
Neuroplastin in Neuropsychiatric Diseases




Abstract
:1. Introduction
2. Molecular Characteristics of Neuroplastin
2.1. Structure and Expression of Neuroplastin
2.2. Interactions and Binding Partners of Neuroplastin in the Nervous System
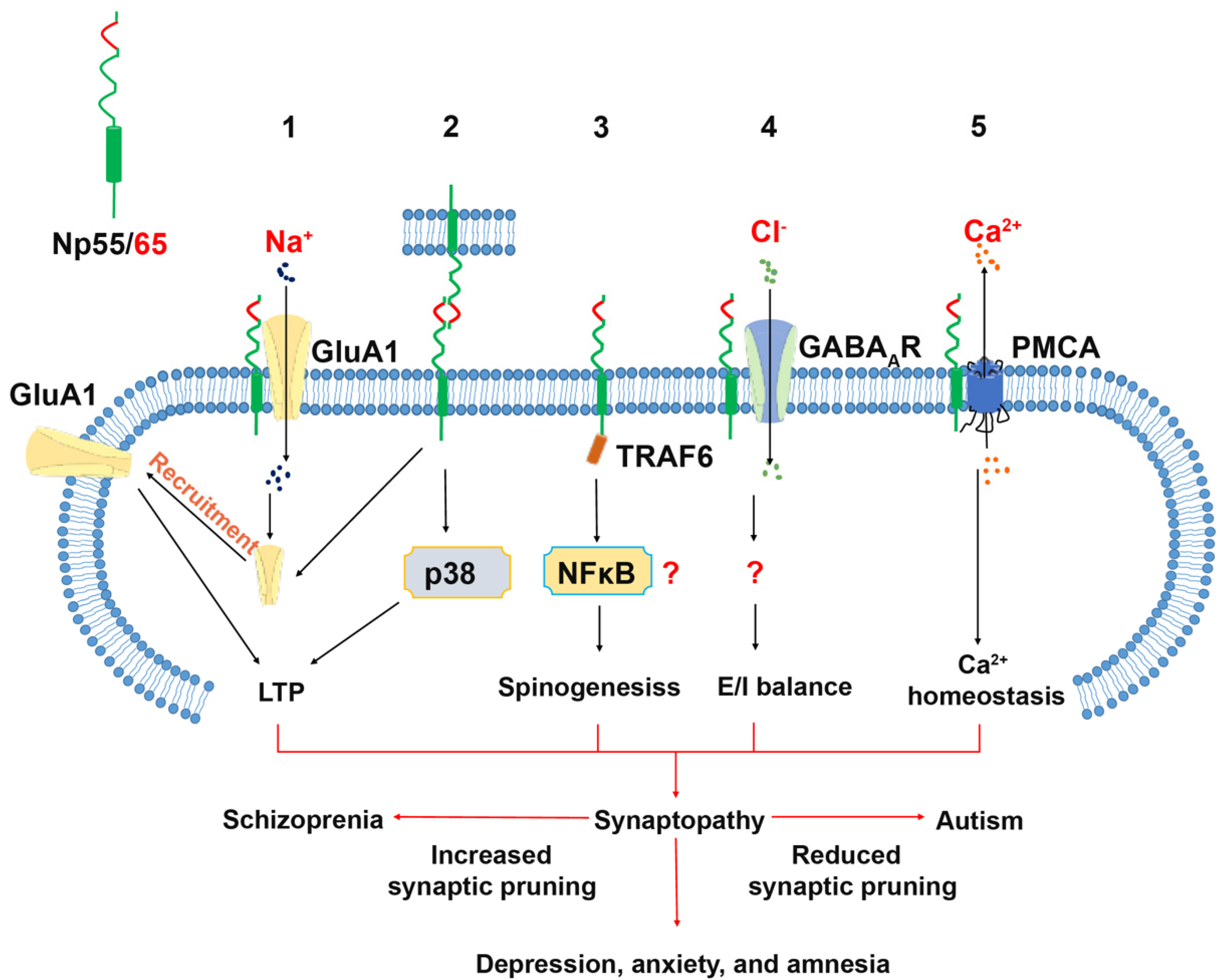
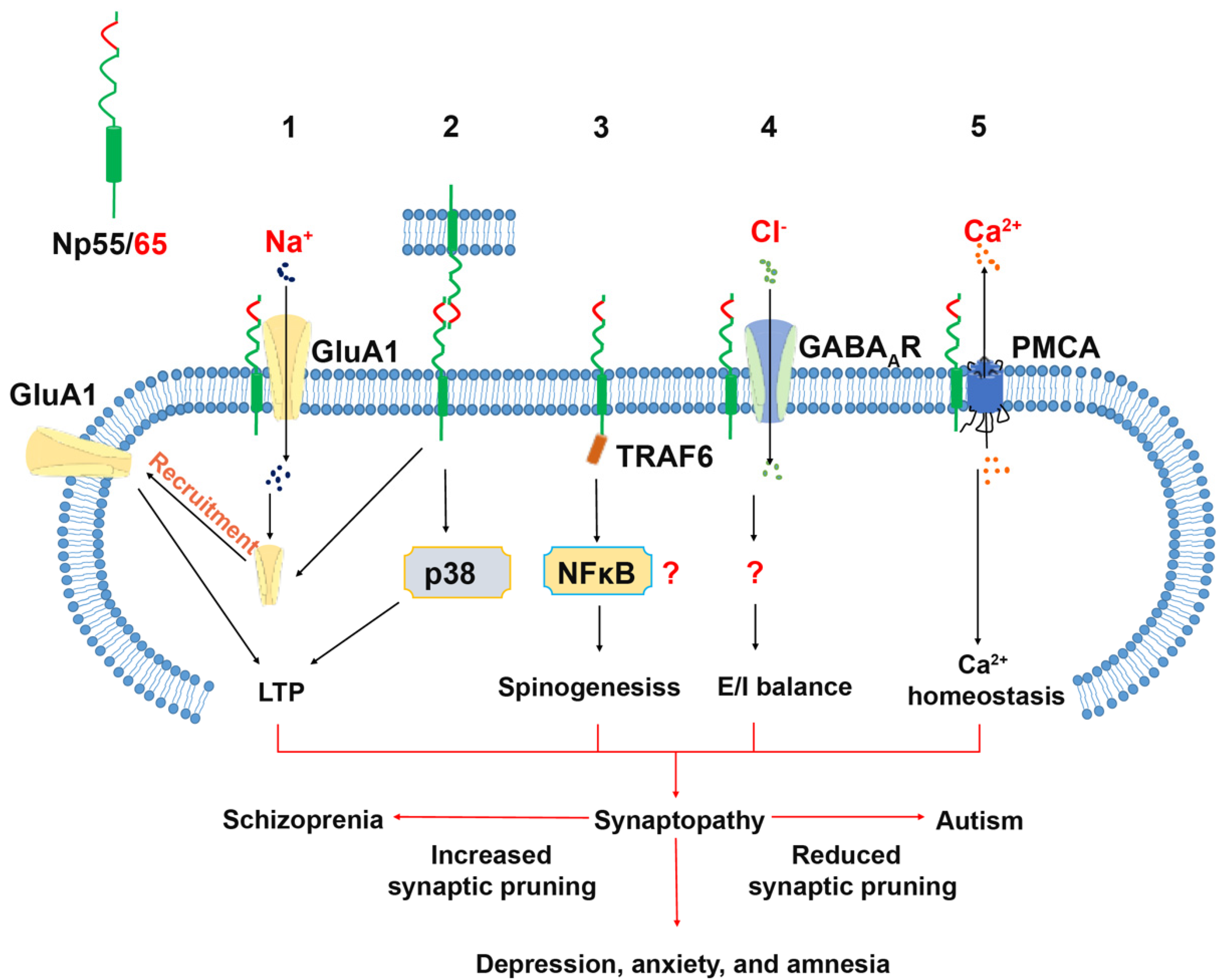
2.2.1. Neuroplastin Homophilic Binding and AMPA Receptor Subunit GluA1
2.2.2. Neuroplastin and GABAA Receptor (GABAAR)
2.2.3. Neuroplastin Binding to TRAF6
2.2.4. Neuroplastin Binding to Plasma Membrane Ca2+ ATPases (PMCA)
3. Neuroplastin in Neurological Diseases
3.1. Schizophrenia (SZ) and Autism Spectrum Disorder (ASD)
3.1.1. Neuroplastin Relation to Schizophrenia
3.1.2. Autism Spectrum Disorder (ASD)
3.2. Depression and Anxiety Disorder
3.3. Alzheimer’s (AD) Disease
3.4. Cognition, Antero- and Retrograde Amnesia
3.5. Other Diseases Related to Neuroplastin
3.5.1. Deafness
3.5.2. Cancer
3.5.3. Various Diseases
4. Future Research Directions
5. Conclusions and Perspectives
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Vos, T.; Abajobir, A.A.; Abbafati, C.; Abbas, K.M.; Abate, K.A.; Abd-Allah, F.; Abdulle, A.M.; Abebo, T.A.; Abera, S.F.; Aboyans, V.; et al. Global, regional, and national incidence, prevalence, and years lived with disability for 328 diseases and injuries for 195 countries, 1990–2016: A systematic analysis for the Global Burden of Disease Study 2016. Lancet 2017, 390, 1211–1259. [Google Scholar] [CrossRef] [Green Version]
- Zablotsky, B.; Black, L.I.; Maenner, M.J.; Schieve, L.A.; Danielson, M.L.; Bitsko, R.H.; Blumberg, S.J.; Kogan, M.D.; Boyle, C.A. Prevalence and Trends of Developmental Disabilities among Children in the United States: 2009–2017. Pediatrics 2019, 144, e20190811. [Google Scholar] [CrossRef]
- Maenner, M.J.; Shaw, K.A.; Baio, J.; Washington, A.; Patrick, M.; DiRienzo, M.; Christensen, D.L.; Wiggins, L.D.; Pettygrove, S.; Andrews, J.G.; et al. Prevalence of Autism Spectrum Disorder Among Children Aged 8 Years—Autism and Developmental Disabilities Monitoring Network, 11 Sites, United States, 2016. MMWR Surveill. Summ. 2020, 69, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Sloane, P.D.; Zimmerman, S.; Suchindran, C.; Reed, P.; Wang, L.; Boustani, M.; Sudha, S. The public health impact of Alzheimer’s disease, 2000–2050: Potential implication of treatment advances. Annu. Rev. Public Health 2002, 23, 213–231. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gustavsson, A.; Svensson, M.; Jacobi, F.; Allgulander, C.; Alonso, J.; Beghi, E.; Dodel, R.; Ekman, M.; Faravelli, C.; Fratiglioni, L.; et al. Cost of disorders of the brain in Europe 2010. Eur. Neuropsychopharmacol. 2011, 21, 718–779. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chong, H.Y.; Teoh, S.L.; Wu, D.B.; Kotirum, S.; Chiou, C.F.; Chaiyakunapruk, N. Global economic burden of schizophrenia: A systematic review. Neuropsychiatr. Dis. Treat. 2016, 12, 357–373. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rogge, N.; Janssen, J. The Economic Costs of Autism Spectrum Disorder: A Literature Review. J. Autism Dev. Disord. 2019, 49, 2873–2900. [Google Scholar] [CrossRef]
- Baresic, A.; Nash, A.J.; Dahoun, T.; Howes, O.; Lenhard, B. Understanding the genetics of neuropsychiatric disorders: The poten-tial role of genomic regulatory blocks. Mol. Psychiatry 2020, 25, 6–18. [Google Scholar] [CrossRef]
- Sakurai, K.; Migita, O.; Toru, M.; Arinami, T. An association between a missense polymorphism in the close homologue of L1 (CHL1, CALL) gene and schizophrenia. Mol. Psychiatry 2002, 7, 412–415. [Google Scholar] [CrossRef] [Green Version]
- Munafo, M.R.; Attwood, A.S.; Flint, J. Neuregulin 1 genotype and schizophrenia. Schizophr. Bull. 2008, 34, 9–12. [Google Scholar] [CrossRef] [Green Version]
- Bernstein, H.G.; Bogerts, B. Neuregulin-1 alpha, the underestimated molecule: Emerging new roles in normal brain function and the pathophysiology of schizophrenia? Genome 2013, 56, 703–704. [Google Scholar] [CrossRef]
- Ripke, S.; Neale, B.M.; Corvin, A.; Walters, J.T.R.; Farh, K.; Holmans, P.A.; Lee, P.; Bulik-Sullivan, B.; Collier, D.A.; Huang, H.; et al. Biological insights from 108 schizophrenia-associated genetic loci. Nature 2014, 511, 421–427. [Google Scholar]
- Dennison, C.A.; Legge, S.E.; Pardinas, A.F.; Walters, J.T.R. Genome-wide association studies in schizophrenia: Recent advances, challenges and future perspective. Schizophr. Res. 2020, 217, 4–12. [Google Scholar] [CrossRef] [PubMed]
- Jamain, S.; Quach, H.; Betancur, C.; Rastam, M.; Colineaux, C.; Gillberg, I.C.; Soderstrom, H.; Giros, B.; Leboyer, M.; Gillberg, C.; et al. Mutations of the X-linked genes encoding neuroligins NLGN3 and NLGN4 are associated with autism. Nat. Genet. 2003, 34, 27–29. [Google Scholar] [CrossRef] [Green Version]
- Yan, J.; Oliveira, G.; Coutinho, A.; Yang, C.; Feng, J.; Katz, C.; Sram, J.; Bockholt, A.; Jones, I.R.; Craddock, N.; et al. Analysis of the neuroligin 3 and 4 genes in autism and other neuropsychiatric patients. Mol. Psychiatry 2005, 10, 329–332. [Google Scholar] [CrossRef] [Green Version]
