
Pattern of New Gene Origination in a Special Fish Lineage, the Flatfishes


{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Data Acquisition
2.2. Phylogenetic Reconstruction
2.3. Identification of New Genes
2.4. Inference of the Origin Mechanism
2.5. Selection Analysis of New Genes
2.6. Gene Expression in Tissues of Starry Flounder
2.7. Asymmetrical Expression Analysis
3. Results
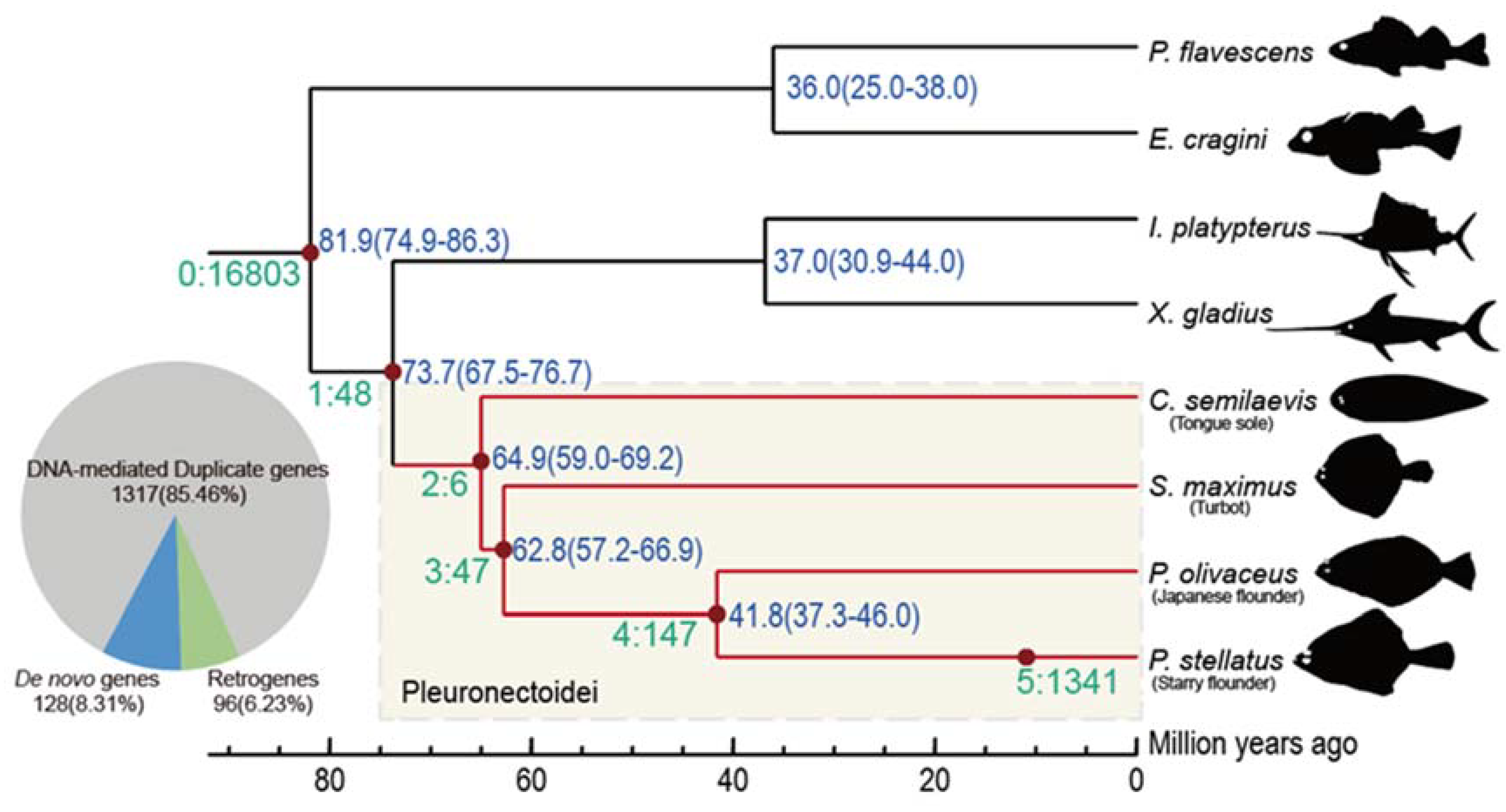
3.1. New Genes Emerged in Pleuronectoidei
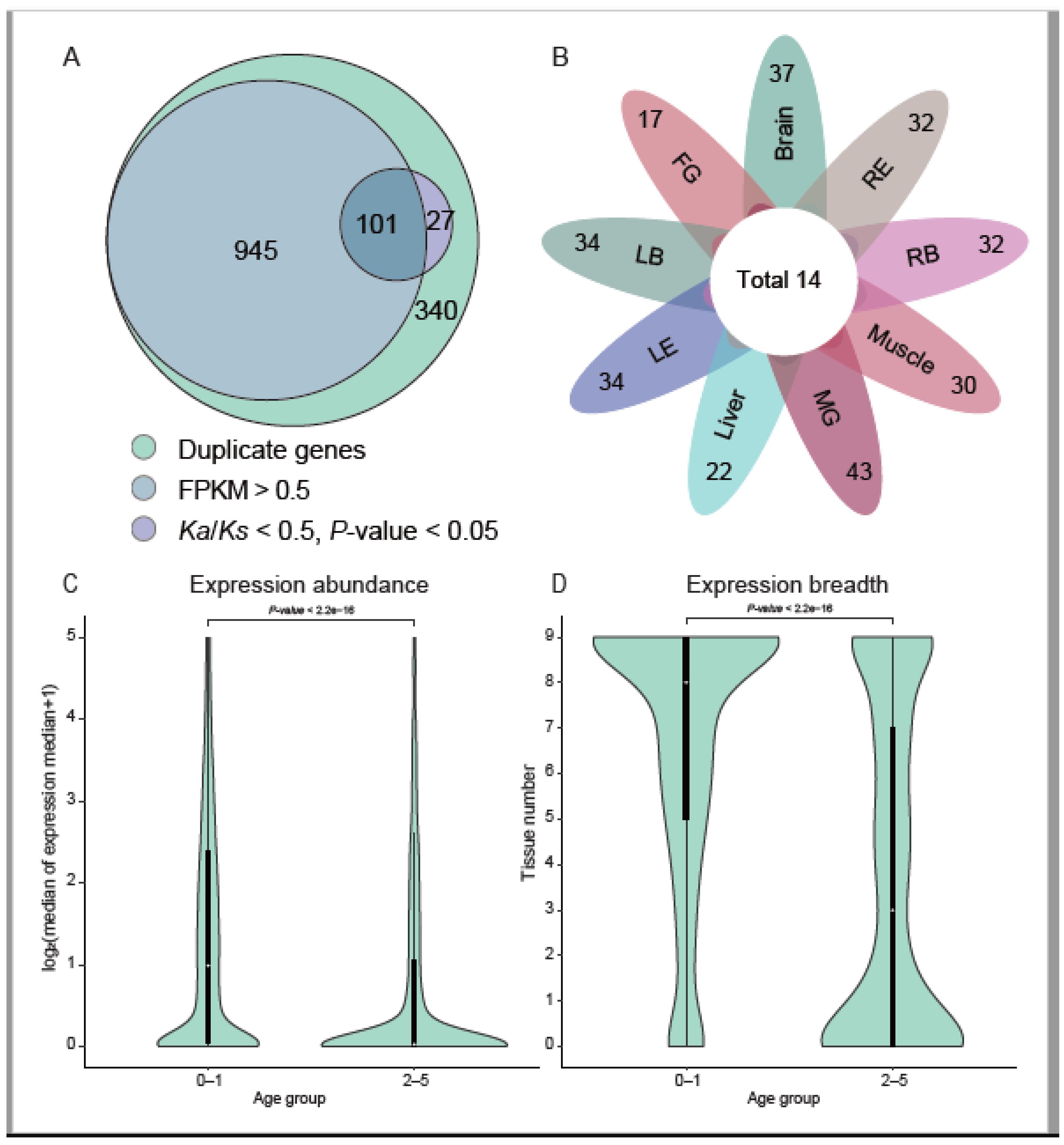
3.2. Most New Genes Are Expressed and Some under Natural Selection
3.3. Asymmetric Expression of Some New Genes May Play Roles in the Formation of the Unique Body Plan of Flatfishes
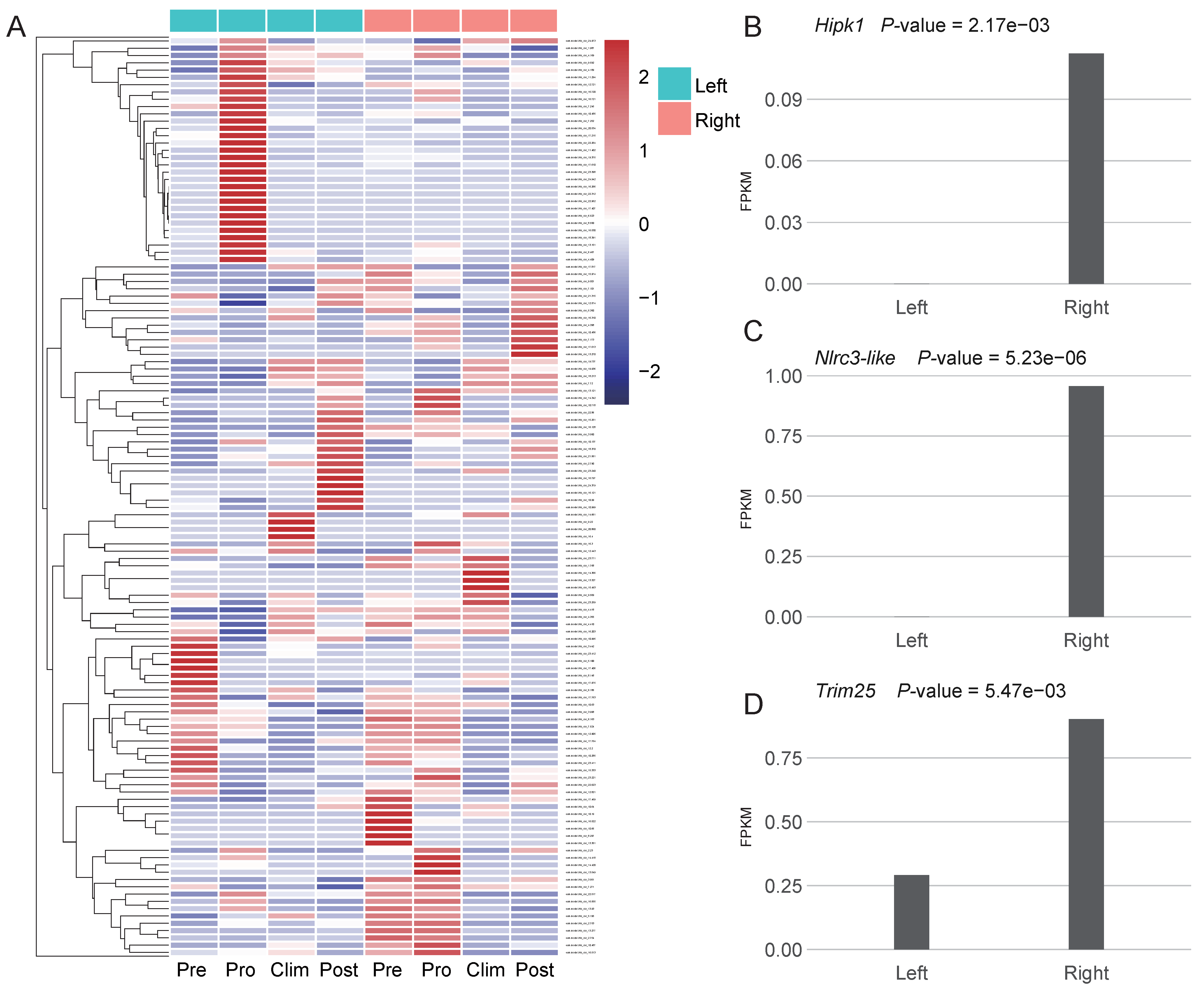
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Conflicts of Interest
References
- Zhang, Y.E.; Long, M.Y. New genes contribute to genetic and phenotypic novelties in human evolution. Curr. Opin. Genet. Dev. 2014, 29, 90–96. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kaessmann, H. Origins, evolution, and phenotypic impact of new genes. Genome Res. 2010, 20, 1313–1326. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Long, M.; Betrán, E.; Thornton, K.; Wang, W. The origin of new genes: Glimpses from the young and old. Nat. Rev. Genet. 2003, 4, 865–875. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.E.; Landback, P.; Vibranovski, M.; Long, M.Y. New genes expressed in human brains: Implications for annotating evolving genomes. Bioessays 2012, 34, 982–991. [Google Scholar] [CrossRef] [PubMed]
- Cardoso-Moreira, M.; Long, M. The origin and evolution of new genes. Methods Mol. Biol. Clifton N. J. 2012, 856, 161–186. [Google Scholar] [CrossRef]
- Ranz, J.M.; Parsch, J. Newly evolved genes: Moving from comparative genomics to functional studies in model systems. Bioessays 2012, 34, 477–483. [Google Scholar] [CrossRef]
