
X-Linked Osteogenesis Imperfecta Possibly Caused by a Novel Variant in PLS3

 ,
,  , , ,
, , , 
Abstract
:1. Introduction
2. Materials and Methods
2.1. Subjects
2.2. Clinical Examination
2.3. Genetic Testing
2.4. Bioinformatics Analysis of Gene Variants of Unknown Significance
3. Results
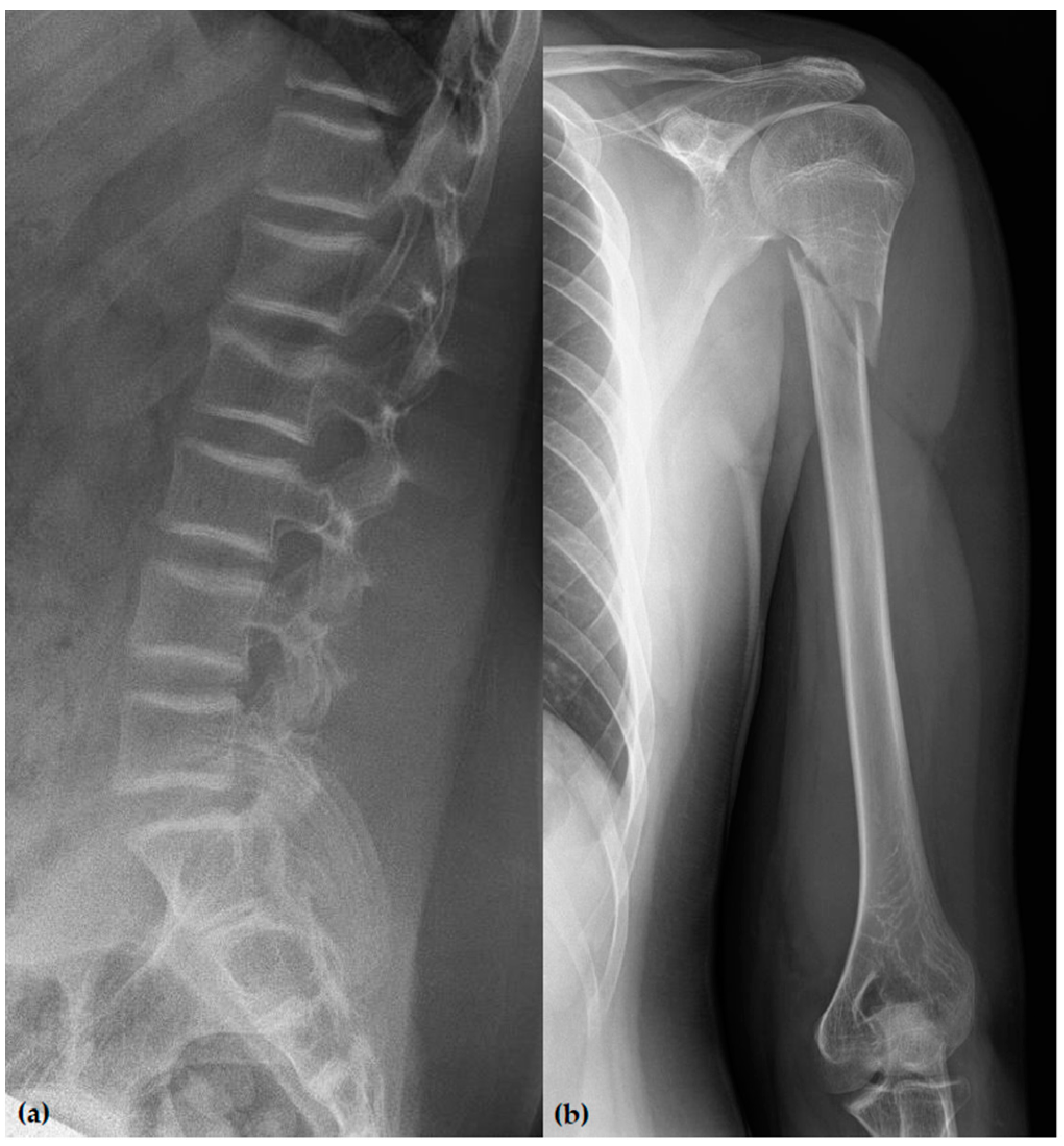
3.1. Phenotypes of the Patients
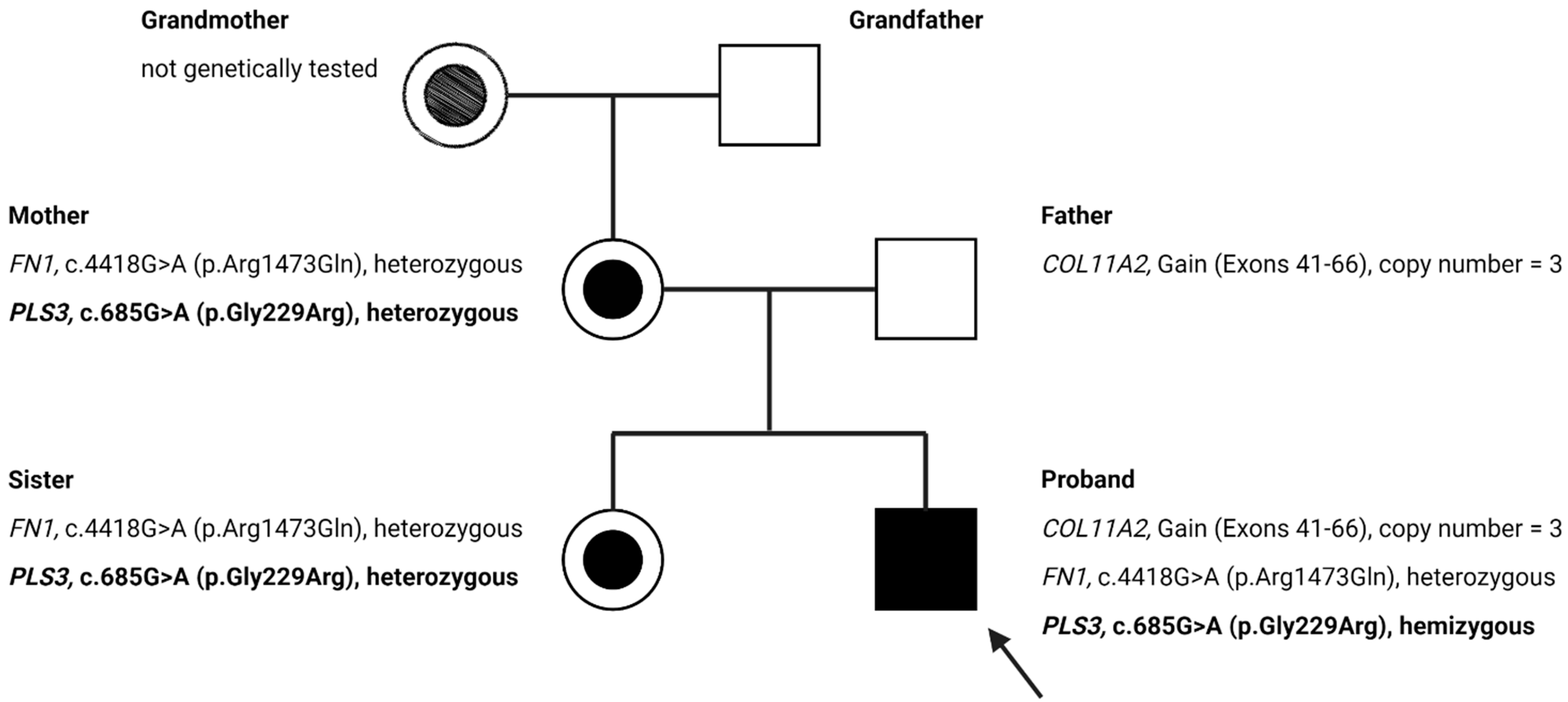
3.2. Genetic Findings
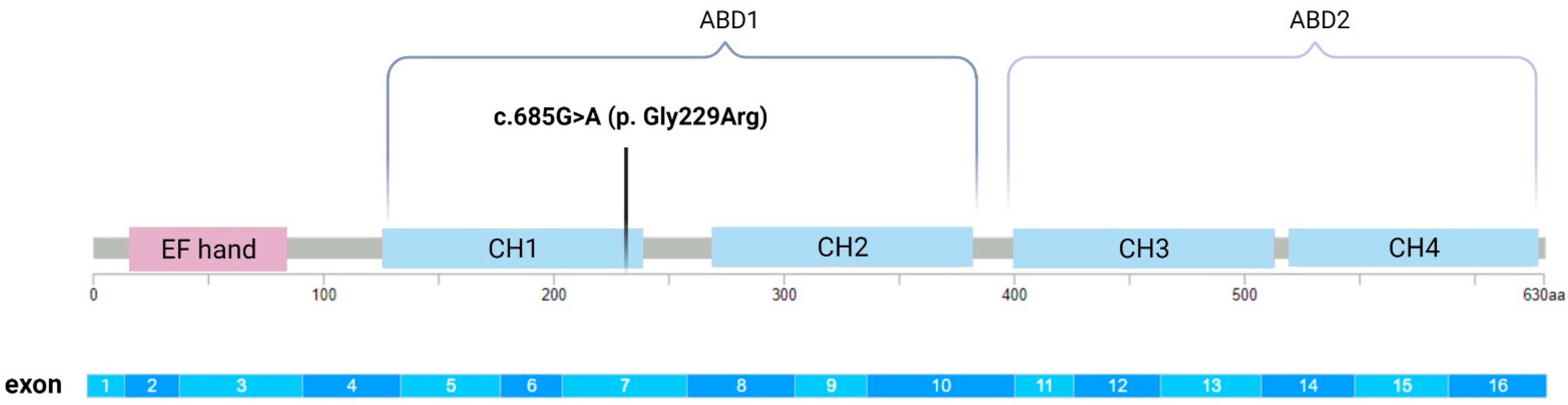
3.3. Bioinformatics Analysis
3.4. The Effect of Vitamin D Treatment
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Jovanovic, M.; Guterman-Ram, G.; Marini, J.C. Osteogenesis Imperfecta: Mechanisms and Signaling Pathways Connecting Classical and Rare OI Types. Endocr. Rev. 2021, bnab017. [Google Scholar] [CrossRef] [PubMed]
- Marini, J.C.; Dang Do, A.N. Osteogenesis Imperfecta. In Endotext; Feingold, K.R., Anawalt, B., Boyce, A., Chrousos, G., de Herder, W.W., Dhatariya, K., Dungan, K., Grossman, A., Hershman, J.M., Hofland, J., et al., Eds.; MDText.com, Inc.: South Dartmouth, MA, USA, 2020. [Google Scholar]
- Primorac, D.; Rowe, D.W.; Mottes, M.; Barisić, I.; Anticević, D.; Mirandola, S.; Lira, M.G.; Kalajzić, I.; Kusec, V.; Glorieux, F.H. Osteogenesis imperfecta at the beginning of bone and joint decade. Croat. Med. J. 2001, 42, 392–414. [Google Scholar]
- Marom, R.; Rabenhorst, B.M.; Morello, R. Management of Endocrine Disease: Osteogenesis imperfecta: An update on clinical features and therapies. Eur. J. Endocrinol. 2020, 183, R95–R106. [Google Scholar] [CrossRef] [PubMed]
- Agarwal, M.; Goheen, M.; Jia, S.; Ling, S.; White, E.S.; Kim, K.K. Type I Collagen Signaling Regulates Opposing Fibrotic Pathways through α2β1 Integrin. Am. J. Respir. Cell Mol. Biol. 2020, 63, 613–622. [Google Scholar] [CrossRef] [PubMed]
- Stover, M.L.; Primorac, D.; Liu, S.C.; McKinstry, M.B.; Rowe, D.W. Defective Splicing of mRNA from One COL1A Allele of Type I Collagen in Nondeforming (Type I) Osteogenesis Imperfecta. J. Clin. Investig. 1993, 92, 1994–2002. [Google Scholar] [CrossRef]
- Johnson, C.V.; Primorac, D.; McKinstry, M.; Rowe, D.W.; Lawrence, J.B. Tracking COL1A1 RNA in Osteogenesis Imperfecta: Splice-defective Transcripts Initiate Transport from the Gene but are Retained within the SC35 Domain. J. Cell Biol. 2000, 150, 417–432. [Google Scholar] [CrossRef] [Green Version]
- Fratzl-Zelman, N.; Wesseling-Perry, K.; Mäkitie, R.E.; Blouin, S.; Hartmann, M.A.; Zwerina, J.; Välimäki, V.-V.; Laine, C.M.; Välimäki, M.J.; Pereira, R.C.; et al. Bone material properties and response to teriparatide in osteoporosis due to WNT1 and PLS3 mutations. Bone 2021, 146, 115900. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.-M.; Lin, C.; Stavre, Z.; Greenblatt, M.B.; Shim, J.-H. Osteoblast-Osteoclast Communication and Bone Homeostasis. Cells 2020, 9, 2073. [Google Scholar] [CrossRef]
