
Species-Specific Duplication and Adaptive Evolution of a Candidate Sex Pheromone Receptor Gene in Weather Loach


Abstract
:1. Introduction
2. Materials and Methods
2.1. Sample Collection
2.2. Transcriptome Sequencing and Analyses
2.2.1. Tissue Sampling and RNA Extraction
2.2.2. Library Construction and Sequencing
2.2.3. Bioinformatic Analyses
Sequence Assembly and Annotation
DEG Detection
Ortholog Identification
2.3. Genomic PCR, Sequencing and Related Analyses
2.4. Quantitative RT-PCR Assays
2.5. Analysis of Adaptive Evolution
3. Results and Discussion
3.1. Identification of a Male-Biasedly Expressed or Gene
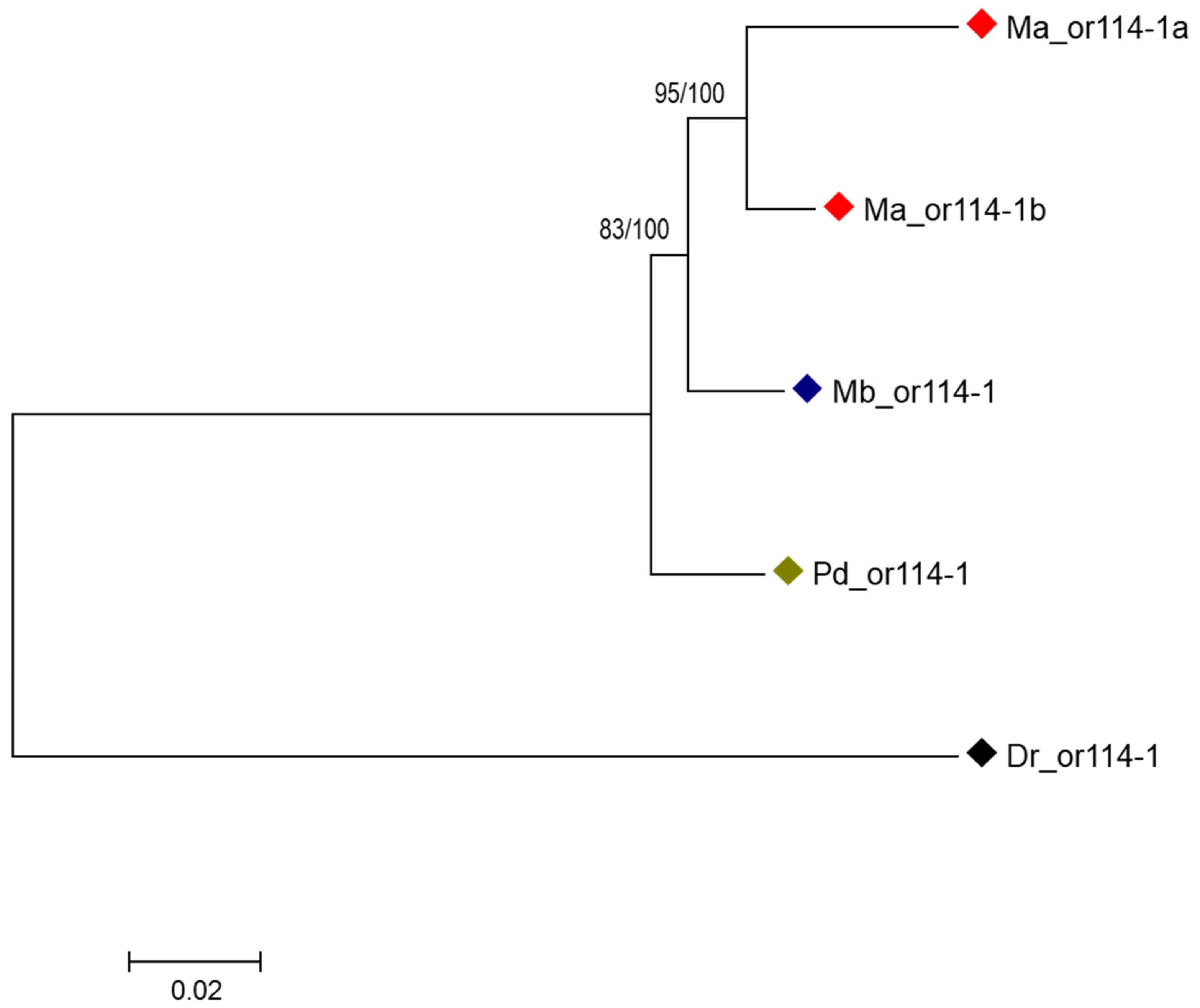
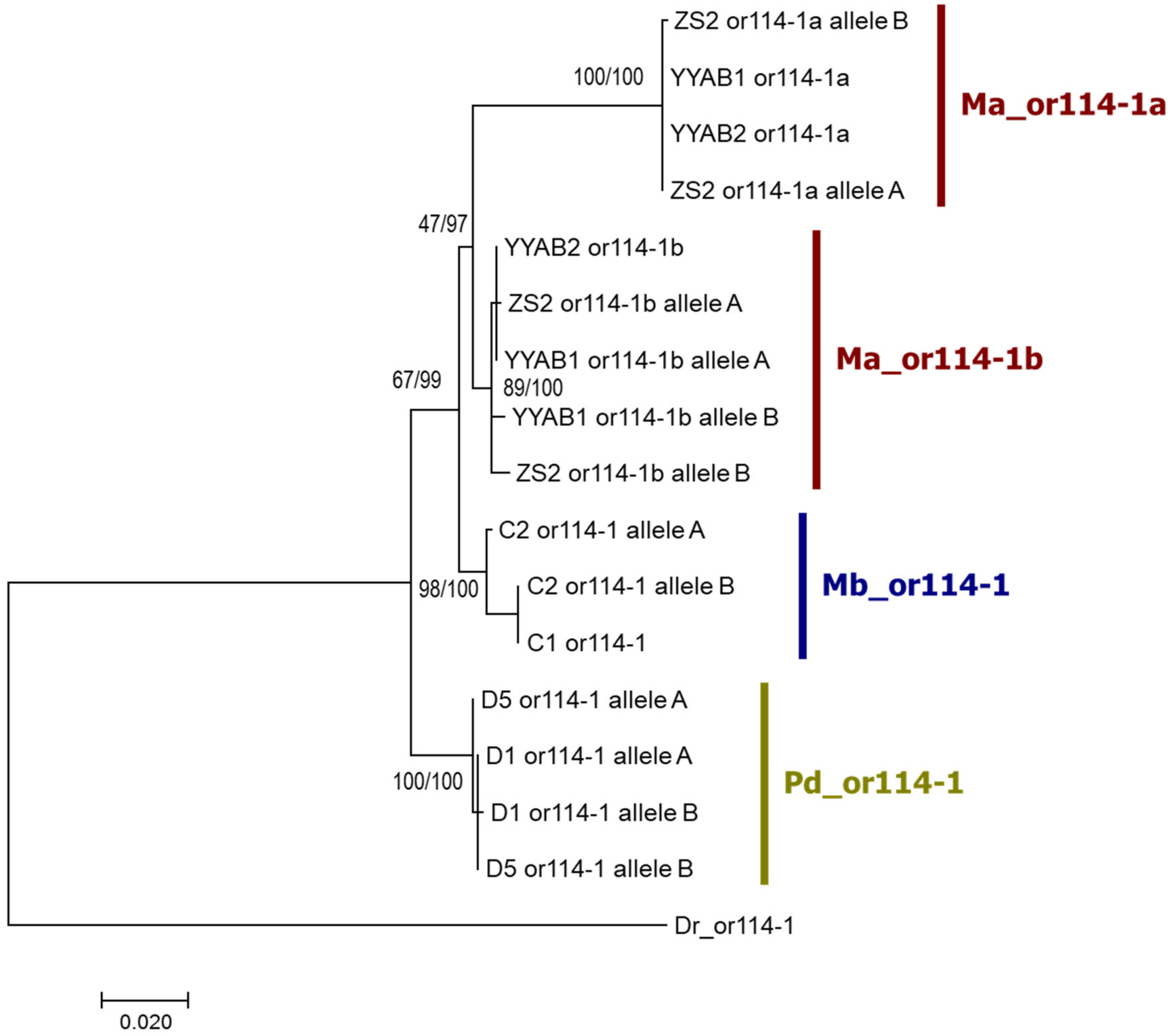
3.2. Species-Specific Duplication of or114-1
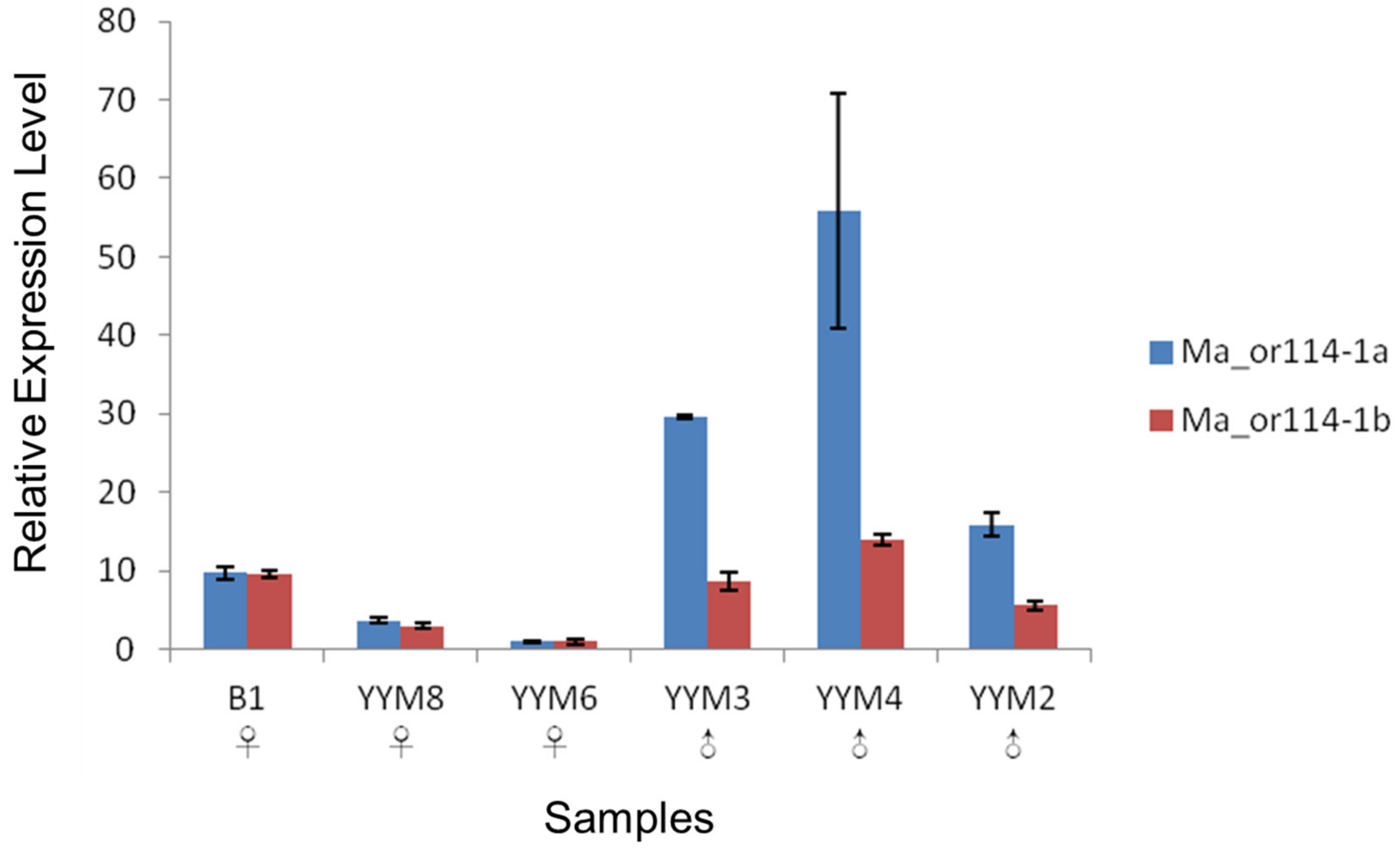
3.3. qRT-PCR Verification of the Sex-Differential Expression
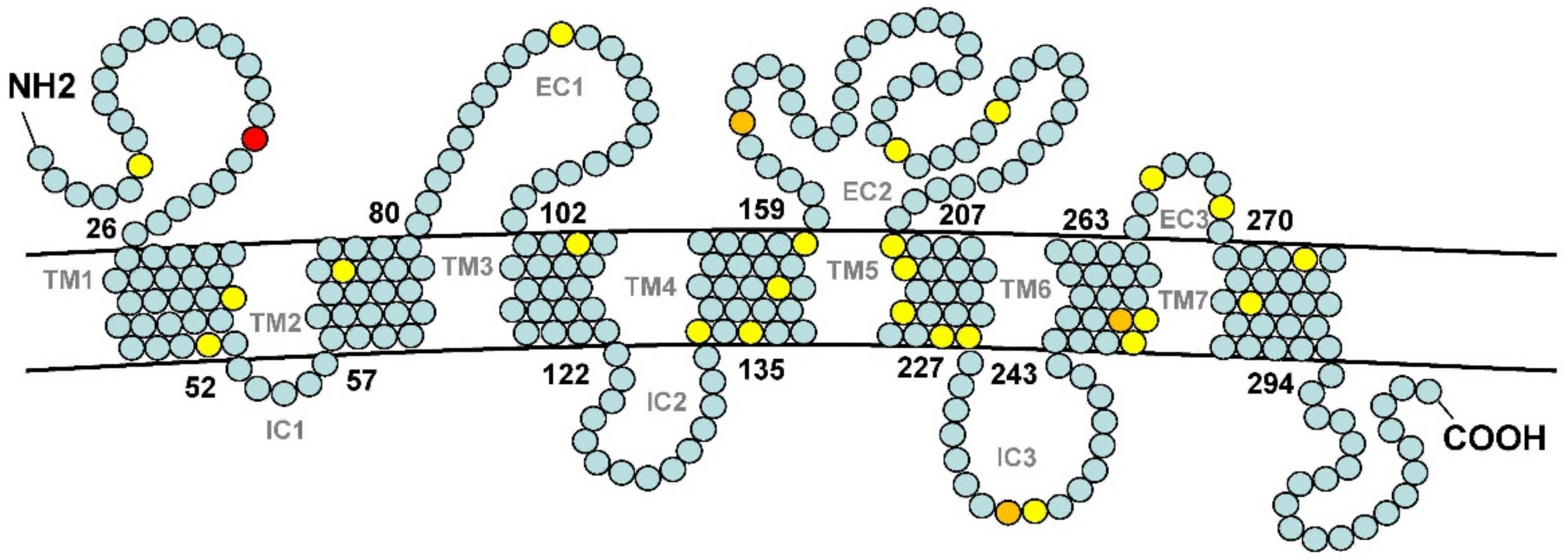
3.4. Adaptive Evolution of or114-1 in Weather Loach
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Conflicts of Interest
References
- Smadja, C.; Butlin, R.K. On the scent of speciation: The chemosensory system and its role in premating isolation. Heredity 2009, 102, 77–97. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Seehausen, O.; Terai, Y.; Magalhaes, I.S.; Carleton, K.L.; Mrosso, H.D.; Miyagi, R.; van der Sluijs, I.; Schneider, M.V.; Maan, M.E.; Tachida, H.; et al. Speciation through sensory drive in cichlid fish. Nature 2008, 455, 620–626. [Google Scholar] [CrossRef] [PubMed]
- Zhang, B.; Zhang, W.; Nie, R.E.; Li, W.Z.; Segraves, K.A.; Yang, X.K.; Xue, H.J. Comparative transcriptome analysis of chemosensory genes in two sister leaf beetles provides insights into chemosensory speciation. Insect. Biochem. Mol. 2016, 79, 108–118. [Google Scholar] [CrossRef] [PubMed]
- Symonds, M.R.E.; Elgar, M.A. The evolution of pheromone diversity. Trends Ecol. Evol. 2008, 23, 220–228. [Google Scholar] [CrossRef]