- Shao, Y.; Chen, C.Y.; Shen, H.; He, B.Z.; Yu, D.Q.; Jiang, S.; Zhao, S.L.; Gao, Z.Q.; Zhu, Z.L.; Chen, X.; et al. GenTree, an integrated resource for analyzing the evolution and function of primate-specific coding genes. Genome Res. 2019, 29, 682–696. [Google Scholar] [CrossRef]
- Domazet-Loso, T.; Tautz, D. Phylostratigraphic tracking of cancer genes suggests a link to the emergence of multicellularity in metazoa. BMC Biol. 2010, 8, 66. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Capra, J.A.; Williams, A.G.; Pollard, K.S. ProteinHistorian: Tools for the comparative analysis of eukaryote protein origin. PLoS Comput. Biol. 2012, 8, e1002567. [Google Scholar] [CrossRef]
- Villanueva-Canas, J.L.; Ruiz-Orera, J.; Agea, M.I.; Gallo, M.; Andreu, D.; Alba, M.M. New Genes and Functional Innovation in Mammals. Genome Biol. Evol. 2017, 9, 1886–1900. [Google Scholar] [CrossRef] [Green Version]
- Huminiecki, L.; Wolfe, K.H. Divergence of spatial gene expression profiles following species-specific gene duplications in human and mouse. Genome Res. 2004, 14, 1870–1879. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, W.; Landback, P.; Gschwend, A.R.; Shen, B.; Long, M. New genes drive the evolution of gene interaction networks in the human and mouse genomes. Genome Biol. 2015, 16, 202. [Google Scholar] [CrossRef] [Green Version]
- Shiao, M.S.; Khil, P.; Camerini-Otero, R.D.; Shiroishi, T.; Moriwaki, K.; Yu, H.T.; Long, M. Origins of new male germ-line functions from X-derived autosomal retrogenes in the mouse. Mol. Biol. Evol. 2007, 24, 2242–2253. [Google Scholar] [CrossRef]
- Cheng, Y.; Shang, D.; Luo, M.; Huang, C.; Lai, F.; Wang, X.; Xu, X.; Ying, R.; Wang, L.; Zhao, Y.; et al. Whole genome-wide chromosome fusion and new gene birth in the Monopterus albus genome. Cell Biosci. 2020, 10, 67. [Google Scholar] [CrossRef]
- Meyer, A.; Schartl, M. Gene and genome duplications in vertebrates: The one-to-four (-to-eight in fish) rule and the evolution of novel gene functions. Curr. Opin. Cell Biol. 1999, 11, 699–704. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.B.; Liu, T.K.; Jiang, J.; Shi, J.; Liu, Y.; Li, S.; Gui, J.F. Identification of a Novel Gig2 Gene Family Specific to Non-Amniote Vertebrates. PLoS ONE 2013, 8, 14. [Google Scholar] [CrossRef]