- Guasto, A.; Cormier-Daire, V. Signaling Pathways in Bone Development and Their Related Skeletal Dysplasia. Int. J. Mol. Sci. 2021, 22, 4321. [Google Scholar] [CrossRef]
- El-Gazzar, A.; Högler, W. Mechanisms of Bone Fragility: From Osteogenesis Imperfecta to Secondary Osteoporosis. Int. J. Mol. Sci. 2021, 22, 625. [Google Scholar] [CrossRef]
- Etich, J.; Rehberg, M.; Eckes, B.; Sengle, G.; Semler, O.; Zaucke, F. Signaling pathways affected by mutations causing osteogenesis imperfecta. Cell. Signal. 2020, 76, 109789. [Google Scholar] [CrossRef]
- Rossi, V.; Lee, B.; Marom, R. Osteogenesis imperfecta: Advancements in genetics and treatment. Curr. Opin. Pediatr. 2019, 31, 708–715. [Google Scholar] [CrossRef]
- Hu, J.; Li, L.; Zheng, W.; Zhao, D.; Wang, O.; Jiang, Y.; Xing, X.; Li, M.; Xia, W. A novel mutation in PLS3 causes extremely rare X-linked osteogenesis imperfecta. Mol. Genet. Genom. Med. 2020, 8, e1525. [Google Scholar] [CrossRef]
- Wolff, L.; Strathmann, E.A.; Müller, I.; Mählich, D.; Veltman, C.; Niehoff, A.; Wirth, B. Plastin 3 in health and disease: A matter of balance. Cell. Mol. Life Sci. 2021, 78, 5275–5301. [Google Scholar] [CrossRef]
- Schwebach, C.L.; Kudryashova, E.; Kudryashov, D.S. Plastin 3 in X-Linked Osteoporosis: Imbalance of Ca2+-Dependent Regulation Is Equivalent to Protein Loss. Front. Cell Dev. Biol. 2021, 8, 1885. [Google Scholar] [CrossRef] [PubMed]
- Chen, T.; Wu, H.; Zhang, C.; Feng, J.; Chen, L.; Xie, R.; Wang, F.; Chen, X.; Zhou, H.; Sun, H.; et al. Clinical, Genetics, and Bioinformatic Characterization of Mutations Affecting an Essential Region of PLS3 in Patients with BMND18. Int. J. Endocrinol. 2018, 2018, 8953217. [Google Scholar] [CrossRef]
- Truty, R.; Paul, J.; Ms, M.K.; Bs, S.E.L.; Olivares, E.; Nussbaum, R.L.; Aradhya, S. Prevalence and properties of intragenic copy-number variation in Mendelian disease genes. Genet. Med. 2019, 21, 114–123. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lincoln, S.E.; Hambuch, T.; Zook, J.M.; Bristow, S.L.; Hatchell, K.; Truty, R.; Kennemer, M.; Shirts, B.H.; Fellowes, A.; Chowdhury, S.; et al. One in seven pathogenic variants can be challenging to detect by NGS: An analysis of 450,000 patients with implications for clinical sensitivity and genetic test implementation. Genet. Med. 2021, 23, 1673–1680. [Google Scholar] [CrossRef] [PubMed]
- Basta, M.; Pandya, A.M. Genetics, X-Linked Inheritance. In StatPearls; StatPearls Publishing: Treasure Island, FL, USA, 2021. [Google Scholar]
- Mäkitie, R.E.; Niinimäki, T.; Suo-Palosaari, M.; Kämpe, A.; Costantini, A.; Toiviainen-Salo, S.; Niinimäki, J.; Mäkitie, O. PLS3 Mutations Cause Severe Age and Sex-Related Spinal Pathology. Front. Endocrinol. 2020, 11, 393. [Google Scholar] [CrossRef] [PubMed]