- Poncelet, G.; Shimeld, S.M. The evolutionary origins of the vertebrate olfactory system. Open Biol. 2020, 10, 200330. [Google Scholar] [CrossRef]
- Touhara, K.; Vosshall, L.B. Sensing Odorants and Pheromones with Chemosensory Receptors. Annu. Rev. Physiol. 2009, 71, 307–332. [Google Scholar] [CrossRef]
- Gomez-Diaz, C.; Benton, R. The joy of sex pheromones. Embo Rep. 2013, 14, 874–883. [Google Scholar] [CrossRef] [Green Version]
- Niehuis, O.; Buellesbach, J.; Gibson, J.D.; Pothmann, D.; Hanner, C.; Mutti, N.S.; Judson, A.K.; Gadau, J.; Ruther, J.; Schmitt, T. Behavioural and genetic analyses of Nasonia shed light on the evolution of sex pheromones. Nature 2013, 494, 345–348. [Google Scholar] [CrossRef]
- Doyle, W.I.; Meeks, J.P. Excreted Steroids in Vertebrate Social Communication. J. Neurosci. 2018, 38, 3377–3387. [Google Scholar] [CrossRef]
- Bear, D.M.; Lassance, J.M.; Hoekstra, H.E.; Datta, S.R. The Evolving Neural and Genetic Architecture of Vertebrate Olfaction. Curr. Biol. 2016, 26, R1039–R1049. [Google Scholar] [CrossRef] [Green Version]
- Haga-Yamanaka, S.; Ma, L.M.; He, J.; Qiu, Q.; Lavis, L.D.; Looger, L.L.; Yu, C.R. Integrated action of pheromone signals in promoting courtship behavior in male mice. Elife 2014, 3, e03025. [Google Scholar] [CrossRef]
- Sorensen, P.W.; Stacey, N.E. Brief review of fish pheromones and discussion of their possible uses in the control of non-indigenous teleost fishes. N. Z. J. Mar. Fresh 2004, 38, 399–417. [Google Scholar] [CrossRef]
- Appelt, C.W.; Sorensen, P.W. Female goldfish signal spawning readiness by altering when and where they release a urinary pheromone. Anim. Behav. 2007, 74, 1329–1338. [Google Scholar] [CrossRef]
- Plenderleith, M.; van Oosterhout, C.; Robinson, R.L.; Turner, G.F. Female preference for conspecific males based on olfactory cues in a Lake Malawi cichlid fish. Biol. Lett. 2005, 1, 411–414. [Google Scholar] [CrossRef] [Green Version]
