
A Two-Year Clinical Description of a Patient with a Rare Type of Low-GGT Cholestasis Caused by a Novel Variant of USP53

 , ,
, , 
Abstract
:1. Introduction
2. Materials and Methods
3. Results
3.1. Clinical Evaluation
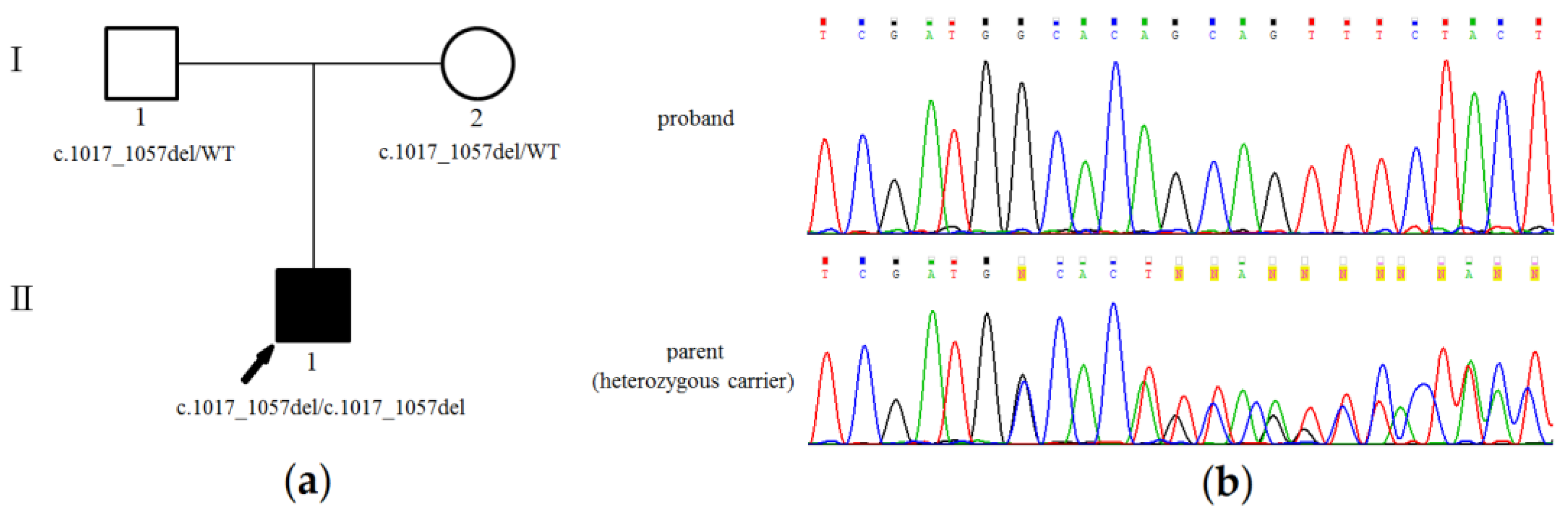
3.2. Genetic Analysis
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Feldman, A.G.; Sokol, R.J. Neonatal Cholestasis. NeoReviews 2013, 14, e63–e73. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gotze, T.; Blessing, H.; Grillhosl, C.; Gerner, P.; Hoerning, A. Neonatal Cholestasis—Differential Diagnoses, Current Diagnostic Procedures, and Treatment. Front. Pediatrics 2015, 3, 43. [Google Scholar]
- Lane, E.; Murray, K.F. Neonatal Cholestasis. Pediatric Clin. N. Am. 2017, 64, 621–639. [Google Scholar] [CrossRef] [PubMed]
- McKiernan, P.J. Neonatal cholestasis. Semin. Neonatol. 2002, 7, 153–165. [Google Scholar] [CrossRef] [PubMed]
- Maddirevula, S.; Alhebbi, H.; Alqahtani, A.; Algoufi, T.; Alsaif, H.S.; Ibrahim, N.; Abdulwahab, F.; Barr, M.; Alzaidan, H.; Almehaideb, A.; et al. Identification of novel loci for pediatric cholestatic liver disease defined by KIF12, PPM1F, USP53, LSR, and WDR83OS pathogenic variants. Genet. Med. Off. J. Am. Coll. Med Genet. 2019, 21, 1164–1172. [Google Scholar] [CrossRef]
- Alhebbi, H.; Peer-Zada, A.A.; Al-Hussaini, A.A.; Algubaisi, S.; Albassami, A.; AlMasri, N.; Alrusayni, Y.; Alruzug, I.M.; Alharby, E.; Samman, M.A.; et al. New paradigms of USP53 disease: Normal GGT cholestasis, BRIC, cholangiopathy, and responsiveness to rifampicin. J. Hum. Genet. 2021, 66, 151–159. [Google Scholar] [CrossRef]
- Zhang, J.; Yang, Y.; Gong, J.Y.; Li, L.T.; Li, J.Q.; Zhang, M.H.; Lu, Y.; Xie, X.B.; Hong, Y.R.; Yu, Z.; et al. Low-GGT intrahepatic cholestasis associated with biallelic USP53 variants: Clinical, histological and ultrastructural characterization. Liver Int. Off. J. Int. Assoc. Study Liver 2020, 40, 1142–1150. [Google Scholar] [CrossRef] [PubMed]
- Bull, L.N.; Ellmers, R.; Foskett, P.; Strautnieks, S.; Sambrotta, M.; Czubkowski, P.; Jankowska, I.; Wagner, B.; Deheragoda, M.; Thompson, R.J. Cholestasis Due to USP53 Deficiency. J. Pediatric Gastroenterol. Nutr. 2021, 72, 667–673. [Google Scholar] [CrossRef] [PubMed]
- Ryzhkova, O.P.; Kardymon, O.L.; Prohorchuk, E.B.; Konovalov, F.A.; Maslennikov, A.B.; Stepanov, V.A.; Afanasyev, A.A.; Zaklyazminskaya, E.V.; Rebrikov, D.V.; Savostianov, K.V.; et al. Guidelines for the interpretation of massive parallel sequencing variants (update 2018, v2). Med Genet. 2019, 18, 3–24. [Google Scholar]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. Off. J. Am. Coll. Med Genet. 2015, 17, 405–424. [Google Scholar] [CrossRef] [PubMed]