- Friedman, M. The evolutionary origin of flatfish asymmetry. Nature 2008, 454, 209–212. [Google Scholar] [CrossRef] [PubMed]
- Schreiber, A.M. Flatfish: An asymmetric perspective on metamorphosis. Curr. Top. Dev. Biol. 2013, 103, 167–194. [Google Scholar] [CrossRef]
- Lu, Z.M.; Gong, L.; Ren, Y.D.; Chen, Y.J.; Wang, Z.K.; Liu, L.Q.; Li, H.R.; Chen, X.Q.; Li, Z.Z.; Luo, H.R.; et al. Large-scale sequencing of flatfish genomes provides insights into the polyphyletic origin of their specialized body plan. Nat. Genet. 2021, 53, 742–751. [Google Scholar] [CrossRef]
- Wu, B.; Feng, C.; Zhu, C.; Xu, W.; Yuan, Y.; Hu, M.; Yuan, K.; Li, Y.; Ren, Y.; Zhou, Y.; et al. The Genomes of Two Billfishes Provide Insights into the Evolution of Endothermy in Teleosts. Mol. Biol. Evol. 2021, 38, 2413–2427. [Google Scholar] [CrossRef]
- Figueras, A.; Robledo, D.; Corvelo, A.; Hermida, M.; Pereiro, P.; Rubiolo, J.A.; Gómez-Garrido, J.; Carreté, L.; Bello, X.; Gut, M.; et al. Whole genome sequencing of turbot (Scophthalmus maximus; Pleuronectiformes): A fish adapted to demersal life. DNA Res. 2016, 23, 181–192. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Feron, R.; Zahm, M.; Cabau, C.; Klopp, C.; Roques, C.; Bouchez, O.; Eché, C.; Valière, S.; Donnadieu, C.; Haffray, P.; et al. Characterization of a Y-specific duplication/insertion of the anti-Mullerian hormone type II receptor gene based on a chromosome-scale genome assembly of yellow perch, Perca flavescens. Mol. Ecol. Resour. 2020, 20, 531–543. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reid, B.N.; Moran, R.L.; Kopack, C.J.; Fitzpatrick, S.W. Rapture-ready darters: Choice of reference genome and genotyping method (whole-genome or sequence capture) influence population genomic inference in Etheostoma. Mol. Ecol. Resour. 2021, 21, 404–420. [Google Scholar] [CrossRef] [PubMed]