- Shi, P.; Zhang, J. Extraordinary Diversity of Chemosensory Receptor Gene Repertoires Among Vertebrates. In Chemosensory Systems in Mammals, Fishes, and Insects; Meyerhof, W., Korsching, S., Eds.; Springer: Berlin/Heidelberg, Germany, 2009; pp. 57–75. [Google Scholar]
- Nei, M.; Niimura, Y.; Nozawa, M. The evolution of animal chemosensory receptor gene repertoires: Roles of chance and necessity. Nat. Rev. Genet. 2008, 9, 951–963. [Google Scholar] [CrossRef]
- Churcher, A.M.; Hubbard, P.C.; Marques, J.P.; Canario, A.V.M.; Huertas, M. Deep sequencing of the olfactory epithelium reveals specific chemosensory receptors are expressed at sexual maturity in the European eel Anguilla anguilla. Mol. Ecol. 2015, 24, 822–834. [Google Scholar] [CrossRef]
- Yabuki, Y.; Koide, T.; Miyasaka, N.; Wakisaka, N.; Masuda, M.; Ohkura, M.; Nakai, J.; Tsuge, K.; Tsuchiya, S.; Sugimoto, Y.; et al. Olfactory receptor for prostaglandin F-2 α mediates male fish courtship behavior. Nat. Neurosci. 2016, 19, 897–904. [Google Scholar] [CrossRef]
- Behrens, M.; Frank, O.; Rawel, H.; Ahuja, G.; Potting, C.; Hofmann, T.; Meyerhof, W.; Korsching, S. ORA1, a Zebrafish Olfactory Receptor Ancestral to All Mammalian V1R Genes, Recognizes 4-Hydroxyphenylacetic Acid, a Putative Reproductive Pheromone. J. Biol. Chem. 2014, 289, 19778–19788. [Google Scholar] [CrossRef] [Green Version]
- Cong, X.; Zheng, Q.; Ren, W.; Cheron, J.B.; Fiorucci, S.; Wen, T.; Zhang, C.; Yu, H.; Golebiowski, J.; Yu, Y. Zebrafish olfactory receptors ORAs differentially detect bile acids and bile salts. J. Biol. Chem. 2019, 294, 6762–6771. [Google Scholar] [CrossRef]
- Saraiva, L.R.; Ahuja, G.; Ivandic, I.; Syed, A.S.; Marioni, J.C.; Korsching, S.I.; Logan, D.W. Molecular and neuronal homology between the olfactory systems of zebrafish and mouse. Sci. Rep. 2015, 5, 11487. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Jiang, H.; Yang, L. Transcriptome Analysis of Zebrafish Olfactory Epithelium Reveal Sexual Differences in Odorant Detection. Genes 2020, 11, 592. [Google Scholar] [CrossRef] [PubMed]
