendola, L.M.; Jarvik, G.P.; Leo, M.C.; McLaughlin, H.M.; Akkari, Y.; Amaral, M.D.; Berg, J.S.; Biswas, S.; Bowling, K.M.; Conlin, L.K.; et al. Performance of ACMG-AMP Variant-Interpretation Guidelines among Nine Laboratories in the Clinical Sequencing Exploratory Research Consortium. Am. J. Hum. Genet. 2016, 98, 1067–1076. [Google Scholar] [CrossRef] [Green Version]
- Pepin, M.G.; Murray, M.L.; Bailey, S.; Leistritz-Kessler, D.; Schwarze, U.; Byers, P.H. The challenge of comprehensive and consistent sequence variant interpretation between clinical laboratories. Genet. Med. 2016, 18, 20–24. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nykamp, K.; Anderson, M.; Powers, M.; Garcia, J.; Herrera, B.; Ho, Y.Y.; Kobayashi, Y.; Patil, N.; Thusberg, J.; Westbrook, M.; et al. Sherloc: A comprehensive refinement of the ACMG-AMP variant classification criteria. Genet. Med. 2017, 19, 1105–1117. [Google Scholar] [CrossRef] [Green Version]
- Karbassi, I.; Maston, G.A.; Love, A.; Divincenzo, C.; Braastad, C.D.; Elzinga, C.D.; Bright, A.R.; Previte, D.; Zhang, K.; Rowland, C.M.; et al. A standardized DNA variant scoring system for pathogenicity assessments in Mendelian disorders. Hum. Mutat. 2016, 37, 127–134. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jarvik, G.P.; Browning, B.L. Consideration of cosegregation in the pathogenicity classification of genomic variants. Am. J. Hum. Genet. 2016, 98, 1077–1081. [Google Scholar] [CrossRef] [Green Version]
- Tavtigian, S.V.; Greenblatt, M.S.; Harrison, S.M.; Nussbaum, R.L.; Prabhu, S.A.; Boucher, K.M.; Biesecker, L.G. Modeling the ACMG/AMP variant classification guidelines as a Bayesian classification framework. Genet. Med. 2018, 20, 1054–1060. [Google Scholar] [CrossRef] [Green Version]
- Abou Tayoun, A.N.; Pesaran, T.; DiStefano, M.T.; Oza, A.; Rehm, H.L.; Biesecker, L.G.; Harrison, S.M. Recommendations for interpreting the loss of function PVS1 ACMG/AMP variant criterion. Hum. Mutat. 2018, 39, 1517–1524. [Google Scholar] [CrossRef]
- Ghosh, R.; Harrison, S.M.; Rehm, H.L.; Plon, S.E.; Biesecker, L.G. Updated recommendation for the benign stand-alone ACMG/AMP criterion. Hum. Mutat. 2018, 39, 1525–1530. [Google Scholar] [CrossRef]
- Biesecker, L.G.; Harrison, S.M. The ACMG/AMP reputable source criteria for the interpretation of sequence variants. Genet. Med. 2018, 20, 1687–1688. [Google Scholar] [CrossRef]
- Brnich, S.E.; Tayoun, A.N.A.; Couch, F.J.; Cutting, G.R.; Greenblatt, M.S.; Heinen, C.D.; Kanavy, D.M.; Luo, X.; McNulty, S.M.; Starita, L.M.; et al. Recommendations for application of the functional evidence PS3/BS3 criterion using the ACMG/AMP sequence variant interpretation framework. bioRxiv 2019, 709428. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oza, A.M.; DiStefano, M.T.; Hemphill, S.E.; Cushman, B.J.; Grant, A.R.; Siegert, R.K.; Shen, J.; Chapin, A.; Boczek, N.J.; Schimmenti, L.A.; et al. Expert specification of the ACMG/AMP variant interpretation guidelines for genetic hearing loss. Hum. Mutat. 2018, 39, 1593–1613. [Google Scholar] [CrossRef]
- Gelb, B.D.; Cavé, H.; Dillon, M.W.; Gripp, K.W.; Lee, J.A.; Mason-Suares, H.; Rauen, K.A.; Williams, B.; Zenker, M.; Vincent, L.M. ClinGen’s RASopathy Expert Panel consensus methods for variant interpretation. Genet. Med. 2018, 20, 1334–1345. [Google Scholar] [CrossRef] [Green Version]
- Lee, K.; Krempely, K.; Roberts, M.E.; Anderson, M.J.; Carneiro, F.; Chao, E.; Dixon, K.; Figueiredo, J.; Ghosh, R.; Huntsman, D.; et al. Specifications of the ACMG/AMP variant curation guidelines for the analysis of germline CDH1 sequence variants. Hum. Mutat. 2018, 39, 1553–1568. [Google Scholar] [CrossRef]