- Sudhof, T.C. Neuroligins and neurexins link synaptic function to cognitive disease. Nature 2008, 455, 903–911. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, Y.; Yao, X.; March, M.E.; Meng, X.; Li, J.; Wei, Z.; Sleiman, P.M.A.; Hakonarson, H.; Xia, Q.; Li, J. Target Genes of Autism Risk Loci in Brain Frontal Cortex. Front. Genet. 2019, 10, 707. [Google Scholar] [CrossRef] [Green Version]
- Anney, R.J.L.; Ripke, S.; Anttila, V.; J Grove, J.; Holmans, P.; Huang, H.; Klei, L.; Lee, P.H.; Medland, S.E.; Neale, B.; et al. Me-ta-analysis of GWAS of over 16,000 individuals with autism spectrum disorder highlights a novel locus at 10q24.32 and a significant overlap with schizophrenia. Mol. Autism 2017, 8, 21. [Google Scholar]
- Hung, A.Y.; Futai, K.; Sala, C.; Valtschanoff, J.G.; Ryu, J.; Woodworth, M.A.; Kidd, F.L.; Sung, C.C.; Miyakawa, T.; Bear, M.F.; et al. Smaller dendritic spines, weaker synaptic transmission, but enhanced spatial learning in mice lacking Shank1. J. Neurosci. 2008, 28, 1697–1708. [Google Scholar] [CrossRef] [Green Version]
- Peca, J.; Feliciano, C.; Ting, J.T.; Wang, W.; Wells, M.F.; Venkatraman, T.N.; Lascola, C.D.; Fu, Z.; Feng, G. Shank3 mutant mice display autistic-like behaviours and striatal dysfunction. Nature 2011, 472, 437–442. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.; McCoy, P.A.; Rodriguiz, R.M.; Pan, Y.; Je, H.S.; Roberts, A.C.; Kim, C.J.; Berrios, J.; Colvin, J.S.; Bousquet-Moore, D.; et al. Synaptic dysfunction and abnormal behaviors in mice lacking major isoforms of Shank3. Hum. Mol. Genet. 2011, 20, 3093–3108. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schmeisser, M.J.; Ey, E.; Wegener, S.; Bockmann, J.; Stempel, A.V.; Kuebler, A.; Janssen, A.L.; Udvardi, P.T.; Shiban, E.; Spilker, C.; et al. Autistic-like behaviours and hyperactivity in mice lacking ProSAP1/Shank2. Nature 2012, 486, 256–260. [Google Scholar] [CrossRef] [PubMed]
- Won, H.; Lee, H.R.; Gee, H.Y.; Mah, W.; Kim, J.I.; Lee, J.; Ha, S.; Chung, C.; Jung, E.S.; Cho, Y.S.; et al. Autistic-like social behav-iour in Shank2-mutant mice improved by restoring NMDA receptor function. Nature 2012, 486, 261–265. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Christian, K.M.; Song, H.; Ming, G.L. Synaptic dysfunction in complex psychiatric disorders: From genetics to mecha-nisms. Genome Med. 2018, 10, 9. [Google Scholar] [CrossRef] [Green Version]
- Penzes, P.; Buonanno, A.; Passafaro, M.; Sala, C.; Sweet, R.A. Developmental vulnerability of synapses and circuits associated with neuropsychiatric disorders. J. Neurochem. 2013, 126, 165–182. [Google Scholar] [CrossRef]
- Lopatina, O.L.; Malinovskaya, N.A.; Komleva, Y.K.; Gorina, Y.V.; Shuvaev, A.N.; Olovyannikova, R.Y.; Belozor, O.S.; Belova, O.A.; Higashida, H.; Salmina, A.B. Excitation/inhibition imbalance and impaired neurogenesis in neurodevelopmental and neurodegenerative disorders. Rev. Neurosci. 2019, 30, 807–820. [Google Scholar] [CrossRef] [PubMed]
- Anderson, R.M.; Hadjichrysanthou, C.; Evans, S.; Wong, M.M. Why do so many clinical trials of therapies for Alzheimer’s disease fail? Lancet 2017, 390, 2327–2329. [Google Scholar] [CrossRef]
- Dhillon, S. Aducanumab: First Approval. Drugs 2021, 81, 1437–1443. [Google Scholar] [CrossRef]
- Paik, J. Olanzapine/Samidorphan: First Approval. Drugs 2021, 81, 1431–1436. [Google Scholar] [CrossRef]
- LeClerc, S.; Easley, D. Pharmacological therapies for autism spectrum disorder: A review. Pharm. Ther. 2015, 40, 389–397. [Google Scholar]
- Baribeau, D.; Anagnostou, E. Novel treatments for autism spectrum disorder based on genomics and systems biology. Pharmacol. Ther. 2021, 107939. [Google Scholar] [CrossRef]
- Doherty, J.L.; Owen, M.J. Genomic insights into the overlap between psychiatric disorders: Implications for research and clinical practice. Genome Med. 2014, 6, 29. [Google Scholar] [CrossRef] [Green Version]
- Beesley, P.W.; Herrera-Molina, R.; Smalla, K.H.; Seidenbecher, C. The Neuroplastin adhesion molecules: Key regulators of neu-ronal plasticity and synaptic function. J. Neurochem. 2014, 131, 268–283. [Google Scholar] [CrossRef]
- Hill, I.E.; Selkirk, C.P.; Hawkes, R.B.; Beesley, P.W. Characterization of novel glycoprotein components of synaptic membranes and postsynaptic densities, gp65 and gp55, with a monoclonal antibody. Brain Res. 1988, 461, 27–43. [Google Scholar] [CrossRef]
- Herrera-Molina, R.; Mlinac-Jerkovic, K.; Ilic, K.; Stober, F.; Vemula, S.K.; Sandoval, M.; Milosevic, N.J.; Simic, G.; Smalla, K.H.; Goldschmidt, J.; et al. Neuroplastin deletion in glutamatergic neurons impairs selective brain functions and calcium regula-tion: Implication for cognitive deterioration. Sci. Rep. 2017, 7, 7273. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Langnaese, K.; Beesley, P.W.; Gundelfinger, E.D. Synaptic membrane glycoproteins gp65 and gp55 are new members of the im-munoglobulin superfamily. J. Biol. Chem. 1997, 272, 821–827. [Google Scholar] [CrossRef] [Green Version]