- Kolmakov, N.N.; Kube, M.; Reinhardt, R.; Canario, A.V. Analysis of the goldfish Carassius auratus olfactory epithelium transcriptome reveals the presence of numerous non-olfactory GPCR and putative receptors for progestin pheromones. BMC Genom. 2008, 9, 429. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fatsini, E.; Bautista, R.; Manchado, M.; Duncan, N.J. Transcriptomic profiles of the upper olfactory rosette in cultured and wild Senegalese sole (Solea senegalensis) males. Comp. Biochem. Physiol. Part D Genom. Proteom. 2016, 20, 125–135. [Google Scholar] [CrossRef] [PubMed]
- Palstra, A.P.; Fukaya, K.; Chiba, H.; Dirks, R.P.; Planas, J.V.; Ueda, H. The Olfactory Transcriptome and Progression of Sexual Maturation in Homing Chum Salmon Oncorhynchus keta. PLoS ONE 2015, 10, e0137404. [Google Scholar]
- Zhu, G.; Wang, L.; Tang, W.; Liu, D.; Yang, J. De novo transcriptomes of olfactory epithelium reveal the genes and pathways for spawning migration in japanese grenadier anchovy (Coilia nasus). PLoS ONE 2014, 9, e103832. [Google Scholar]
- Liu, H.; Chen, C.; Lv, M.; Liu, N.; Hu, Y.; Zhang, H.; Enbody, E.D.; Gao, Z.; Andersson, L.; Wang, W. A chromosome-level assembly of blunt snout bream (Megalobrama amblycephala) reveals an expansion of olfactory receptor genes in freshwater fish. Mol. Biol. Evol. 2021, 38, 4238–4251. [Google Scholar] [CrossRef]
- Honda, H. Female sex pheromone of the loach, Misgurnus anguillicaudatus, involved in courtship behavior. Bull. Jpn. Soc. Sci. Fish. 1980, 46, 1223–1225. [Google Scholar] [CrossRef] [Green Version]
- Kitamura, S.; Ogata, H.; Takashima, F. Activities of F-type prostaglandins as releaser sex pheromones in cobitide loach, Msgurnus anguillicaudatus. Comp. Biochem. Physiol. Part A Physiol. 1994, 107, 161–169. [Google Scholar] [CrossRef]