- Balasubramanian, M.; Fratzl-Zelman, N.; O’Sullivan, R.; Bull, M.; Peel, N.F.; Pollitt, R.C.; Jones, R.; Milne, E.; Smith, K.; Roschger, P.; et al. Novel PLS3 variants in X-linked osteoporosis: Exploring bone material properties. Am. J. Med Genet. Part A 2018, 176, 1578–1586. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Bian, X.; Cheng, G.; Zhao, P.; Xiang, X.; Tian, W.; Li, T.; Zhai, Q. A novel nonsense variant in PLS3 causes X-linked osteoporosis in a Chinese family. Ann. Hum. Genet. 2020, 84, 92–96. [Google Scholar] [CrossRef] [PubMed]
- Velthaus, A.; Cornils, K.; Hennigs, J.K.; Grüb, S.; Stamm, H.; Wicklein, D.; Bokemeyer, C.; Heuser, M.; Windhorst, S.; Fiedler, W.; et al. The Actin Binding Protein Plastin-3 Is Involved in the Pathogenesis of Acute Myeloid Leukemia. Cancers 2019, 11, 1663. [Google Scholar] [CrossRef] [Green Version]
- Schwebach, C.L.; Kudryashova, E.; Zheng, W.; Orchard, M.; Smith, H.; Runyan, L.A.; Egelman, E.H.; Kudryashov, D.S. Osteogenesis imperfecta mutations in plastin 3 lead to impaired calcium regulation of actin bundling. Bone Res. 2020, 8, 21. [Google Scholar] [CrossRef] [PubMed]
- Yorgan, T.A.; Sari, H.; Rolvien, T.; Windhorst, S.; Failla, A.V.; Kornak, U.; Oheim, R.; Amling, M.; Schinke, T. Mice lacking plastin-3 display a specific defect of cortical bone acquisition. Bone 2020, 130, 115062. [Google Scholar] [CrossRef]
- Wang, W.; Sarazin, B.A.; Kornilowicz, G.; Lynch, M. Mechanically-Loaded Breast Cancer Cells Modify Osteocyte Mechanosensitivity by Secreting Factors That Increase Osteocyte Dendrite Formation and Downstream Resorption. Front. Endocrinol. 2018, 9, 352. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Dijk, F.S.; Zillikens, M.C.; Micha, D.; Riessland, M.; Marcelis, C.L.; De Die-Smulders, C.E.; Milbradt, J.; Franken, A.A.; Harsevoort, A.J.; Lichtenbelt, K.D.; et al. PLS3 Mutations in X-Linked Osteoporosis with Fractures. N. Engl. J. Med. 2013, 369, 1529–1536. [Google Scholar] [CrossRef] [Green Version]
- Fahiminiya, S.; Majewski, J.; Al-Jallad, H.; Moffatt, P.; Mort, J.; Glorieux, F.H.; Roschger, P.; Klaushofer, K.; Rauch, F. Osteoporosis Caused by Mutations inPLS3: Clinical and Bone Tissue Characteristics. J. Bone Miner. Res. 2014, 29, 1805–1814. [Google Scholar] [CrossRef]
- Neugebauer, J.; Heilig, J.; Hosseini-Barkooie, S.; Ross, B.C.; Mendoza-Ferreira, N.; Nolte, F.; Peters, M.; Hölker, I.; Hupperich, K.; Tschanz, T.; et al. Plastin 3 influences bone homeostasis through regulation of osteoclast activity. Hum. Mol. Genet. 2018, 27, 4249–4262. [Google Scholar] [CrossRef]
- Roschger, P.; Paschalis, E.; Fratzl, P.; Klaushofer, K. Bone mineralization density distribution in health and disease. Bone 2008, 42, 456–466. [Google Scholar] [CrossRef]