- Langnaese, K.; Mummery, R.; Gundelfinger, E.D.; Beesley, P.W. Immunoglobulin superfamily members gp65 and gp55: Tissue distribution of glycoforms. FEBS Lett. 1998, 429, 284–288. [Google Scholar] [CrossRef] [Green Version]
- Sakaguchi, M.; Yamamoto, M.; Miyai, M.; Maeda, T.; Hiruma, J.; Murata, H.; Kinoshita, R.; Winarsa Ruma, I.M.; Putranto, E.W.; Inoue, Y.; et al. Identification of an S100A8 Receptor Neuroplastin-beta and its Heterodimer Formation with EMMPRIN. J. Invest. Derm. 2016, 136, 2240–2250. [Google Scholar] [CrossRef] [PubMed]
- Smalla, K.H.; Matthies, H.; Langnase, K.; Shabir, S.; Bockers, T.M.; Wyneken, U.; Staak, S.; Krug, M.; Beesley, P.W.; Gundelfinger, E.D. The synaptic glycoprotein neuroplastin is involved in long-term potentiation at hippocampal CA1 synapses. Proc. Natl. Acad. Sci. USA 2000, 97, 4327–4332. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Herrera-Molina, R.; Sarto-Jackson, I.; Montenegro-Venegas, C.; Heine, M.; Smalla, K.H.; Seidenbecher, C.I.; Beesley, P.W.; Gun-delfinger, E.D.; Montag, D. Structure of excitatory synapses and GABAA receptor localization at inhibitory synapses are regulated by neuroplastin-65. J. Biol. Chem. 2014, 289, 8973–8988. [Google Scholar] [CrossRef] [Green Version]
- Kreutz, M.R.; Langnaese, K.; Dieterich, D.C.; Seidenbecher, C.I.; Zuschratter, W.; Beesley, P.W.; Gundelfinger, E.D. Distribution of transcript and protein isoforms of the synaptic glycoprotein neuroplastin in rat retina. Invest. Ophthalmol. Vis. Sci. 2001, 42, 1907–1914. [Google Scholar] [PubMed]
- Bernstein, H.G.; Smalla, K.H.; Bogerts, B.; Gordon-Weeks, P.R.; Beesley, P.W.; Gundelfinger, E.D.; Kreutz, M.R. The immuno-localization of the synaptic glycoprotein neuroplastin differs substantially between the human and the rodent brain. Brain Res. 2007, 1134, 107–112. [Google Scholar] [CrossRef] [PubMed]
- Lin, X.; Brunk, M.G.K.; Yuanxiang, P.; Curran, A.W.; Zhang, E.; Stober, F.; Goldschmidt, J.; Gundelfinger, E.D.; Vollmer, M.; Hap-pel, M.F.K.; et al. Neuroplastin expression is essential for hearing and hair cell PMCA expression. Brain Struct. Funct. 2021, 226, 1533–1551. [Google Scholar] [CrossRef] [PubMed]
- Saito, A.; Fujikura-Ouchi, Y.; Kuramasu, A.; Shimoda, K.; Akiyama, K.; Matsuoka, H.; Ito, C. Association study of putative pro-moter polymorphisms in the neuroplastin gene and schizophrenia. Neurosci. Lett. 2007, 411, 168–173. [Google Scholar] [CrossRef] [PubMed]
- Owczarek, S.; Kiryushko, D.; Larsen, M.H.; Kastrup, J.S.; Gajhede, M.; Sandi, C.; Berezin, V.; Bock, E.; Soroka, V. Neuroplastin-55 binds to and signals through the fibroblast growth factor receptor. FASEB J. 2010, 24, 1139–1150. [Google Scholar] [CrossRef]
- Owczarek, S.; Soroka, V.; Kiryushko, D.; Larsen, M.H.; Yuan, Q.; Sandi, C.; Berezin, V.; Bock, E. Neuroplastin-65 and a mimetic peptide derived from its homophilic binding site modulate neuritogenesis and neuronal plasticity. J. Neurochem. 2011, 117, 984–994. [Google Scholar] [CrossRef]
- Sarto-Jackson, I.; Milenkovic, I.; Smalla, K.H.; Gundelfinger, E.D.; Kaehne, T.; Herrera-Molina, R.; Thomas, S.; Kiebler, M.A.; Sieghart, W. The cell adhesion molecule neuroplastin-65 is a novel interaction partner of gamma-aminobutyric acid type A receptors. J. Biol. Chem. 2012, 287, 14201–14214. [Google Scholar] [CrossRef] [Green Version]
- Wilson, M.C.; Kraus, M.; Marzban, H.; Sarna, J.R.; Wang, Y.; Hawkes, R.; Halestrap, A.P.; Beesley, P.W. The neuroplastin adhesion molecules are accessory proteins that chaperone the monocarboxylate transporter MCT2 to the neuronal cell surface. PLoS ONE 2013, 8, e78654. [Google Scholar]
- Desrivieres, S.; Lourdusamy, A.; Tao, C.; Toro, R.; Jia, T.; Loth, E.; Medina, L.M.; Kepa, A.; Fernandes, A.; Ruggeri, B.; et al. Single nucleotide polymorphism in the neuroplastin locus associates with cortical thickness and intellectual ability in adolescents. Mol. Psychiatry 2015, 20, 263–274. [Google Scholar] [CrossRef] [Green Version]
- Bhattacharya, S.; Herrera-Molina, R.; Sabanov, V.; Ahmed, T.; Iscru, E.; Stober, F.; Richter, K.; Fischer, K.D.; Angenstein, F.; Gold-schmidt, J.; et al. Genetically Induced Retrograde Amnesia of Associative Memories After Neuroplastin Ablation. Biol. Psy-chiatry 2017, 81, 124–135. [Google Scholar] [CrossRef] [Green Version]
- Carrott, L.; Bowl, M.R.; Aguilar, C.; Johnson, S.L.; Chessum, L.; West, M.; Morse, S.; Dorning, J.; Smart, E.; Hardisty-Hughes, R.; et al. Absence of Neuroplastin-65 Affects Synaptogenesis in Mouse Inner Hair Cells and Causes Profound Hearing Loss. J. Neurosci. 2016, 36, 222–234. [Google Scholar] [CrossRef] [Green Version]
- Zeng, W.Z.; Grillet, N.; Dewey, J.B.; Trouillet, A.; Krey, J.F.; Barr-Gillespie, P.G.; Oghalai, J.S.; Muller, U. Neuroplastin Isoform Np55 Is Expressed in the Stereocilia of Outer Hair Cells and Required for Normal Outer Hair Cell Function. J. Neurosci. 2016, 36, 9201–9216. [Google Scholar] [CrossRef] [Green Version]
- Suzuki, J.; Imanishi, E.; Nagata, S. Xkr8 phospholipid scrambling complex in apoptotic phosphatidylserine exposure. Proc. Natl. Acad. Sci. USA 2016, 113, 9509–9514. [Google Scholar] [CrossRef] [Green Version]