- Bajenaru, N.; Balaban, V.; Savulescu, F.; Campeanu, I.; Patrascu, T. Hepatic hemangioma—review. J. Med. Life 2015, 8, 4–11. [Google Scholar]
- Kim, H.; Pawlikowsk, A.L.; Su, H.; Young, W.L. Genetics and Vascular Biology of Angiogenesis and Vascular Malformations. In Stroke, 6th ed.; Pathophysiology, Diagnosis, and Management; Elsevier: Amsterdam, The Netherlands, 2016; pp. 149–162.e7. [Google Scholar]

{kind=link}
| Normal | 8 m | 1 y 2 m | 1 y 6 m | 1 y 8 m | 2 y 1 m | ||||
| Hemoglobin | 110–140 g/L | 115 | 139 | 122 | 134 | 112 | |||
| red blood cells | 3.5–4.5 × 1012/L | - | 5.3 | 5.3 | 5.28 | 5.74 | |||
| white blood cells | 6–17.5 × 109/L | 17.4 | 19.7 | 12.6 | 16.4 | 12 | |||
| platelets | 160–390 × 109/L | 339 | 297 | 232 | 311 | 262 | |||
| Normal | 3 m | 4 m | 8 m | 11 m | 1 y 2 m | 1 y 6 m | 1 y 8 m | 2 y 1 m | |
| total bilirubin | 5–21 µmol/L | 147 | 245 | 157 | 22.9 | 38.8 | 26.5 | 33.6 | 6.6 |
| direct bilirubin | < 3.4 µmol/L | 124.5 | 120 | 86 | 14.7 | 18.2 | 16.7 | 15.2 | 2.1 |
| ALT 1 | 0–40 U/L | 185 | 237 | 359 | 49.5 | 46.4 | 57.9 | 63.3 | 31.6 |
| AST 2 | 0–40 U/L | 133.5 | 122 | 338 | 64.2 | 72.9 | 73.9 | 36.2 | |
| ALP 3 | 82–383 U/L | 1596 | 1283 | 404 | 492.9 | 472 | 453.8 | 458.8 | |
| GGT 4 | 0–6 m: < 204; 6–12 m: < 34; 1–3 y: < 18 U/L | 54 | 61 | 38.7 | - | 23.3 | 16.9 | 21.1 | - |
| glucose | µmol/L | - | - | - | - | 2.9 | - | 4.16 | 4.27 |
| urea | 2.8–7.2 µmol/L | - | - | 3.7 | 3.2 | 5 | 4.1 | 5.1 | 4.3 |
| cholesterol | 3.2–5.2 µmol/L | - | 4.9 | 3.3 | - | - | 2.32 | 2.89 | 3.01 |
| total protein | 64–83 g/L | - | 65 | 65.2 | - | 74 | - | 65.6 | - |
| albumin | 35–52 g/L | - | - | 37.1 | - | 39.7 | - | 39.6 | - |
| AFP 5 | 0.5–50000 IU/ml | - | 281 | 2459 | - | 5.97 | - | 1.95 | - |
| calcium | 2.25–2.75 µmol/L | - | - | - | - | 2.59 | - | 2.69 | - |
| Normal | 3 m | 8 m | 1 y 2 m | 1 y 6 m | 1 y 8 m | 2 y 1 m | |||
| fibrinogen | 2–4 g/L | 1.06 | 2.5 | 2.7 | 1.59 | 2.62 | 2.56 | ||
| prothrombin index | 81–138% | - | 52 | 85 | 96.7 | 90 | 98.3 | ||
| aPTT 6 | 25–35 sec. | - | - | 35 | 35.6 | - | 32.6 | ||