- Blouin, S.; Fratzl-Zelman, N.; Glorieux, F.H.; Roschger, P.; Klaushofer, K.; Marini, J.C.; Rauch, F. Hypermineralization and High Osteocyte Lacunar Density in Osteogenesis Imperfecta Type V Bone Indicate Exuberant Primary Bone Formation. J. Bone Miner. Res. 2017, 32, 1884–1892. [Google Scholar] [CrossRef] [Green Version]
- Kämpe, A.J.; Costantini, A.; Levy-Shraga, Y.; Zeitlin, L.; Roschger, P.; Taylan, F.; Lindstrand, A.; Paschalis, E.P.; Gamsjaeger, S.; Raas-Rothschild, A.; et al. PLS3 Deletions Lead to Severe Spinal Osteoporosis and Disturbed Bone Matrix Mineralization. J. Bone Miner. Res. 2017, 32, 2394–2404. [Google Scholar] [CrossRef] [Green Version]
- Kamioka, H.; Sugawara, Y.; Honjo, T.; Yamashiro, T.; Takano-Yamamoto, T. Terminal Differentiation of Osteoblasts to Osteocytes Is Accompanied by Dramatic Changes in the Distribution of Actin-Binding Proteins. J. Bone Miner. Res. 2004, 19, 471–478. [Google Scholar] [CrossRef] [PubMed]
- Yamauchi, M.; Sricholpech, M.; Terajima, M.; Tomer, K.B.; Perdivara, I. Glycosylation of Type I Collagen. Springer Protoc. Handb. 2019, 1934, 127–144. [Google Scholar]
- Hanagata, N. IFITM5 mutations and osteogenesis imperfecta. J. Bone Miner. Metab. 2016, 34, 123–131. [Google Scholar] [CrossRef] [PubMed]
- Al-Jallad, H.; Palomo, T.; Roughley, P.; Glorieux, F.H.; McKee, M.D.; Moffatt, P.; Rauch, F. The effect of SERPINF1 in-frame mutations in osteogenesis imperfecta type VI. Bone 2015, 76, 115–120. [Google Scholar] [CrossRef] [PubMed]
- Komori, T. Runx2, an inducer of osteoblast and chondrocyte differentiation. Histochem. Cell Biol. 2018, 149, 313–323. [Google Scholar] [CrossRef]
- Liu, S.-C.; Sun, Q.-Z.; Qiao, X.-F.; Li, X.-G.; Yang, J.-H.; Wang, T.-Q.; Xiao, Y.-J.; Qiao, J.-M. LncRNA TUG1 influences osteoblast proliferation and differentiation through the Wnt/β-catenin signaling pathway. Eur. Rev. Med. Pharmacol. Sci. 2019, 23, 4584–4590. [Google Scholar]
- Chang, M.-K.; Kramer, I.; Keller, H.; Gooi, J.H.; Collett, C.; Jenkins, D.; Ettenberg, S.A.; Cong, F.; Halleux, C.; Kneissel, M. Reversing LRP5-Dependent Osteoporosis and SOST Deficiency-Induced Sclerosing Bone Disorders by Altering WNT Signaling Activity. J. Bone Miner. Res. 2014, 29, 29–42. [Google Scholar] [CrossRef]
- Yasuda, H. Discovery of the RANKL/RANK/OPG system. J. Bone Miner. Metab. 2021, 39, 2–11. [Google Scholar] [CrossRef]
- Lee, S.-Y.; Long, F. Notch signaling suppresses glucose metabolism in mesenchymal progenitors to restrict osteoblast differentiation. J. Clin. Investig. 2018, 128, 5573–5586. [Google Scholar] [CrossRef] [Green Version]
- Götherström, C.; Walther-Jallow, L. Stem Cell Therapy as a Treatment for Osteogenesis Imperfecta. Curr. Osteoporos. Rep. 2020, 18, 337–343. [Google Scholar] [CrossRef] [PubMed]
- Dwan, K.; Phillipi, C.A.; Steiner, R.; Basel, D. Bisphosphonate therapy for osteogenesis imperfecta. Cochrane Database Syst. Rev. 2016, 10, CD005088. [Google Scholar] [CrossRef] [PubMed]