- Hu, Y.; Zhan, Q.; Zhang, H.; Liu, X.; Huang, L.; Li, H.; Yuan, Q. Increased Susceptibility to Ischemic Brain Injury in Neuroplastin 65-Deficient Mice Likely via Glutamate Excitotoxicity. Front. Cell Neurosci. 2017, 11, 110. [Google Scholar] [CrossRef]
- Korthals, M.; Langnaese, K.; Smalla, K.H.; Kahne, T.; Herrera-Molina, R.; Handschuh, J.; Lehmann, A.C.; Mamula, D.; Naumann, M.; Seidenbecher, C.; et al. A complex of Neuroplastin and Plasma Membrane Ca(2+) ATPase controls T cell activation. Sci. Rep. 2017, 7, 8358. [Google Scholar] [CrossRef]
- Schmidt, N.; Kollewe, A.; Constantin, C.E.; Henrich, S.; Ritzau-Jost, A.; Bildl, W.; Saalbach, A.; Hallermann, S.; Kulik, A.; Fakler, B.; et al. Neuroplastin and Basigin Are Essential Auxiliary Subunits of Plasma Membrane Ca(2+)-ATPases and Key Regula-tors of Ca(2+) Clearance. Neuron 2017, 96, 827–838 e829. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gong, D.; Chi, X.; Ren, K.; Huang, G.; Zhou, G.; Yan, N.; Lei, J.; Zhou, Q. Structure of the human plasma membrane Ca(2+)-ATPase 1 in complex with its obligatory subunit neuroplastin. Nat. Commun. 2018, 9, 3623. [Google Scholar] [CrossRef] [PubMed]
- Ilic, K.; Mlinac-Jerkovic, K.; Jovanov-Milosevic, N.; Simic, G.; Habek, N.; Bogdanovic, N.; Kalanj-Bognar, S. Hippocampal expres-sion of cell-adhesion glycoprotein neuroplastin is altered in Alzheimer’s disease. J. Cell. Mol. Med. 2019, 23, 1602–1607. [Google Scholar] [CrossRef]
- Li, H.; Liu, Y.; Gao, X.; Liu, L.; Amuti, S.; Wu, D.; Jiang, F.; Huang, L.; Wang, G.; Zeng, J.; et al. Neuroplastin 65 modulates anxiety- and depression-like behavior likely through adult hippocampal neurogenesis and central 5-HT activity. FEBS J. 2019, 286, 3401–3415. [Google Scholar] [CrossRef] [PubMed]
- Vemula, S.K.; Malci, A.; Junge, L.; Lehmann, A.C.; Rama, R.; Hradsky, J.; Matute, R.A.; Weber, A.; Prigge, M.; Naumann, M.; et al. The Interaction of TRAF6 With Neuroplastin Promotes Spinogenesis During Early Neuronal Development. Front. Cell Dev. Biol. 2020, 8, 579513. [Google Scholar] [CrossRef]
- Yagi, T.; Asada, R.; Kanekura, K.; Eesmaa, A.; Lindahl, M.; Saarma, M.; Urano, F. Neuroplastin Modulates Anti-inflammatory Effects of MANF. iScience 2020, 23, 101810. [Google Scholar] [CrossRef]
- Jiang, C.H.; Wei, M.; Zhang, C.; Shi, Y.S. The amino-terminal domain of GluA1 mediates LTP maintenance via interaction with neuroplastin-65. Proc.Natl. Acad. Sci. USA 2021, 118, e2019194118. [Google Scholar] [CrossRef] [PubMed]
- Balog, M.; Blazetic, S.; Ivic, V.; Labak, I.; Krajnik, B.; Marin, R.; Canerina-Amaro, A.; de Pablo, D.P.; Bardak, A.; Gaspar, R.; et al. Disarranged neuroplastin environment upon aging and chronic stress recovery in female Sprague Dawley rats. Eur. J. Neu-rosci. 2021, 1–17. [Google Scholar] [CrossRef]
- Empson, R.M.; Buckby, L.E.; Kraus, M.; Bates, K.J.; Crompton, M.R.; Gundelfinger, E.D.; Beesley, P.W. The cell adhesion molecule neuroplastin-65 inhibits hippocampal long-term potentiation via a mitogen-activated protein kinase p38-dependent reduc-tion in surface expression of GluR1-containing glutamate receptors. J. Neurochem. 2006, 99, 850–860. [Google Scholar] [CrossRef]
- Ge, Y.; Wang, Y.T. GluA1-homomeric AMPA receptor in synaptic plasticity and neurological diseases. Neuropharmacology 2021, 197, 108708. [Google Scholar] [CrossRef]
- Gugustea, R.; Jia, Z. Genetic manipulations of AMPA glutamate receptors in hippocampal synaptic plasticity. Neuropharmacology 2021, 194, 108630. [Google Scholar] [CrossRef]
- Zamanillo, D.; Sprengel, R.; Hvalby, O.; Jensen, V.; Burnashev, N.; Rozov, A.; Kaiser, K.M.; Koster, H.J.; Borchardt, T.; Worley, P.; et al. Importance of AMPA receptors for hippocampal synaptic plasticity but not for spatial learning. Science 1999, 284, 1805–1811. [Google Scholar] [CrossRef] [PubMed]
- Sanderson, D.J.; Sprengel, R.; Seeburg, P.H.; Bannerman, D.M. Deletion of the GluA1 AMPA receptor subunit alters the expression of short-term memory. Learn. Mem. 2011, 18, 128–131. [Google Scholar] [CrossRef] [Green Version]
- Wiedholz, L.M.; Owens, W.A.; Horton, R.E.; Feyder, M.; Karlsson, R.M.; Hefner, K.; Sprengel, R.; Celikel, T.; Daws, L.C.; Holmes, A. Mice lacking the AMPA GluR1 receptor exhibit striatal hyperdopaminergia and ‘schizophrenia-related’ behaviors. Mol. Psychiatry 2008, 13, 631–640. [Google Scholar] [CrossRef] [PubMed]
- Barkus, C.; Feyder, M.; Graybeal, C.; Wright, T.; Wiedholz, L.; Izquierdo, A.; Kiselycznyk, C.; Schmitt, W.; Sanderson, D.J.; Rawlins, J.N.; et al. Do GluA1 knockout mice exhibit behavioral abnormalities relevant to the negative or cognitive symp-toms of schizophrenia and schizoaffective disorder? Neuropharmacology 2012, 62, 1263–1272. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Minano-Molina, A.J.; Espana, J.; Martin, E.; Barneda-Zahonero, B.; Fado, R.; Sole, M.; Trullas, R.; Saura, C.A.; Rodriguez-Alvarez, J. Soluble oligomers of amyloid-beta peptide disrupt membrane trafficking of al-pha-amino-3-hydroxy-5-methylisoxazole-4-propionic acid receptor contributing to early synapse dysfunction. J. Biol. Chem. 2011, 286, 27311–27321. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moriguchi, S. Pharmacological study on Alzheimer’s drugs targeting calcium/calmodulin-dependent protein kinase II. J. Pharm. Sci. 2011, 117, 6–11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roman-Albasini, L.; Diaz-Veliz, G.; Olave, F.A.; Aguayo, F.I.; Garcia-Rojo, G.; Corrales, W.A.; Silva, J.P.; Avalos, A.M.; Rojas, P.S.; Aliaga, E.; et al. Antidepressant-relevant behavioral and synaptic molecular effects of long-term fasudil treatment in chron-ically stressed male rats. Neurobiol. Stress 2020, 13, 100234. [Google Scholar] [CrossRef] [PubMed]