- Chen, J. A study on the classification of the subfamily cobitinae of china. Trans. Chin. Ichthyol. Soc. 1981, 1, 21–32. [Google Scholar]
- Fujimoto, T.; Nishimura, T.; Goto-Kazeto, R.; Kawakami, Y.; Yamaha, E.; Arai, K. Sexual dimorphism of gonadal structure and gene expression in germ cell-deficient loach, a teleost fish. Proc. Natl. Acad. Sci. USA 2010, 107, 17211–17216. [Google Scholar] [CrossRef] [Green Version]
- Yu, Y.Y.; Abbas, K.; Wang, W.M.; Zhou, X.Y. Geographical distribution of ploidy level variation of loach Misgurnus anguillicaudatus in China. Pak. J. Agr. Sci. 2014, 51, 273–281. [Google Scholar]
- Zhong, J.; Yi, S.; Ma, L.; Wang, W. Evolution and phylogeography analysis of diploid and polyploid Misgurnus anguillicaudatus populations across China. Proc. Biol. Sci. 2019, 286, 20190076. [Google Scholar] [CrossRef] [Green Version]
- Grabherr, M.G.; Haas, B.J.; Yassour, M.; Levin, J.Z.; Thompson, D.A.; Amit, I.; Adiconis, X.; Fan, L.; Raychowdhury, R.; Zeng, Q.; et al. Full-length transcriptome assembly from RNA-Seq data without a reference genome. Nat. Biotechnol. 2011, 29, 644–652. [Google Scholar] [CrossRef] [Green Version]
- Pertea, G.; Huang, X.; Liang, F.; Antonescu, V.; Sultana, R.; Karamycheva, S.; Lee, Y.; White, J.; Cheung, F.; Parvizi, B.; et al. TIGR Gene Indices clustering tools (TGICL): A software system for fast clustering of large EST datasets. Bioinformatics 2003, 19, 651–652. [Google Scholar] [CrossRef] [Green Version]
- Audic, S.; Claverie, J.M. The significance of digital gene expression profiles. Genome Res. 1997, 7, 986–995. [Google Scholar] [CrossRef]
- Kumar, S.; Stecher, G.; Tamura, K. MEGA7: Molecular Evolutionary Genetics Analysis Version 7.0 for Bigger Datasets. Mol. Biol. Evol. 2016, 33, 1870–1874. [Google Scholar] [CrossRef] [Green Version]