- Johnston, J.J.; Dirksen, R.T.; Girard, T.; Gonsalves, S.G.; Hopkins, P.M.; Riazi, S.; Saddic, L.A.; Sambuughin, N.; Saxena, R.; Stowell, K.; et al. Variant curation expert panel recommendations for RYR1 pathogenicity classifications in malignant hyperthermia susceptibility. Genet. Med. 2021, 17, 1288–1295. [Google Scholar] [CrossRef] [PubMed]
- McCormick, E.M.; Lott, M.T.; Dulik, M.C.; Shen, L.; Attimonelli, M.; Vitale, O.; Karaa, A.; Bai, R.; Pineda-Alvarez, D.E.; Singh, L.N.; et al. Specifications of the ACMG/AMP standards and guidelines for mitochondrial DNA variant interpretation. Hum. Mutat. 2020, 41, 2028–2057. [Google Scholar] [CrossRef] [PubMed]
- Shen, J.; Oza, A.M.; del Castillo, I.; Duzkale, H.; Matsunaga, T.; Pandya, A.; Kang, H.P.; Mar-Heyming, R.; Guha, S.; Moyer, K.; et al. Consensus interpretation of the p.Met34Thr and p.Val37Ile variants in GJB2 by the ClinGen Hearing Loss Expert Panel. Genet. Med. 2019, 21, 2442–2452. [Google Scholar] [CrossRef]
- Harrison, S.M.; Dolinsky, J.S.; Knight Johnson, A.E.; Pesaran, T.; Azzariti, D.R.; Bale, S.; Chao, E.C.; Das, S.; Vincent, L.; Rehm, H.L. Clinical laboratories collaborate to resolve differences in variant interpretations submitted to ClinVar. Genet. Med. 2017, 19, 1096–1104. [Google Scholar] [CrossRef] [Green Version]
- Harrison, S.M.; Dolinksy, J.S.; Chen, W.; Collins, C.D.; Das, S.; Deignan, J.L.; Garber, K.B.; Garcia, J.; Jarinova, O.; Knight Johnson, A.E.; et al. Scaling resolution of variant classification differences in ClinVar between 41 clinical laboratories through an outlier approach. Hum. Mutat. 2018, 39, 1641–1649. [Google Scholar] [CrossRef]
- Kopanos, C.; Tsiolkas, V.; Kouris, A.; Chapple, C.E.; Albarca Aguilera, M.; Meyer, R.; Massouras, A. VarSome: The human genomic variant search engine. Bioinformatics 2019, 35, 1978–1980. [Google Scholar] [CrossRef] [PubMed]
- Sorrentino, E.; Cristofoli, F.; Modena, C.; Marceddu, G. Integrate VarSome API for automate ACMG clinical variant interpretation. Acta Biomed. 2021. accepted. [Google Scholar]
- Marceddu, G.; Dallavilla, T.; Guerri, G.; Manara, E.; Chiurazzi, P.; Bertelli, M. PipeMagi: An integrated and validated workflow for analysis of NGS data for clinical diagnostics. Eur. Rev. Med. Pharmacol. Sci. 2019, 12, 6753–6765. [Google Scholar]
- Marceddu, G.; Dallavilla, T.; Xhuvani, A.; Daja, M.; De Antoni, L.; Casadei, A.; Bertelli, M. AppMAGI: A complete laboratory information management system for clinical diagnostics. Acta Biomed. 2020, 91, 1–7. [Google Scholar] [CrossRef]
- Liu, X.; Li, C.; Mou, C.; Dong, Y.; Tu, Y. dbNSFP v4: A comprehensive database of transcript-specific functional predictions and annotations for human nonsynonymous and splice-site SNVs. Genome Med. 2020, 12, 1–8. [Google Scholar] [CrossRef] [PubMed]
- McLaren, W.; Gil, L.; Hunt, S.E.; Riat, H.S.; Ritchie, G.R.S.; Thormann, A.; Flicek, P.; Cunningham, F. The Ensembl Variant Effect Predictor. Genome Biol. 2016, 17, 1–14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Karczewski, K.J.; Francioli, L.C.; Tiao, G.; Cummings, B.B.; Alföldi, J.; Wang, Q.; Collins, R.L.; Laricchia, K.M.; Ganna, A.; Birnbaum, D.P.; et al. The mutational constraint spectrum quantified from variation in 141,456 humans. Nature 2020, 581, 434. [Google Scholar] [CrossRef] [PubMed]
- Ellard, S.; Baple, E.L.; Berry, I.; Forrester, N.; Turnbull, C.; Owens, M.; Eccles, D.M.; Abbs, S.; Scott, R.; Deans, Z.C.; et al. ACGS Best Practice Guidelines for Variant Classification 2019. 2019. Available online: https://www.acgs.uk.com/news/acgs-best-practice-guidelines-for-variant-classification-2019/ (accessed on 15 April 2021).