- Shao, S.; Yao, D.; Li, S.; Li, J.; Si, Y.; Zhang, H.; Zhu, Z.; Song, D.; Li, H. N-cadherin regulates GluA1-mediated depressive-like behavior in adolescent female rat offspring following prenatal stress. Neuroendocrinology 2021. [Google Scholar] [CrossRef] [PubMed]
- Leal, R.B.; Lopes, M.W.; Formolo, D.A.; de Carvalho, C.R.; Hoeller, A.A.; Latini, A.; Sousa, D.S.; Wolf, P.; Prediger, R.D.; Bortolot-to, Z.A.; et al. Amygdala levels of the GluA1 subunit of glutamate receptors and its phosphorylation state at serine 845 in the anterior hippocampus are biomarkers of ictal fear but not anxiety. Mol. Psychiatry 2020, 25, 655–665. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- An, S.; Wang, J.; Zhang, X.; Duan, Y.; Xu, Y.; Lv, J.; Wang, D.; Zhang, H.; Richter-Levin, G.; Klavir, O.; et al. alphaCaMKII in the lateral amygdala mediates PTSD-Like behaviors and NMDAR-Dependent LTD. Neurobiol. Stress 2021, 15, 100359. [Google Scholar] [CrossRef]
- Zhu, S.; Noviello, C.M.; Teng, J.; Walsh, R.M., Jr.; Kim, J.J.; Hibbs, R.E. Structure of a human synaptic GABAA receptor. Nature 2018, 559, 67–72. [Google Scholar] [CrossRef]
- Castellano, D.; Shepard, R.D.; Lu, W. Looking for Novelty in an “Old” Receptor: Recent Advances Toward Our Understanding of GABAARs and Their Implications in Receptor Pharmacology. Front. Neurosci. 2020, 14, 616298. [Google Scholar] [CrossRef]
- Gonzalez-Burgos, G.; Cho, R.Y.; Lewis, D.A. Alterations in cortical network oscillations and parvalbumin neurons in schizo-phrenia. Biol. Psychiatry 2015, 77, 1031–1040. [Google Scholar] [CrossRef] [Green Version]
- Marin, O. Interneuron dysfunction in psychiatric disorders. Nat. Rev. Neurosci. 2012, 13, 107–120. [Google Scholar] [CrossRef]
- Mori, T.; Mori, K.; Fujii, E.; Toda, Y.; Miyazaki, M.; Harada, M.; Hashimoto, T.; Kagami, S. Evaluation of the GABAergic nervous system in autistic brain: (123)I-iomazenil SPECT study. Brain Dev. 2012, 34, 648–654. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; van Gerven, J.; Cohen, A.; Jacobs, G. Human pharmacology of positive GABA-A subtype-selective receptor modulators for the treatment of anxiety. Acta Pharm. Sin. 2019, 40, 571–582. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, N.; Kadono, Y.; Naito, A.; Matsumoto, K.; Yamamoto, T.; Tanaka, S.; Inoue, J. Segregation of TRAF6-mediated signal-ing pathways clarifies its role in osteoclastogenesis. EMBO J. 2001, 20, 1271–1280. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xie, P. TRAF molecules in cell signaling and in human diseases. J. Mol. Signal 2013, 8, 7. [Google Scholar] [CrossRef]
- Walsh, M.C.; Lee, J.; Choi, Y. Tumor necrosis factor receptor- associated factor 6 (TRAF6) regulation of development, function, and homeostasis of the immune system. Immunol. Rev. 2015, 266, 72–92. [Google Scholar] [CrossRef]
- Dou, Y.; Tian, X.; Zhang, J.; Wang, Z.; Chen, G. Roles of TRAF6 in Central Nervous System. Curr. Neuropharmacol. 2018, 16, 1306–1313. [Google Scholar] [CrossRef]
- Lomaga, M.A.; Yeh, W.C.; Sarosi, I.; Duncan, G.S.; Furlonger, C.; Ho, A.; Morony, S.; Capparelli, C.; Van, G.; Kaufman, S.; et al. TRAF6 deficiency results in osteopetrosis and defective interleukin-1, CD40, and LPS signaling. Genes Dev. 1999, 13, 1015–1024. [Google Scholar] [CrossRef] [Green Version]
- Boda, B.; Dubos, A.; Muller, D. Signaling mechanisms regulating synapse formation and function in mental retardation. Curr. Opin. Neurobiol. 2010, 20, 519–527. [Google Scholar] [CrossRef]
- Lima Caldeira, G.; Peca, J.; Carvalho, A.L. New insights on synaptic dysfunction in neuropsychiatric disorders. Curr. Opin. Neuro-biol. 2019, 57, 62–70. [Google Scholar] [CrossRef]
- Armstrong, A.P.; Tometsko, M.E.; Glaccum, M.; Sutherland, C.L.; Cosman, D.; Dougall, W.C. A RANK/TRAF6-dependent signal transduction pathway is essential for osteoclast cytoskeletal organization and resorptive function. J. Biol. Chem. 2002, 277, 44347–44356. [Google Scholar] [CrossRef] [Green Version]
- Krebs, J. The plethora of PMCA isoforms: Alternative splicing and differential expression. Biochim. Biophys. Acta 2015, 1853, 2018–2024. [Google Scholar] [CrossRef] [Green Version]
- Lopreiato, R.; Giacomello, M.; Carafoli, E. The plasma membrane calcium pump: New ways to look at an old enzyme. J. Biol. Chem. 2014, 289, 10261–10268. [Google Scholar] [CrossRef] [Green Version]