- Stamatakis, A. RAxML version 8: A tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics 2014, 30, 1312–1313. [Google Scholar] [CrossRef]
- Yang, Z.H. PAML 4: Phylogenetic analysis by maximum likelihood. Mol. Biol. Evol. 2007, 24, 1586–1591. [Google Scholar] [CrossRef] [Green Version]
- Harris, R.S. Improved Pairwise Alignment of Genomic DNA. Ph.D. Thesis, The Pennsylvania State University, State College, PA, USA, 2007. [Google Scholar]
- Zhang, Y.E.; Vibranovski, M.D.; Krinsky, B.H.; Long, M.Y. Age-dependent chromosomal distribution of male-biased genes in Drosophila. Genome Res. 2010, 20, 1526–1533. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.E.; Vibranovski, M.D.; Landback, P.; Marais, G.A.; Long, M. Chromosomal redistribution of male-biased genes in mammalian evolution with two bursts of gene gain on the X chromosome. PLoS Biol. 2010, 8, e1000494. [Google Scholar] [CrossRef]
- Edgar, R.C. MUSCLE: Multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 2004, 32, 1792–1797. [Google Scholar] [CrossRef] [Green Version]
- Suyama, M.; Torrents, D.; Bork, P. PAL2NAL: Robust conversion of protein sequence alignments into the corresponding codon alignments. Nucleic Acids Res. 2006, 34, W609–W612. [Google Scholar] [CrossRef] [Green Version]
- Betran, E.; Thornton, K.; Long, M. Retroposed new genes out of the X in Drosophila. Genome Res. 2002, 12, 1854–1859. [Google Scholar] [CrossRef]
- Slater, G.S.; Birney, E. Automated generation of heuristics for biological sequence comparison. BMC Bioinform. 2005, 6, 31. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kechin, A.; Boyarskikh, U.; Kel, A.; Filipenko, M. cutPrimers: A New Tool for Accurate Cutting of Primers from Reads of Targeted Next Generation Sequencing. J. Comput. Biol. 2017, 24, 1138–1143. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.; Paggi, J.M.; Park, C.; Bennett, C.; Salzberg, S.L. Graph-based genome alignment and genotyping with HISAT2 and HISAT-genotype. Nat. Biotechnol. 2019, 37, 907–915. [Google Scholar] [CrossRef] [PubMed]