- Cremers, F.P.M.; Lee, W.; Collin, R.W.J.; Allikmets, R. Clinical spectrum, genetic complexity and therapeutic approaches forretinal disease caused by ABCA4 mutations. Prog. Retin. Eye Res. 2020, 79, 100861. [Google Scholar] [CrossRef]
- Zernant, J.; Lee, W.; Collison, F.T.; Fishman, G.A.; Sergeev, Y.V.; Schuerch, K.; Sparrow, J.R.; Tsang, S.H.; Allikmets, R. Frequent hypomorphic alleles account for a significant fraction of ABCA4 disease and distinguish it from age-related macular degeneration. J. Med. Genet. 2017, 54, 404. [Google Scholar] [CrossRef] [PubMed]
- Fukai, K.; Holmes, S.A.; Lucchese, N.J.; Siu, V.M.; Weleber, R.G.; Schnur, R.E.; Spritz, R.A. Autosomal recessive ocular albinism associated with a functionally significant tyrosinase gene polymorphism. Nat. Genet. 1995, 9, 92–95. [Google Scholar] [CrossRef]
- Simeonov, D.R.; Wang, X.; Wang, C.; Sergeev, Y.; Dolinska, M.; Bower, M.; Fischer, R.; Winer, D.; Dubrovsky, G.; Balog, J.Z.; et al. DNA Variations in oculocutaneous albinism: An updated mutation list and current outstanding issues in molecular diagnostics. Hum. Mutat. 2013, 34, 827–835. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xiang, J.; Peng, J.; Baxter, S.; Peng, Z. AutoPVS1: An automatic classification tool for PVS1 interpretation of null variants. Hum. Mutat. 2020, 41, 1488–1498. [Google Scholar] [CrossRef]
- Ellard, S.; Baple, E.L.; Callaway, A.; Berry, I.; Forrester, N.; Turnbull, C.; Owens, M.; Eccles, D.M.; Abbs, S.; Scott, R.; et al. ACGS Best Practice Guidelines for Variant Classification in Rare Disease 2020. 2020. Available online: https://www.acgs.uk.com/media/11631/uk-practice-guidelines-for-variant-classification-v4-01-2020.pdf (accessed on 15 April 2021).
- Houge, G.; Laner, A.; Cirak, S.; de Leeuw, N.; Scheffer, H.; den Dunnen, J.T. Stepwise ABC system for classification of any type of genetic variant. Eur. J. Hum. Genet. 2021, 1–10. [Google Scholar] [CrossRef] [PubMed]
- MacDonald, I.M.; Lee, T.; Lawrence, J. Bestrophinopathies; Adam, M.P., Ardinger, H.H., Pagon, R.A., Wallace, S.E., Bean, L.J.H., Stephens, K., Amemiya, A., Eds.; University of Washington: Seattle, WA, USA, 2020. [Google Scholar]
- Majewski, I.J.; Kluijt, I.; Cats, A.; Scerri, T.S.; De Jong, D.; Kluin, R.J.C.; Hansford, S.; Hogervorst, F.B.L.; Bosma, A.J.; Hofland, I.; et al. An α-E-catenin (CTNNA1) mutation in hereditary diffuse gastric cancer. J. Pathol. 2013, 229, 621–629. [Google Scholar] [CrossRef]
- Clark, D.F.; Michalski, S.T.; Tondon, R.; Nehoray, B.; Ebrahimzadeh, J.; Hughes, S.K.; Soper, E.R.; Domchek, S.M.; Rustgi, A.K.; Pineda-Alvarez, D.; et al. Loss-of-function variants in CTNNA1 detected on multigene panel testing in individuals with gastric or breast cancer. Genet. Med. 2020, 22, 840–846. [Google Scholar] [CrossRef] [Green Version]
- Saksens, N.T.M.; Krebs, M.P.; Schoenmaker-Koller, F.E.; Hicks, W.; Yu, M.; Shi, L.; Rowe, L.; Collin, G.B.; Charette, J.R.; Letteboer, S.J.; et al. Mutations in CTNNA1 cause butterfly-shaped pigment dystrophy and perturbed retinal pigment epithelium integrity. Nat. Genet. 2016, 48, 144–151. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tanner, A.; Chan, H.W.; Pulido, J.S.; Arno, G.; Ba-Abbad, R.; Jurkute, N.; Robson, A.G.; Egan, C.A.; Knight, H.; Calcagni, A.; et al. Clinical and genetic findings in CTNNA1-associated macular pattern dystrophy. Ophthalmology 2021, 128, 952–955. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
| Pathogenic | (i) 1 Very strong (PVS1) AND (a) ≥1 Strong (PS1–PS4) OR (b) ≥2 Moderate (PM1–PM6) OR (c) 1 Moderate (PM1–PM6) AND at least * 1 Supporting (PP1–PP5) OR (d) ≥2 Supporting (PP1–PP5) OR (e)≥1 Very strong (PVS1) * (ii) ≥2 Strong (PS1–PS4) OR (iii) 1 Strong (PS1–PS4) AND (a)≥3 Moderate (PM1–PM6) OR (b)2 Moderate (PM1–PM6) AND ≥ 2 Supporting (PP1–PP5) OR (c)1 Moderate (PM1–PM6) AND ≥ 4 supporting (PP1–PP5) |