| Study | Patient | Sex | Age at Presentation | Cholestasis | Itch | Hepatomegaly | Hepatosplenomegaly | Fibrosis in the Liver | Coagulopathy | Cholangiopathy | Deafness | Additional Phenotype | Variant USP53 (NM_019050.2) | Age of Sampling | Total Bilirubin (µmol/L) | Direct Bilirubin (µmol/L) | ALT (U/L) 1 | AST (U/L) 2 | GGT (U/L) 3 | ALP (U/L) 4 | Clinical Course (Current Age) |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| this study a | 1 | M | Neonatal | L/N-GGT 5 | + | + | + | + | - | - | - | Hepatic hemangiomas | c.1017_1057del; p.(Cys339Trpfs) | 3 m | 147 | 124 | 185 | 133 | 54 | 159 | ANL 8 (3 y) |
| Hamoud Al Habibi et al. b | 1 IV:4 | M | 4 m | L/N-GGT | - | - | - | NK 7 | + | - | - | - | c.951delT; p.(Phe317fs) | 4 m | 172 | 158 | 30 | 37 | 23 | 553 | ANL (2 y) |
| 1a IV:5 | F | 15 m | L/N-GGT | - | - | - | NK | - | + | + | - | c.951delT; p.(Phe317fs) | 15 m | 159 | 132 | 97 | 82 | 35 | 6316 | APLT 9 (24 y) | |
| 1a IV:8 | F | 5 m | BRIC 6 | - | - | - | NK | - | - | + | - | c.951delT; p.(Phe317fs) | 5 m | 26 | 20 | 25 | 29 | 39 | 4432 | ANL (17 y) | |
| 1a IV:1 | M | 1 y | L/N-GGT | - | - | - | NK | - | - | - | - | c.951delT; p.(Phe317fs) | 1 y | 402 | 298 | 136 | 352 | 30 | 2557 | ANL (6 y) | |
| 2 III:1 | M | 18 m | BRIC | - | + | + | + | - | - | - | Speech and developmental delay | c.951delT; p.(Phe317fs) | 18 m | 155 | 85 | 51 | 61 | 24 | 719 | ANL (7 y) | |
| 3 V:1 | F | 16 m | L/N-GGT | + | - | - | NK | - | - | - | - | c.1744C > T; p.(Arg582Ter) | 16 m | 146 | 142 | 36 | 69 | 23 | 504 | ANL (1 y) | |
| 3 IV:3 | M | 18 y | BRIC | - | + | + | + | - | - | - | - | c.1744C > T; p.(Arg582Ter) | 18 y | 876 | 680 | 45 | 45 | 39 | 939 | ANL (35 y) | |
| 3 IV:2 | F | 16 y | BRIC | - | - | - | - | - | - | - | Hypothyroidism | c.1744C > T; p.(Arg582Ter) | 16 y | 179 | 128 | 63 | 80 | 23 | 727 | ANL (18 y) | |
| Jing Zhang et al. c | P1 | F | 3 d | L/N-GGT | NK | NK | NK | + | NK | NK | - | - | c.1012C > T; p.(Arg338Ter) | 12 m | 23 | 19 | 184 | 215 | 23 | 330 | ANL (2 y) |
| P2 | M | 2 d | L/N-GGT | NK | NK | NK | + | NK | NK | - | - | c.169C > T; p.(Arg57Ter) + c.831_832insAG; p.(Val279GlufsTer16) | 4 m | 90 | 65 | 70 | 71 | 72 | 548 | ANL (3 y) | |
| P3 | F | 6 m | L/N-GGT | NK | NK | NK | + | NK | NK | - | - | c.569 + 2T > C + c.878G > T; p.(Gly293Val) | 8 m | 212 | 159 | 103 | 121 | 34 | NK | ANL (5 y) | |
| P4 | M | 5 m | L/N-GGT | NK | NK | NK | + | NK | NK | - | - | c.581del; p.(Arg195GlufsTer38) + c.1012C > T; p.(Arg338Ter) | 9 m | 308 | 167 | 32 | 84 | 39 | 636 | ANL (17 y) | |
| P5 | F | 1 m | L/N-GGT | NK | NK | NK | + | NK | NK | - | - | c.1012C > T; p.(Arg338Te)r + c.1426C > T; p.(Arg476Ter) | 4 m | 275 | 216 | 28 | 51 | 40 | 543 | LF 10 | |