- Pertea, M.; Pertea, G.M.; Antonescu, C.M.; Chang, T.C.; Mendell, J.T.; Salzberg, S.L. StringTie enables improved reconstruction of a transcriptome from RNA-seq reads. Nat. Biotechnol. 2015, 33, 290–295. [Google Scholar] [CrossRef] [Green Version]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 38. [Google Scholar] [CrossRef] [Green Version]
- Shao, C.; Bao, B.; Xie, Z.; Chen, X.; Li, B.; Jia, X.; Yao, Q.; Ortí, G.; Li, W.; Li, X.; et al. The genome and transcriptome of Japanese flounder provide insights into flatfish asymmetry. Nat. Genet. 2017, 49, 119–124. [Google Scholar] [CrossRef]
- Magadum, S.; Banerjee, U.; Murugan, P.; Gangapur, D.; Ravikesavan, R. Gene duplication as a major force in evolution. J. Genet. 2013, 92, 155–161. [Google Scholar] [CrossRef]
- Blomme, T.; Vandepoele, K.; De Bodt, S.; Simillion, C.; Maere, S.; Van de Peer, Y. The gain and loss of genes during 600 million years of vertebrate evolution. Genome Biol. 2006, 7, 12. [Google Scholar] [CrossRef] [Green Version]
- Lu, J.; Peatman, E.; Tang, H.; Lewis, J.; Liu, Z. Profiling of gene duplication patterns of sequenced teleost genomes: Evidence for rapid lineage-specific genome expansion mediated by recent tandem duplications. BMC Genom. 2012, 13, 246. [Google Scholar] [CrossRef] [Green Version]
- Inoue, T.; Kagawa, T.; Inoue-Mochita, M.; Isono, K.; Ohtsu, N.; Nobuhisa, I.; Fukushima, M.; Tanihara, H.; Taga, T. Involvement of the Hipk family in regulation of eyeball size, lens formation and retinal morphogenesis. FEBS Lett. 2010, 584, 3233–3238. [Google Scholar] [CrossRef] [Green Version]
- Zha, L.H.; Zhou, J.; Tan, Y.; Guo, S.; Zhang, M.Q.; Li, S.; Yan, P.; Yu, Z.X. NLRC3 inhibits PDGF-induced PASMCs proliferation via PI3K-mTOR pathway. J. Cell. Physiol. 2020, 235, 9557–9567. [Google Scholar] [CrossRef] [PubMed]
- Takayama, K.I.; Suzuki, T.; Tanaka, T.; Fujimura, T.; Takahashi, S.; Urano, T.; Ikeda, K.; Inoue, S. TRIM25 enhances cell growth and cell survival by modulating p53 signals via interaction with G3BP2 in prostate cancer. Oncogene 2018, 37, 2165–2180. [Google Scholar] [CrossRef] [PubMed]