| Likely Pathogenic | (i) 1 Very strong (PVS1) AND 1 moderate (PM1– PM6) OR (ii) 1 Strong (PS1–PS4) AND 1–2 moderate (PM1–PM6) OR (iii) 1 Strong (PS1–PS4) AND ≥ 2 supporting (PP1–PP5) OR (iv) ≥3 Moderate (PM1–PM6) OR (v) 2 Moderate (PM1–PM6) AND ≥ 2 supporting (PP1–PP5) OR (vi) 1 Moderate (PM1–PM6) AND ≥ 3 supporting (PP1–PP5) |
| Benign | (i) 1 Stand-alone (BA1) OR (ii) ≥2 Strong (BS1–BS4) |
| Likely benign | (i) 1 Strong (BS1–BS4) ° OR (ii) ≥2 Supporting (BP1–BP7) |
| Uncertain significance | (i) Other criteria shown above are not met OR (ii) the criteria for benign and pathogenic are contradictory |
| BENIGN CRITERIA | ||
|---|---|---|
| ACMG CRITERIA | ASSIGNED BY VARSOME | EXCEPTIONS AND INDICATIONS |
| BA1 Allele frequency > 5% in Exome Sequencing Project, 1000 Genomes Project or Exome Aggregation Consortium | YES | Variants recommended in Ghosh et al. 2018 - Hypomorphic variants: NM_000350 (ABCA4):c.5603A>T (p.Asn1868Ile) NM_000372 (TYR):c.1205G>A (p.Arg402Gln) |
| BS1 Allele frequency greater than expected for disorder | YES (not evaluated if BA1 or PM2 are activated) | Use STRONG as default. For AD diseases with high penetrance, the criterion can be used as STAND-ALONE evidence (sufficient to classify a variant as likely benign). |
| BS2 Observed in a healthy adult for a recessive (homozygous), dominant (heterozygous), or X-linked (hemizygous) disorder, with full penetrance expected at an early age | YES (not evaluated if BA1 or PM2 are activated) | Since VarSome retrieves information from the CGD database, in cases of known inheritance discrepancies, use the following gnomAD cutoffs * to include other variants in the selection: - AR/XL model: <3 homozygotes/hemizygotes in gnomAD exomes&genomes - AD model: <5 heterozygotes in gnomAD exomes&genomes * These rules are used to decide whether variants in genes with AD/AR inheritance should be reported in “Primary” or “Secondary” results in the clinical report (“Primary”: any P/LP variant in genes associated with AD or AR diseases, or VUS in AD genes; “Secondary”: any VUS in AR genes or VUS in AD/AR genes with ≥5 heterozygotes in gnomAD). |
| BS3 Well-established in vitro or in vivo functional studies show no damaging effect on protein function or splicing | VARIABLE | Consult PUBMED, LOVD and other available databases (Mastermind, LitVar, etc.) to find functional evidence [13]. |
| BS4 Lack of segregation in affected members of a family | NO | Segregation analysis required. |
| BP1 Missense variant in a gene for which primarily truncating variants are known to cause disease | YES (Mutually exclusive vs. PP2) | Use SUPPORTING as default. |
| BP2 Observed in trans with a pathogenic variant for a fully penetrant dominant gene/disorder or observed in cis with a pathogenic variant in any inheritance pattern | NO | Segregation analysis required, use SUPPORTING as default. Use STRONG if the condition is confirmed in many individuals (literature or internal evidence) or with different variants. |
| BP3 In-frame deletions/insertions in a repetitive region without a known function | YES | Use SUPPORTING as default. |
| BP4 Multiple lines of computational evidence suggest no impact on gene or gene product (conservation, evolutionary, splicing impact, etc.) | YES | - Prediction must be based on at least 3 total and concordant predictors, otherwise exclude the criterion. - Do not assign to any variant with PVS1 activated. - The criterion must not be assigned to canonical splicing variants (±1–2) if PVS1 is assigned. If available, ADA and RF scores § can be used to assign the criterion to intronic variants. - Do not use ADA and RF scores to assign the criterion to synonymous variants if BP7 is already assigned. |
| BP5 Variant found in a case with an alternative molecular basis for disease | NO | Segregation analysis or literature evidence required. Use SUPPORTING as default. |