| P6 | M | 5 m | L/N-GGT | NK | NK | NK | + | NK | NK | + | - | c.1558C > T; p.(Arg520Ter) + c.395A > G; p.(His132Arg) | 6 m | 85 | 72 | 26 | 41 | 27 | 342 | ANL (1 y); CI 11 (1 y) | |
| P7 | M | 1 m | L/N-GGT | NK | NK | NK | + | NK | NK | - | - | c.297G > T; p.(Arg99Ser) + c.1012C > T; p.(Arg338Ter) | 8 m | 153 | 137 | 18 | 225 | 22 | 283 | ANL (1 y) | |
| Laura N Bull et al. d | 1 | F | 3 m | L/N-GGT | - | - | + | + | NK | NK | - | - | Deletion of first coding exon | 3 m | 583 | 539 | 163 | NK | 41 | NK | ANL (11 y) |
| 2 | M | 2 m | L/N-GGT | - | - | upper limit of normal | + | NK | NK | - | - | Deletion of first coding exon | 2 m | 459 | 335 | 169 | 222 | 62 | NK | ANL (8 y) | |
| 3 | F | 5 m | L/N-GGT | - | - | + | NK | NK | NK | - | - | Deletion of first coding exon | 7 m | 1626 | 1290 | 61 | 103 | 46 | NK | ANL (2 y) | |
| 4 | F | 7 y | L/N-GGT | - | - | - | NK | NK | NK | NK | - | c.145-11_167del | NK | NK | NK | NK | NK | Normal | NK | ANL (10 y) | |
| 5 | M | Neonatal | L/N-GGT | - | - | + | + | NK | NK | NK | - | c.145-11_167del | 13 y | 344 | 167 | 22 | 38 | 35 | NK | ANL (13 y) | |
| 6 | M | 15 y | L/N-GGT | - | - | + | + | NK | NK | NK | - | c.725C > T; p.(Pro242Leu) | 15 y | 300 | NK | 78 | 74 | 25 | NK | ANL (18 y) | |
| 7 | M | 4 y | L/N-GGT | + | - | - | NK | NK | NK | NK | - | c.510del; p.(Ser171ArgfsTer62) | 21 y | 557 | 247 | 84 | 46 | 34 | NK | ANL (21 y) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Shatokhina, O.; Semenova, N.; Demina, N.; Dadali, E.; Polyakov, A.; Ryzhkova, O. A Two-Year Clinical Description of a Patient with a Rare Type of Low-GGT Cholestasis Caused by a Novel Variant of USP53. Genes 2021, 12, 1618. https://doi.org/10.3390/genes12101618
Shatokhina O, Semenova N, Demina N, Dadali E, Polyakov A, Ryzhkova O. A Two-Year Clinical Description of a Patient with a Rare Type of Low-GGT Cholestasis Caused by a Novel Variant of USP53. Genes. 2021; 12(10):1618. https://doi.org/10.3390/genes12101618
Chicago/Turabian StyleShatokhina, Olga, Natalia Semenova, Nina Demina, Elena Dadali, Alexander Polyakov, and Oxana Ryzhkova. 2021. "A Two-Year Clinical Description of a Patient with a Rare Type of Low-GGT Cholestasis Caused by a Novel Variant of USP53" Genes 12, no. 10: 1618. https://doi.org/10.3390/genes12101618
APA StyleShatokhina, O., Semenova, N., Demina, N., Dadali, E., Polyakov, A., & Ryzhkova, O. (2021). A Two-Year Clinical Description of a Patient with a Rare Type of Low-GGT Cholestasis Caused by a Novel Variant of USP53. Genes, 12(10), 1618. https://doi.org/10.3390/genes12101618