- Ronquist, F.; Huelsenbeck, J.P. MrBayes 3: Bayesian phylogenetic inference under mixed models. Bioinformatics 2003, 19, 1572–1574. [Google Scholar] [CrossRef] [Green Version]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef]
- Yang, Z.H. PAML 4: Phylogenetic analysis by maximum likelihood. Mol. Biol. Evol. 2007, 24, 1586–1591. [Google Scholar] [CrossRef] [Green Version]
- Wertheim, J.O.; Murrell, B.; Smith, M.D.; Pond, S.L.K.; Scheffler, K. RELAX: Detecting Relaxed Selection in a Phylogenetic Framework. Mol. Biol. Evol. 2015, 32, 820–832. [Google Scholar] [CrossRef] [Green Version]
- Smith, M.D.; Wertheim, J.O.; Weaver, S.; Murrell, B.; Scheffler, K.; Kosakovsky Pond, S.L. Less is more: An adaptive branch-site random effects model for efficient detection of episodic diversifying selection. Mol. Biol. Evol. 2015, 32, 1342–1353. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Murrell, B.; Weaver, S.; Smith, M.D.; Wertheim, J.O.; Murrell, S.; Aylward, A.; Eren, K.; Pollner, T.; Martin, D.P.; Smith, D.M.; et al. Gene-wide identification of episodic selection. Mol. Biol. Evol. 2015, 32, 1365–1371. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hofmann, K. TMBASE—A database of membrane spanning protein segments. Biol Chem. 1993, 374, 1–3. [Google Scholar]
- Alioto, T.S.; Ngai, J. The odorant receptor repertoire of teleost fish. BMC Genom. 2005, 6, 173. [Google Scholar] [CrossRef] [Green Version]
- Bushdid, C.; de March, C.A.; Fiorucci, S.; Matsunami, H.; Golebiowski, J. Agonists of G-Protein-Coupled Odorant Receptors Are Predicted from Chemical Features. J. Phys. Chem. Lett. 2018, 9, 2235–2240. [Google Scholar] [CrossRef]
- Zhang, J.Z. Evolution by gene duplication: An update. Trends Ecol. Evol. 2003, 18, 292–298. [Google Scholar] [CrossRef] [Green Version]
- Ortiz-Barrientos, D.; Counterman, B.A.; Noor, M.A. The genetics of speciation by reinforcement. PLoS Biol. 2004, 2, e416. [Google Scholar] [CrossRef] [Green Version]