- Biggin, A.; Munns, C.F. Long-Term Bisphosphonate Therapy in Osteogenesis Imperfecta. Curr. Osteoporos. Rep. 2017, 15, 412–418. [Google Scholar] [CrossRef]
- Boyce, A.M. Denosumab: An Emerging Therapy in Pediatric Bone Disorders. Curr. Osteoporos. Rep. 2017, 15, 283–292. [Google Scholar] [CrossRef] [PubMed]
- Delgado-Calle, J.; Sato, A.Y.; Bellido, T. Role and mechanism of action of sclerostin in bone. Bone 2017, 96, 29–37. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Paik, J.; Scott, L.J. Romosozumab: A Review in Postmenopausal Osteoporosis. Drugs Aging 2020, 37, 845–855. [Google Scholar] [CrossRef] [PubMed]
- Iolascon, G.; Moretti, A.; Toro, G.; Gimigliano, F.; Liguori, S.; Paoletta, M. Pharmacological Therapy of Osteoporosis: What’s New? Clin. Interv. Aging 2020, 15, 485–491. [Google Scholar] [CrossRef] [Green Version]
- Chen, G.; Deng, C.; Li, Y.-P. TGF-β and BMP Signaling in Osteoblast Differentiation and Bone Formation. Int. J. Biol. Sci. 2012, 8, 272–288. [Google Scholar] [CrossRef] [Green Version]
- Jeleč, Ž.; Primorac, D.; Antičević, D. Personalized surgery approach in severe form of osteogenesis imperfecta type III: Point of view. J. Pediatr. Orthop. B 2019, 28, 505–508. [Google Scholar] [CrossRef]
- Wang, X.; Thomsen, P. Mesenchymal stem cell–derived small extracellular vesicles and bone regeneration. Basic Clin. Pharmacol. Toxicol. 2021, 128, 18–36. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
| GENE | TRANSCRIPT | GENE | TRANSCRIPT | GENE | TRANSCRIPT | GENE | TRANSCRIPT |
|---|---|---|---|---|---|---|---|
| ACAN | NM_013227.3 | CDKN1C | NM_000076.2 | DHCR24 | NM_014762.3 | C2CD3 | NM_015531.5 |
| ACP5 | NM_001111035.2 | CDT1 | NM_030928.3 | DIP2C | NM_014974.2 | CA2 | NM_000067.2 |
| ACVR1 | NM_001105.4 | CENPJ | NM_018451.4 | DLL3 | NM_016941.3 | CANT1 | NM_138793.3 |
| ADAMTS10 | NM_030957.3 | CEP120 | NM_153223.3 | DLX3 | NM_005220.2 | CASR | NM_000388.3 |
| ADAMTS17 | NM_139057.3 | CEP135 | NM_025009.4 | DMRT2 | NM_006557.6 | CCDC8 | NM_032040.4 |
| AFF4 | NM_014423.3 | CEP152 | NM_014985.3 | DNA2 | NM_001080449.2 | CDC45 | NM_001178010.2 |
| AGA | NM_000027.3 | CEP63 | NM_025180.3 | DONSON | NM_017613.3 | CDC6 | NM_001254.3 |
| AGPS | NM_003659.3 | CFAP410 | NM_004928.2 | DVL1 | NM_004421.2 | FN1 | NM_212482.2 |
| AIFM1 | NM_004208.3 | CHST14 | NM_130468.3 | DVL3 | NM_004423.3 | FTO | NM_001080432.2 |
| ALPL | NM_000478.5 | CHST3 | NM_004273.4 | DYM | NM_017653.3 | FUCA1 | NM_000147.4 |
| AMER1 | NM_152424.3 | CHUK | NM_001278.4 | DYNC2H1 | NM_001080463.1 | FZD2 | NM_001466.3 |
| ANKH | NM_054027.4 | CLCN7 | NM_001287.5 | DYNC2LI1 | NM_016008.3 | GALNS | NM_000512.4 |
| ANO5 | NM_213599.2 | COG1 | NM_018714.2 | EBP | NM_006579.2 | GALNT3 | NM_004482.3 |