- Palmieri, L.; Papaleo, V.; Porcelli, V.; Scarcia, P.; Gaita, L.; Sacco, R.; Hager, J.; Rousseau, F.; Curatolo, P.; Manzi, B.; et al. Altered calcium homeostasis in autism-spectrum disorders: Evidence from biochemical and genetic studies of the mitochondrial aspartate/glutamate carrier AGC1. Mol. Psychiatry 2010, 15, 38–52. [Google Scholar] [CrossRef] [Green Version]
- Popugaeva, E.; Pchitskaya, E.; Bezprozvanny, I. Dysregulation of neuronal calcium homeostasis in Alzheimer’s disease—A ther-apeutic opportunity? Biochem. Biophys. Res. Commun. 2017, 483, 998–1004. [Google Scholar] [CrossRef] [Green Version]
- Chami, M.; Checler, F. Alterations of the Endoplasmic Reticulum (ER) Calcium Signaling Molecular Components in Alzheimer’s Disease. Cells 2020, 9, 2577. [Google Scholar] [CrossRef]
- Berrocal, M.; Marcos, D.; Sepulveda, M.R.; Perez, M.; Avila, J.; Mata, A.M. Altered Ca2+ dependence of synaptosomal plasma membrane Ca2+-ATPase in human brain affected by Alzheimer’s disease. FASEB J. 2009, 23, 1826–1834. [Google Scholar] [CrossRef]
- Mata, A.M. Functional interplay between plasma membrane Ca(2+)-ATPase, amyloid beta-peptide and tau. Neurosci. Lett. 2018, 663, 55–59. [Google Scholar] [CrossRef]
- Berrocal, M.; Saez, L.; Mata, A.M. Sorcin Activates the Brain PMCA and Blocks the Inhibitory Effects of Molecular Markers of Alzheimer’s Disease on the Pump Activity. Int. J. Mol. Sci. 2021, 22, 6055. [Google Scholar] [CrossRef] [PubMed]
- Berrocal, M.; Caballero-Bermejo, M.; Gutierrez-Merino, C.; Mata, A.M. Methylene Blue Blocks and Reverses the Inhibitory Effect of Tau on PMCA Function. Int. J. Mol. Sci. 2019, 20, 3521. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brini, M.; Carafoli, E.; Cali, T. The plasma membrane calcium pumps: Focus on the role in (neuro)pathology. Biochem. Biophys. Res. Commun. 2017, 483, 1116–1124. [Google Scholar]
- Zheng, Z.; Zheng, P.; Zou, X. Association between schizophrenia and autism spectrum disorder: A systematic review and me-ta-analysis. Autism Res. 2018, 11, 1110–1119. [Google Scholar] [CrossRef] [PubMed]
- Grant, S.G. Synaptopathies: Diseases of the synaptome. Curr. Opin. Neurobiol. 2012, 22, 522–529. [Google Scholar] [CrossRef] [PubMed]
- Brose, N.; O’Connor, V.; Skehel, P. Synaptopathy: Dysfunction of synaptic function? Biochem. Soc. Trans. 2010, 38, 443–444. [Google Scholar] [CrossRef]
- Eltokhi, A.; Janmaat, I.E.; Genedi, M.; Haarman, B.C.M.; Sommer, I.E.C. Dysregulation of synaptic pruning as a possible link between intestinal microbiota dysbiosis and neuropsychiatric disorders. J. Neurosci. Res. 2020, 98, 1335–1369. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Courchesne, E.; Carper, R.; Akshoomoff, N. Evidence of brain overgrowth in the first year of life in autism. JAMA 2003, 290, 337–344. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Walker, L.; Gozzi, M.; Lenroot, R.; Thurm, A.; Behseta, B.; Swedo, S.; Pierpaoli, C. Diffusion tensor imaging in young children with autism: Biological effects and potential confounds. Biol. Psychiatry 2012, 72, 1043–1051. [Google Scholar] [CrossRef] [Green Version]
- Tang, G.; Gudsnuk, K.; Kuo, S.H.; Cotrina, M.L.; Rosoklija, G.; Sosunov, A.; Sonders, M.S.; Kanter, E.; Castagna, C.; Yamamoto, A.; et al. Loss of mTOR-dependent macroautophagy causes autistic-like synaptic pruning deficits. Neuron 2014, 83, 1131–1143. [Google Scholar] [CrossRef] [Green Version]
- Belmonte, M.K.; Allen, G.; Beckel-Mitchener, A.; Boulanger, L.M.; Carper, R.A.; Webb, S.J. Autism and abnormal development of brain connectivity. J. Neurosci. 2004, 24, 9228–9231. [Google Scholar] [CrossRef]
- Schumann, C.M.; Bloss, C.S.; Barnes, C.C.; Wideman, G.M.; Carper, R.A.; Akshoomoff, N.; Pierce, K.; Hagler, D.; Schork, N.; Lord, C.; et al. Longitudinal magnetic resonance imaging study of cortical development through early childhood in autism. J. Neurosci. 2010, 30, 4419–4427. [Google Scholar] [CrossRef]
- Gogtay, N. Cortical brain development in schizophrenia: Insights from neuroimaging studies in childhood-onset schizophrenia. Schizophr. Bull. 2008, 34, 30–36. [Google Scholar] [CrossRef] [Green Version]
- Keshavan, M.S.; Anderson, S.; Pettegrew, J.W. Is schizophrenia due to excessive synaptic pruning in the prefrontal cortex? The Feinberg hypothesis revisited. J. Psychiatr. 1994, 28, 239–265. [Google Scholar] [CrossRef]
- Mallya, A.P.; Deutch, A.Y. (Micro)Glia as Effectors of Cortical Volume Loss in Schizophrenia. Schizophr. Bull. 2018, 44, 948–957. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sellgren, C.M.; Gracias, J.; Watmuff, B.; Biag, J.D.; Thanos, J.M.; Whittredge, P.B.; Fu, T.; Worringer, K.; Brown, H.E.; Wang, J.; et al. Increased synapse elimination by microglia in schizophrenia patient-derived models of synaptic pruning. Nat. Neurosci. 2019, 22, 374–385. [Google Scholar] [CrossRef] [PubMed]
- Ouchi, Y.; Kubota, Y.; Kuramasu, A.; Watanabe, T.; Ito, C. Gene expression profiling in whole cerebral cortices of phencyclidine- or methamphetamine-treated rats. Mol. Brain Res. 2005, 140, 142–149. [Google Scholar] [CrossRef] [PubMed]
- Sato, M.; Chen, C.C.; Akiyama, K.; Otsuki, S. Acute exacerbation of paranoid psychotic state after long-term abstinence in pa-tients with previous methamphetamine psychosis. Biol. Psychiatry 1983, 18, 429–440. [Google Scholar]