| BP6 Reputable source recently reports variant as benign, but the evidence is not available to the laboratory to perform an independent evaluation | YES | Exclude the criterion if the variant has “Review status” 0 stars in ClinVar and there are no other submissions in other clinical databases (e.g., LOVD). Be aware that certain UniProt classifications might be outdated. |
| BP7 A synonymous (silent) variant for which splicing prediction algorithms predict no impact on the splice consensus sequence nor creation of a new splice site AND the nucleotide is not highly conserved | YES | Use SUPPORTING as default. |
| PATHOGENIC CRITERIA | ||
| ACMG CRITERIA | ASSIGNED BY VARSOME | EXCEPTIONS AND INDICATIONS |
| PVS1 Null variant (nonsense, frameshift, canonical ±1 or 2 splice sites, initiation codon, single or multiexon deletion) in a gene where LoF is a known mechanism of disease | YES | - Modify the criterion strength according to Abu Tayoun et al. 2018 [10]. - the criterion does not apply to variants in the first/last base of an exon (not considered canonical in the ACMG guidelines). - use SUPPORTING if NMD is not predicted (variant in the last exon or in the last 50 bps of the second-last exon) AND there are no other P/LP variants downstream. |
| PS1 Same amino acid change as a previously established pathogenic variant regardless of nucleotide change | YES | Use STRONG as default, reduce to SUPPORTING if the alternative variant is classified as likely pathogenic. Always check interpretation of alternative variant. |
| PS2: De novo (confirmed maternity and paternity) in a patient with the disease and no family history | NO | Segregation analysis required. |
| PS3: Well-established in vitro or in vivo functional studies supporting a damaging effect on the gene or gene product | VARIABLE | - Consult PUBMED, LOVD, and other available databases (Mastermind, LitVar, etc.) to identify functional evidence - Modify criterion strength according to evidence relevance [13] |
| PS4 The prevalence of the variant in affected individuals is significantly higher than in controls | NO | - Use the criterion at STRONG level if prevalence data (cases/controls) are available - Use the criterion when the variant has been reported in at least 5 unrelated affected individuals in the laboratory |
| PM1: Located in a mutational hotspot and/or critical and well-established functional domain (e.g., the active site of an enzyme) without benign variation | YES | - Use MODERATE as default. - Reduce to SUPPORTING with <10 variants in the domain |
| PM2: Absent in controls (or extremely low frequency if recessive) in Exome Sequencing Project, 1000 Genomes Project, or Exome Aggregation Consortium | YES | Use MODERATE as default Since VarSome employs the CGD database, in case of known inheritance discrepancies, use the following gnomAD cutoffs * to include other variants in the selection: - AR/XL model: <3 homozygotes/hemizygotes in gnomAD exomes&genomes - AD model: <5 heterozygotes in gnomAD exomes&genomes * These rules are used to decide if variants in genes with AD/AR inheritance should be reported in “Primary” or “Secondary” results (“Primary”: any P/LP variant in genes associated with AD or AR disease, or VUS in AD genes; “Secondary”: any VUS in AR genes, or VUS in AD/AR genes with ≥5 heterozygotes in gnomAD). |
| PM3 For recessive disorders, detected in trans with a pathogenic variant | NO | - Use MODERATE as default - Use SUPPORTING if variant found in trans with only one other LP/P variant in one affected individual or for homozygous genotypes [29] - Upgrade to STRONG if found in trans with multiple different pathogenic variants or in multiple affected individuals (in the literature or in using internal segregation evidence). |
| PM4 Protein length changes as a result of in-frame deletions/insertions in a non-repeat region or stop-loss variants | YES (not applicable if PVS1 is enabled) | Use MODERATE as default. |