| ARCN1 | NM_001655.4 | COL10A1 | NM_000493.3 | EIF2AK3 | NM_004836.6 | GDF5 | NM_000557.4 |
| ARSB | NM_000046.3 | COL11A1 | NM_001854.3 | ESCO2 | NM_001017420.2 | GDF6 | NM_001001557.2 |
| ARSE | NM_000047.2 | COL11A2 | NM_080680.2 | EVC | NM_153717.2 | GHR | NM_000163.4 |
| ASCC1 | NM_001198800.2 | COL1A1 | NM_000088.3 | EVC2 | NM_147127.4 | GHRHR | NM_000823.3 |
| ASPM | NM_018136.4 | COL1A2 | NM_000089.3 | EXOC6B | NM_001321729.1 | GHSR | NM_198407.2 |
| ATR | NM_001184.3 | COL27A1 | NM_032888.3 | EXOSC2 | NM_014285.6 | CSGALNACT1 | NM_001130518.1 |
| B3GALT6 | NM_080605.3 | COL2A1 | NM_001844.4 | EXT1 | NM_000127.2 | CSPP1 | NM_024790.6 |
| B3GAT3 | NM_012200.3 | COL9A1 | NM_001851.4 | EXT2 | NM_207122.1 | CTSA | NM_000308.3 |
| B4GALT7 | NM_007255.2 | COL9A2 | NM_001852.3 | EXTL3 | NM_001440.3 | CTSK | NM_000396.3 |
| BGN | NM_001711.5 | COL9A3 | NM_001853.3 | FAM20C | NM_020223.3 | CUL7 | NM_014780.4 |
| BMP1 | NM_006129.4 | COMP | NM_000095.2 | FAM46A | NM_017633.2 | CWC27 | NM_005869.3 |
| BMP2 | NM_001200.3 | CREB3L1 | NM_052854.3 | FAR1 | NM_032228.5 | DDR2 | NM_006182.2 |
| BMPER | NM_133468.4 | CRTAP | NM_006371.4 | FBN1 | NM_000138.4 | DDRGK1 | NM_023935.2 |
| BMPR1B | NM_001203.2 | CSF1R | NM_005211.3 | FGF23 | NM_020638.2 | IFT43 | NM_052873.2 |
| IFT52 | NM_001303458.2 | IFT122 | NM_052985.3 | LIG4 | NM_002312.3 | MYH3 | NM_002470.3 |
| IFT57 | NM_018010.3 | IFT140 | NM_014714.3 | LMNA | NM_170707.3 | MYO18B | NM_032608.6 |
| IFT74 | NM_001099222.1 | IFT172 | NM_015662.2 | LMX1B | NM_002316.3 | NAGLU | NM_000263.3 |
| IFT80 | NM_020800.2 | IDUA | NM_000203.4 | LONP1 | NM_004793.3 | NANS | NM_018946.3 |
| IFT81 | NM_014055.3 | IFITM5 | NM_001025295.2 | LOXL3 | NM_032603.3 | NBAS | NM_015909.3 |
| IGF1 | NM_000618.4 | IFT122 | NM_052985.3 | LRP4 | NM_002334.3 | NEK1 | NM_012224.2 |
| IGF2 | NM_000612.5 | IFT140 | NM_014714.3 | LRP5 | NM_002335.3 | NEU1 | NM_000434.3 |
| IHH | NM_002181.3 | IFT172 | NM_015662.2 | LRRK1 | NM_024652.4 | NKX3-2 | NM_001189.3 |
| IMPAD1 | NM_017813.4 | IDUA | NM_000203.4 | LTBP2 | NM_000428.2 | NOG | NM_005450.4 |
| FGF9 | NM_002010.2 | IFITM5 | NM_001025295.2 | LTBP3 | NM_001130144.2 | NOTCH2 | NM_024408.3 |
| FGFR1 | NM_023110.2 | PCYT1A | NM_005017.3 | MAFB | NM_005461.4 | NPPC | NM_024409.3 |
| FGFR2 | NM_000141.4 | PDE4D | NM_001104631.1 | MAN2B1 | NM_000528.3 | NPR2 | NM_003995.3 |
| FGFR3 | NM_000142.4 | PEX5 | NM_001131025.1 | MANBA | NM_005908.3 | NPR3 | NM_000908.3 |
| FIG4 | NM_014845.5 | PEX7 | NM_000288.3 | MAP3K7 | NM_145331.2 | NSDHL | NM_015922.2 |
| FKBP10 | NM_021939.3 | PGM3 | NM_001199917.1 | MATN3 | NM_002381.4 | NSMCE2 | NM_173685.2 |
| FLNA | NM_001456.3 | PISD | NM_001326411.1 | MBTPS2 | NM_015884.3 | NXN | NM_022463.4 |
| FLNB | NM_001457.3 | PKDCC | NM_138370.2 | SH3PXD2B | NM_001017995.2 | OBSL1 | NM_015311.2 |