- Javitt, D.C.; Zukin, S.R. Recent advances in the phencyclidine model of schizophrenia. J. Psychiatry 1991, 148, 1301–1308. [Google Scholar]
- Mena, A.; Ruiz-Salas, J.C.; Puentes, A.; Dorado, I.; Ruiz-Veguilla, M.; De la Casa, L.G. Reduced Prepulse Inhibition as a Biomarker of Schizophrenia. Front. Behav. Neurosci. 2016, 10, 202. [Google Scholar] [CrossRef] [Green Version]
- Smith, M.; Spence, M.A.; Flodman, P. Nuclear and mitochondrial genome defects in autisms. Ann. N. Y. Acad. Sci. 2009, 1151, 102–132. [Google Scholar] [CrossRef]
- Liu, Y.; Zhang, Y.; Zarrei, M.; Dong, R.; Yang, X.; Zhao, D.; Scherer, S.W.; Gai, Z. Refining critical regions in 15q24 microdeletion syndrome pertaining to autism. Am. J. Med. Genet. B Neuropsychiatr. Genet. 2020, 183, 217–226. [Google Scholar] [CrossRef]
- Carayol, J.; Sacco, R.; Tores, F.; Rousseau, F.; Lewin, P.; Hager, J.; Persico, A.M. Converging evidence for an association of ATP2B2 allelic variants with autism in male subjects. Biol. Psychiatry 2011, 70, 880–887. [Google Scholar] [CrossRef]
- Blanken, L.M.; Mous, S.E.; Ghassabian, A.; Muetzel, R.L.; Schoemaker, N.K.; El Marroun, H.; van der Lugt, A.; Jaddoe, V.W.; Hof-man, A.; Verhulst, F.C.; et al. Cortical morphology in 6- to 10-year old children with autistic traits: A population-based neuroimaging study. Am. J. Psychiatry 2015, 172, 479–486. [Google Scholar] [CrossRef] [Green Version]
- Persico, A.M.; Bourgeron, T. Searching for ways out of the autism maze: Genetic, epigenetic and environmental clues. Trends Neurosci. 2006, 29, 349–358. [Google Scholar] [CrossRef] [PubMed]
- Amuti, S.; Tang, Y.; Wu, S.; Liu, L.; Huang, L.; Zhang, H.; Li, H.; Jiang, F.; Wang, G.; Liu, X.; et al. Neuroplastin 65 mediates cogni-tive functions via excitatory/inhibitory synapse imbalance and ERK signal pathway. Neurobiol. Learn. Mem. 2016, 127, 72–83. [Google Scholar] [CrossRef] [PubMed]
- Diefenbach, G.J.; McCarthy-Larzelere, M.E.; Williamson, D.A.; Mathews, A.; Manguno-Mire, G.M.; Bentz, B.G. Anxiety, depres-sion, and the content of worries. Depress. Anxiety 2001, 14, 247–250. [Google Scholar] [PubMed]
- Tiller, J.W. Depression and anxiety. Med. J. Aust. 2013, 199, S28–S31. [Google Scholar] [CrossRef]
- Gottschalk, M.G.; Domschke, K. Genetics of generalized anxiety disorder and related traits. Dialogues Clin. Neurosci. 2017, 19, 159–168. [Google Scholar]
- Ilic, K.; Mlinac-Jerkovic, K.; Sedmak, G.; Rosenzweig, I.; Kalanj-Bognar, S. Neuroplastin in human cognition: Review of literature and future perspectives. Transl. Psychiatry 2021, 11, 394. [Google Scholar] [CrossRef]
- Mark, R.J.; Hensley, K.; Butterfield, D.A.; Mattson, M.P. Amyloid beta-peptide impairs ion-motive ATPase activities: Evidence for a role in loss of neuronal Ca2+ homeostasis and cell death. J. Neurosci. 1995, 15, 6239–6249. [Google Scholar] [CrossRef]
- Berrocal, M.; Corbacho, I.; Vazquez-Hernandez, M.; Avila, J.; Sepulveda, M.R.; Mata, A.M. Inhibition of PMCA activity by tau as a function of aging and Alzheimer’s neuropathology. Biochim. Biophys. Acta 2015, 1852, 1465–1476. [Google Scholar] [CrossRef] [Green Version]
- Arancio, A.; Ilya, B.; Berger, T.; Bouteiller, J.M.; Carrillo, M.; Disterhoft, J.; Foskett, K.; Khachaturian, A.S.; LaFerla, F.; Landfield, P.W.; et al. Calcium Hypothesis of Alzheimer’s disease and brain aging: A framework for integrating new evidence into a com-prehensive theory of pathogenesis. Alzheimers Dement. 2017, 13, 178–182.e117. [Google Scholar]
- Chen, G.F.; Xu, T.H.; Yan, Y.; Zhou, Y.R.; Jiang, Y.; Melcher, K.; Xu, H.E. Amyloid beta: Structure, biology and structure-based therapeutic development. Acta Pharm. Sin. 2017, 38, 1205–1235. [Google Scholar] [CrossRef]
- Luo, Y.; Bolon, B.; Kahn, S.; Bennett, B.D.; Babu-Khan, S.; Denis, P.; Fan, W.; Kha, H.; Zhang, J.; Gong, Y.; et al. Mice deficient in BACE1, the Alzheimer’s beta-secretase, have normal phenotype and abolished beta-amyloid generation. Nat. Neurosci. 2001, 4, 231–232. [Google Scholar] [CrossRef]
- Das, B.; Yan, R. A Close Look at BACE1 Inhibitors for Alzheimer’s Disease Treatment. CNS Drugs 2019, 33, 251–263. [Google Scholar] [CrossRef]
- Johnson, J.L.; Chambers, E.; Jayasundera, K. Application of a Bioinformatics-Based Approach to Identify Novel Putative in vivo BACE1 Substrates. Biomed. Eng. Comput. Biol. 2013, 5, 1–15. [Google Scholar] [CrossRef]
- Panza, F.; Lozupone, M.; Solfrizzi, V.; Sardone, R.; Piccininni, C.; Dibello, V.; Stallone, R.; Giannelli, G.; Bellomo, A.; Greco, A.; et al. BACE inhibitors in clinical development for the treatment of Alzheimer’s disease. Expert Rev. Neurother. 2018, 18, 847–857. [Google Scholar] [CrossRef] [PubMed]
- Orwig, W.; Diez, I.; Bueicheku, E.; Vannini, P.; Beaty, R.; Sepulcre, J. Cortical Networks of Creative Ability Trace Gene Expression Profiles of Synaptic Plasticity in the Human Brain. Front. Hum. Neurosci 2021, 15, 694274. [Google Scholar] [CrossRef] [PubMed]
- Young, C.; Butcher, R. Propranolol for Post-Traumatic Stress Disorder: A Review of Clinical Effectiveness; CADTH Rapid Response Re-ports; Canadian Agency for Drugs and Technologies in Health: Ottawa, ON, Canada, 2020. [Google Scholar]