| PM5: Novel missense change at an amino acid residue where a different missense change determined to be pathogenic has been seen before. | YES | Reduce to SUPPORTING in dubious cases, be aware that certain UniProt classification might be outdated, therefore always check interpretation of different missense changes. |
| PM6: Assumed de novo, but without confirmation of paternity and maternity. | NO | It is possible to modify criterion strength according to the compatibility of the proband’s phenotype with the disease associated with the gene and if the variant has been found de novo in other non-consanguineous individuals in the internal database. (Further implementations ongoing to refine grading). |
| PP1: Cosegregation with disease in multiple affected family members in a gene definitively known to cause the disease. | NO | Use: - STRONG: ≥4 total affected persons including the one tested - MODERATE: 2–3 affected persons including the tested individual - SUPPORTING: 1 affected person including the tested individual |
| PP2: Missense variant in a gene that has a low rate of benign missense variation and in which missense variants are a common mechanism of disease. | YES | Use SUPPORTING as default. |
| PP3: Multiple lines of computational evidence support a deleterious effect on the gene or gene product (conservation, evolutionary, splicing impact, etc.) | YES | - Prediction must be based on at least 3 total and concordant predictors, otherwise exclude the criterion. - Do not assign to any variant with PVS1 activated. - The criterion must not be assigned to canonical splicing variants (±1–2) if PVS1 is assigned. If available, ADA and RF scores§ can be used to assign the criterion to intronic variants. |
| PP4: Patient’s phenotype or family history is highly specific for a disease with a single genetic etiology. | NO | - Use SUPPORTING for diseases with no more than 5 associated genes (e.g., Stargardt disease) - Use MODERATE for true single-gene disorders (e.g., CHM for choroideremia). |
| PP5: Reputable source recently reports variant as pathogenic, but the evidence is not available to the laboratory to perform an independent evaluation. | YES | Exclude the criterion if the variant has “Review status” 0 stars in ClinVar and there are no other submissions in other clinical databases (e.g., LOVD). Be aware that certain UniProt classifications might be outdated. |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cristofoli, F.; Sorrentino, E.; Guerri, G.; Miotto, R.; Romanelli, R.; Zulian, A.; Cecchin, S.; Paolacci, S.; Miertus, J.; Bertelli, M.; et al. Variant Selection and Interpretation: An Example of Modified VarSome Classifier of ACMG Guidelines in the Diagnostic Setting. Genes 2021, 12, 1885. https://doi.org/10.3390/genes12121885
Cristofoli F, Sorrentino E, Guerri G, Miotto R, Romanelli R, Zulian A, Cecchin S, Paolacci S, Miertus J, Bertelli M, et al. Variant Selection and Interpretation: An Example of Modified VarSome Classifier of ACMG Guidelines in the Diagnostic Setting. Genes. 2021; 12(12):1885. https://doi.org/10.3390/genes12121885
Chicago/Turabian StyleCristofoli, Francesca, Elisa Sorrentino, Giulia Guerri, Roberta Miotto, Roberta Romanelli, Alessandra Zulian, Stefano Cecchin, Stefano Paolacci, Jan Miertus, Matteo Bertelli, and et al. 2021. "Variant Selection and Interpretation: An Example of Modified VarSome Classifier of ACMG Guidelines in the Diagnostic Setting" Genes 12, no. 12: 1885. https://doi.org/10.3390/genes12121885
APA StyleCristofoli, F., Sorrentino, E., Guerri, G., Miotto, R., Romanelli, R., Zulian, A., Cecchin, S., Paolacci, S., Miertus, J., Bertelli, M., Maltese, P. E., Chiurazzi, P., Stuppia, L., Castori, M., & Marceddu, G. (2021). Variant Selection and Interpretation: An Example of Modified VarSome Classifier of ACMG Guidelines in the Diagnostic Setting. Genes, 12(12), 1885. https://doi.org/10.3390/genes12121885