| MCM5 | NM_006739.3 | PLK4 | NM_014264.4 | SLC17A5 | NM_012434.4 | OCRL | NM_000276.3 |
| MCPH1 | NM_024596.4 | PLOD2 | NM_182943.2 | SLC26A2 | NM_000112.3 | ORC1 | NM_004153.3 |
| MEOX1 | NM_004527.3 | PLS3 | NM_005032.6 | SLC35D1 | NM_015139.2 | ORC4 | NM_002552.4 |
| MESP2 | NM_001039958.1 | POC1A | NM_015426.4 | SLC39A13 | NM_152264.4 | ORC6 | NM_014321.3 |
| MGP | NM_000900.3 | POLR1A | NM_015425.4 | SLCO2A1 | NM_005630.2 | OSTM1 | NM_014028.3 |
| MMP13 | NM_002427.3 | POP1 | NM_015029.2 | SLCO5A1 | NM_030958.2 | P3H1 | NM_022356.3 |
| MMP14 | NM_004995.3 | POR | NM_000941.2 | SMAD4 | NM_005359.5 | P4HB | NM_000918.3 |
| MMP2 | NM_004530.5 | PPIB | NM_000942.4 | SMARCAL1 | NM_014140.3 | PAM16 | NM_016069.9 |
| MMP9 | NM_004994.2 | PPP3CA | NM_000944.4 | SNRPB | NM_198216.1 | PAPSS2 | NM_001015880.1 |
| MNX1 | NM_005515.3 | PRKAR1A | NM_002734.4 | SNX10 | NM_001199835.1 | PCGF2 | NM_007144.2 |
| GJA1 | NM_000165.4 | PTDSS1 | NM_014754.2 | SOX9 | NM_000346.3 | PCNT | NM_006031.5 |
| GLB1 | NM_000404.2 | PTH1R | NM_000316.2 | SP7 | NM_001173467.2 | TRMT10A | NM_152292.4 |
| GMNN | NM_015895.4 | PTHLH | NM_198965.1 | SPARC | NM_003118.3 | TRPS1 | NM_014112.4 |
| GNAS | NM_000516.5 | PTPN11 | NM_002834.3 | SQSTM1 | NM_003900.4 | TRPV4 | NM_021625.4 |
| GNE | NM_001128227.2 | PYCR1 | NM_006907.3 | SRCAP | NM_006662.2 | TTC21B | NM_024753.4 |
| GNPAT | NM_014236.3 | RAB33B | NM_031296.2 | SUCO | NM_014283.4 | TUBGCP6 | NM_020461.3 |
| GNPTAB | NM_024312.4 | RBBP8 | NM_002894.2 | SULF1 | NM_001128205.1 | TYROBP | NM_003332.3 |
| GNPTG | NM_032520.4 | RECQL4 | NM_004260.3 | TAB2 | NM_015093.5 | VAC14 | NM_018052.3 |
| GNS | NM_002076.3 | RIPPLY2 | NM_001009994.2 | TAPT1 | NM_153365.2 | VPS33A | NM_022916.4 |
| GORAB | NM_152281.2 | RMRP | NR_003051.3 | TBCE | NM_003193.4 | WDR19 | NM_025132.3 |
| GPC6 | NM_005708.3 | RNU4ATAC | NR_023343.1 | TBX15 | NM_152380.2 | WDR34 | NM_052844.3 |
| GPX4 | NM_001039848.2 | SFRP4 | NM_003014.3 | TBX3 | NM_005996.3 | WDR35 | NM_001006657.1 |
| GSC | NM_173849.2 | INPPL1 | NM_001567.3 | TBX5 | NM_000192.3 | WDR60 | NM_018051.4 |
| GUSB | NM_000181.3 | JAG1 | NM_000214.2 | TBX6 | NM_004608.3 | WISP3 | NM_003880.3 |
| GZF1 | NM_022482.4 | KAT6B | NM_012330.3 | TBXAS1 | NM_001061.4 | WNT1 | NM_005430.3 |
| HES7 | NM_032580.3 | KIAA0586 | NM_001244189.1 | TCIRG1 | NM_006019.3 | WNT3 | NM_030753.4 |
| HGSNAT | NM_152419.2 | KIAA0753 | NM_014804.2 | TCTEX1D2 | NM_152773.4 | WNT3A | NM_033131.3 |
| HPGD | NM_000860.5 | KIF22 | NM_007317.2 | TCTN3 | NM_015631.5 | WNT5A | NM_003392.4 |
| HSPG2 | NM_005529.6 | KL | NM_004795.3 | TGFB1 | NM_000660.5 | XRCC4 | NM_022406.3 |
| HYAL1 | NM_153281.1 | KMT2A | NM_001197104.1 | TMEM165 | NM_018475.4 | XYLT1 | NM_022166.3 |
| IARS2 | NM_018060.3 | LARP7 | NM_016648.3 | TMEM38B | NM_018112.2 | XYLT2 | NM_022167.3 |