- Spencer, H.G. A further perspective on speciation by reinforcement. Theor. Biol. Forum. 2020, 113, 63–66. [Google Scholar]
- Butlin, R.K.; Smadja, C.M. Coupling, Reinforcement, and Speciation. Am. Nat. 2018, 191, 155–172. [Google Scholar] [CrossRef]
- Servedio, M.R.; Noor, M.A.F. The Role of Reinforcement in Speciation: Theory and Data. Annu. Rev. Ecol. Evol. Syst. 2003, 34, 339–364. [Google Scholar] [CrossRef]
- Matute, D.R.; Ortiz-Barrientos, D. Speciation: The strength of natural selection driving reinforcement. Curr. Biol. 2014, 24, R955–R957. [Google Scholar] [CrossRef] [Green Version]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Species | Sample Number | Sex | Location |
|---|---|---|---|
| Misgurnus anguillicaudatus | B1 | Female | Yueyang |
| B2 | Male | Yueyang | |
| YYAB1 | Female | Yueyang | |
| YYAB2 | Male | Yueyang | |
| YYM2 | Male | Yueyang | |
| YYM3 | Male | Yueyang | |
| YYM4 | Male | Yueyang | |
| YYM6 | Female | Yueyang | |
| YYM8 | Female | Yueyang | |
| ZS2 | Female | Zhoushan | |
| Misgurnus bipartitus | C1 | Female | Daqing |
| C2 | Male | Daqing | |
| DQB52 | Female | Daqing | |
| DQB53 | Female | Daqing | |
| DQB54 | Male | Daqing | |
| DQB56 | Male | Daqing | |
| Paramisgurnus dabryanus | D1 | Female | Honghu |
| D5 | Male | Honghu | |
| D7 | Female | Honghu | |
| YYD1 | Male | Yueyang | |
| YYD2 | Male | Yueyang | |
| YYD3 | Male | Yueyang | |
| YYD5 | Female | Yueyang | |
| YYD6 | Female | Yueyang |
| Primer Name | Primer Sequence (5′-3′) | TM (°C) | Product Length (bp) |
|---|---|---|---|
| β-actin F | GGGTATGGAGTCTTGCGGTA | 58.88 | 131 |
| β-actin R | CAGCAATGCCAGGGTACATG | 59.26 | |
| Ma_or114-1a F | AAGATCTACGTGTGATATGTACC | 59.08 | 140 |
| Ma_or114-1a R | TTAATGCAGATTAACCTTATTAGAAG | 58.20 | |
| Ma_or114-1b F | GAGATCTACGTGTGCTATGTAC | 58.89 | 150 |