- Roed, A.; Brodal, B. Inhibition of sarcolemma ATPases by some membrane-stabilizing drugs. Acta Pharm. Toxicol. 1981, 48, 65–68. [Google Scholar] [CrossRef] [PubMed]
- Bortolozzi, M.; Mammano, F. PMCA2 pump mutations and hereditary deafness. Neurosci. Lett. 2018, 663, 18–24. [Google Scholar] [CrossRef] [PubMed]
- Gaspar, C.; Cardoso, J.; Franken, P.; Molenaar, L.; Morreau, H.; Moslein, G.; Sampson, J.; Boer, J.M.; de Menezes, R.X.; Fodde, R. Cross-species comparison of human and mouse intestinal polyps reveals conserved mechanisms in adenomatous polyposis coli (APC)-driven tumorigenesis. Am. J. Pathol. 2008, 172, 1363–1380. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rodriguez-Pinto, D.; Sparkowski, J.; Keough, M.P.; Phoenix, K.N.; Vumbaca, F.; Han, D.K.; Gundelfinger, E.D.; Beesley, P.; Claffey, K.P. Identification of novel tumor antigens with patient-derived immune-selected antibodies. Cancer Immunol. Im-munother. 2009, 58, 221–234. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sumardika, I.W.; Chen, Y.; Tomonobu, N.; Kinoshita, R.; Ruma, I.M.W.; Sato, H.; Kondo, E.; Inoue, Y.; Yamauchi, A.; Murata, H.; et al. Neuroplastin-beta mediates S100A8/A9-induced lung cancer dissminative progression. Mol. Carcinog. 2019, 58, 980–995. [Google Scholar] [CrossRef] [PubMed]
- Bajkowska, K.; Sumardika, I.W.; Tomonobu, N.; Chen, Y.; Yamamoto, K.I.; Kinoshita, R.; Murata, H.; Gede Yoni Komalasari, N.L.; Jiang, F.; Yamauchi, A.; et al. Neuroplastin beta-mediated upregulation of solute carrier family 22 member 18 antisense (SLC22A18AS) plays a crucial role in the epithelial-mesenchymal transition, leading to lung cancer cells’ enhanced motility. Biochem. Biophys. Rep. 2020, 22, 100768. [Google Scholar] [PubMed]
- Choy, M.K.; Javierre, B.M.; Williams, S.G.; Baross, S.L.; Liu, Y.; Wingett, S.W.; Akbarov, A.; Wallace, C.; Freire-Pritchett, P.; Rugg-Gunn, P.J.; et al. Promoter interactome of human embryonic stem cell-derived cardiomyocytes connects GWAS re-gions to cardiac gene networks. Nat. Commun. 2018, 9, 2526. [Google Scholar] [CrossRef] [PubMed] [Green Version]



{kind=link}
{kind=link}
{kind=link}
| Year | Main Incident | References |
|---|---|---|
| 1988 | Neuroplastins are first described as glycoproteins in synaptic membranes | [34] |
| 1997 | Neuroplastins are Ig superfamily members with similarity to basigin | [36] |
| 2000 | Np65 is involved in LTP | [39] |
| 2001 | Nptn expression in rat retina | [41] |
| 2006 | Np65 activates p38 MAPK and regulates surface GluR1 and LTP | [37] |
| 2007 | NPTN is associated with developmental delay and schizophrenia | [44] |
| 2010 | Np55 interacts with FGFR promoting neurite outgrowth | [45] |
| 2011 | Extracellular Np65 binding to Np65 regulates neuritogenesis | [46] |
| 2012 | Np65 co-localizes with GABAA receptor | [47] |
| 2013 | Neuroplastin might chaperone MCT2 | [48] |
| 2014 | Np65 regulates the number and function of excitatory and inhibitory synapses | [40] |
| 2015 | NPTN is associated with cortical thickness and intellectual ability in adolescents | [49] |
| 2016 | Retrograde amnesia of associative memories and PMCA loss after inducible Nptn deletionNeuroplastin is required for learning and memory | [50] |
| 2016 | Nptn is identified as a deafness gene | [51,52] |
| 2016 | Neuroplastin-kr8 complex in apoptotic phosphatidylserine exposure | [53] |
| 2016 | Np65 as receptor for S100A8/9A signaling via GRB2 and TRAF2 | [38] |
| 2017 | Np65 KO mice are more susceptible to ischemic brain injury | [54] |
| 2017 | Neuroplastin elimination in glutamatergic neurons causes PMCA loss and behavioral alterations in mice | [35] |
| 2017 | Neuroplastin–PMCA complexes | [55,56] |
| 2018 | Cryo-EM structure of Neuroplastin–PMCA1 complex | [57] |
| 2019 | Neuroplastin expression in AD | [58] |
| 2019 | Np65 KO mice exhibit anxiety and depression-like behavior | [59] |
| 2020 | Neuroplastin interacts with TRAF6 to promote spinogenesis | [60] |
| 2020 | Neuroplastin interacts with MANF to regulate inflammatory responses | [61] |
| 2021 | Neuroplastin–GluA1 interaction mediates LTP | [62] |
| 2021 | Neuroplastin is essential for hearing and hair cell PMCA expression | [43] |
| 2021 | Neuroplastin is related to aging and chronic stress | [63] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lin, X.; Liang, Y.; Herrera-Molina, R.; Montag, D. Neuroplastin in Neuropsychiatric Diseases. Genes 2021, 12, 1507. https://doi.org/10.3390/genes12101507
Lin X, Liang Y, Herrera-Molina R, Montag D. Neuroplastin in Neuropsychiatric Diseases. Genes. 2021; 12(10):1507. https://doi.org/10.3390/genes12101507
Chicago/Turabian StyleLin, Xiao, Yi Liang, Rodrigo Herrera-Molina, and Dirk Montag. 2021. "Neuroplastin in Neuropsychiatric Diseases" Genes 12, no. 10: 1507. https://doi.org/10.3390/genes12101507
APA StyleLin, X., Liang, Y., Herrera-Molina, R., & Montag, D. (2021). Neuroplastin in Neuropsychiatric Diseases. Genes, 12(10), 1507. https://doi.org/10.3390/genes12101507