| ICK | NM_016513.4 | LBR | NM_002296.3 | TNFRSF11A | NM_003839.3 | SGSH | NM_000199.3 |
| IDS | NM_000202.6 | LEMD3 | NM_014319.4 | TNFRSF11B | NM_002546.3 | ROR2 | NM_004560.3 |
| IDUA | NM_000203.4 | LFNG | NM_001040167.1 | TNFSF11 | NM_003701.3 | RSPRY1 | NM_133368.2 |
| IFITM5 | NM_001025295.2 | LIFR | NM_002310.5 | MSX2 | NM_002449.4 | RTTN | NM_173630.3 |
| SEC24D | NM_014822.3 | TRAPPC2 | NM_001011658.3 | SERPINH1 | NM_001235.3 | RUNX2 | NM_001024630.3 |
| TRIP11 | NM_004239.4 | TREM2 | NM_018965.3 | SERPINF1 | NM_002615.6 | SC5D | NM_006918.4 |
| SETBP1 | NM_015559.2 | TRIM37 | NM_015294.4 | ZMPSTE24 | NM_005857.4 |
| Proband | Sister | Mother | Father | |
|---|---|---|---|---|
| Age (years) | 15 | 14 | 42 | 41 |
| Height (cm) | 164 | 163 | 155 | 177 |
| Weight (kg) | 65 | 89 | 70 | 107 |
| Vertebral compression fractures | 1 | No | No | No |
| Long-bone fractures | 10 | No | No | No |
| Sclerae | White | White | White | White |
| Subluxation of the joints | 3 | No | No | No |
| Dentinogenesis imperfecta | No | No | No | No |
| Hearing loss | No | No | No | No |
| F1 BDM (g/cm2) | 0.604 | / | / | / |
| F1 BDM T-score | −2.8 | / | / | / |
| F2 BDM (g/cm2) | 0.689 | 0.971 | 0.903 | 1.163 |
| F2 BDM T-score | −2.3 | 0.2 | −0.3 | 0.9 |
| S1 BDM (g/cm2) | 0.587 | / | / | / |
| S1 BDM T-score | −4.6 | / | / | / |
| S2 BDM (g/cm2) | 0.622 | 0.990 | 0.897 | 0.916 |
| S2 BDM T-score | −4.3 | −0.5 | −1.4 | −1.6 |
| Ca (mmol/L) | 2.67 * | 2.61 | 2.43 | 2.58 * |
| Inorganic phosphates (mmol/L) | 1.39 | 0.84 * | 1.22 | 1.07 |
| Osteocalcin (µg/L) | 34.0 * | 18.3 | 7.91 | 3.92 |
| vitamin D (25-OH) (nmol/L) | 83 | 36 * | 39 * | 75 |
| Creatinine (mmol/L) | 12.3 | 4.9 | 13.4 | 15.6 |
| Deoxypyridinoline (nM/mM of creatinine) | 9.1 | 9.9 | 4.4 | 5.0 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Brlek, P.; Antičević, D.; Molnar, V.; Matišić, V.; Robinson, K.; Aradhya, S.; Krpan, D.; Primorac, D. X-Linked Osteogenesis Imperfecta Possibly Caused by a Novel Variant in PLS3. Genes 2021, 12, 1851. https://doi.org/10.3390/genes12121851
Brlek P, Antičević D, Molnar V, Matišić V, Robinson K, Aradhya S, Krpan D, Primorac D. X-Linked Osteogenesis Imperfecta Possibly Caused by a Novel Variant in PLS3. Genes. 2021; 12(12):1851. https://doi.org/10.3390/genes12121851
Chicago/Turabian StyleBrlek, Petar, Darko Antičević, Vilim Molnar, Vid Matišić, Kristina Robinson, Swaroop Aradhya, Dalibor Krpan, and Dragan Primorac. 2021. "X-Linked Osteogenesis Imperfecta Possibly Caused by a Novel Variant in PLS3" Genes 12, no. 12: 1851. https://doi.org/10.3390/genes12121851
APA StyleBrlek, P., Antičević, D., Molnar, V., Matišić, V., Robinson, K., Aradhya, S., Krpan, D., & Primorac, D. (2021). X-Linked Osteogenesis Imperfecta Possibly Caused by a Novel Variant in PLS3. Genes, 12(12), 1851. https://doi.org/10.3390/genes12121851