| Ma_or114-1b R | GGGAAGACCACTGATGTAGAT | 58.78 | |
| Mb_or114-1 F | GCCATCATTTTCCCCTTGCA | 59.10 | 119 |
| Mb_or114-1 R | CCCAGCAAACCAACCATTGA | 58.95 | |
| Pd_or114-1 F | TGTGTTTGTGAACACACATCA | 58.00 | 163 |
| Pd_or114-1 R | CGCCTGAAGGAGAACGAAAT | 59.72 |
| Model | log L | Parameters | LRT |
|---|---|---|---|
| M0 (one-ratio) | −1717.957 | ω = 0.376 | M0 vs. two-ratio: 2△ι = 19.57, df = 1, p < 0.001. two-ratio (ω1 = 1) vs. two-ratio: 2△ι = 0.67, df = 1, p > 0.05 |
| two-ratio model | −1708.171 | ωbackground = 0.136, ωforeground = 1.461 | |
| two-ratio model (ω1 = 1) | −1708.506 | ωbackground = 0.137, ωforeground = 1.000 |
| Model | log L | LRT | Sites under Positive Selection |
|---|---|---|---|
| Model A | −1705.590 | 2△ι = 4.04, df = 1, p < 0.05 | 6 L, 20 S **, 41 S, 50 V, 74 Q, 90 K, 105 M, 136 F, 138 T, 148 F, 158 T, 164 I *, 188 I, 193 R, 208 T, 215 F, 219 V, 225 M, 226 S, 234 W *, 235 H, 247 A, 248 M, 249 F *, 265 S, 269 K, 274 I, 281 V |
| Model A (ω2 = 1) | −1707.570 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhong, L.; Wang, W.; Cao, X. Species-Specific Duplication and Adaptive Evolution of a Candidate Sex Pheromone Receptor Gene in Weather Loach. Genes 2021, 12, 1845. https://doi.org/10.3390/genes12121845
Zhong L, Wang W, Cao X. Species-Specific Duplication and Adaptive Evolution of a Candidate Sex Pheromone Receptor Gene in Weather Loach. Genes. 2021; 12(12):1845. https://doi.org/10.3390/genes12121845
Chicago/Turabian StyleZhong, Lei, Weimin Wang, and Xiaojuan Cao. 2021. "Species-Specific Duplication and Adaptive Evolution of a Candidate Sex Pheromone Receptor Gene in Weather Loach" Genes 12, no. 12: 1845. https://doi.org/10.3390/genes12121845
APA StyleZhong, L., Wang, W., & Cao, X. (2021). Species-Specific Duplication and Adaptive Evolution of a Candidate Sex Pheromone Receptor Gene in Weather Loach. Genes, 12(12), 1845. https://doi.org/10.3390/genes12121845