- Kolde, R. Pheatmap: Pretty Heatmaps. 2015. Available online: https://rdrr.io/cran/pheatmap/ (accessed on 25 February 2021).
- Zhou, Q.; Zhang, G.; Zhang, Y.; Xu, S.; Zhao, R.; Zhan, Z.; Li, X.; Ding, Y.; Yang, S.; Wang, W. On the origin of new genes in Drosophila. Genome Res. 2008, 18, 1446–1455. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schneider, H.; Sampaio, I. The systematics and evolution of New World primates—A review. Mol. Phylogen. Evol. 2015, 82, 348–357. [Google Scholar] [CrossRef]
- Vinckenbosch, N.; Dupanloup, I.; Kaessmann, H. Evolutionary fate of retroposed gene copies in the human genome. Proc. Natl. Acad. Sci. USA 2006, 103, 3220–3225. [Google Scholar] [CrossRef] [Green Version]
- Edmiston, J.S.; Archer, K.J.; Scian, M.J.; Joyce, A.R.; Zedler, B.K.; Murrelle, E.L. Gene expression profiling of peripheral blood leukocytes identifies potential novel biomarkers of chronic obstructive pulmonary disease in current and former smokers. Biomarkers 2010, 15, 715–730. [Google Scholar] [CrossRef]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, H.; Chen, C.; Wang, Z.; Wang, K.; Li, Y.; Wang, W. Pattern of New Gene Origination in a Special Fish Lineage, the Flatfishes. Genes 2021, 12, 1819. https://doi.org/10.3390/genes12111819
Li H, Chen C, Wang Z, Wang K, Li Y, Wang W. Pattern of New Gene Origination in a Special Fish Lineage, the Flatfishes. Genes. 2021; 12(11):1819. https://doi.org/10.3390/genes12111819
Chicago/Turabian StyleLi, Haorong, Chunyan Chen, Zhongkai Wang, Kun Wang, Yongxin Li, and Wen Wang. 2021. "Pattern of New Gene Origination in a Special Fish Lineage, the Flatfishes" Genes 12, no. 11: 1819. https://doi.org/10.3390/genes12111819
APA StyleLi, H., Chen, C., Wang, Z., Wang, K., Li, Y., & Wang, W. (2021). Pattern of New Gene Origination in a Special Fish Lineage, the Flatfishes. Genes, 12(11), 1819. https://doi.org/10.3390/genes12111819