The Identification of a Novel Fucosidosis-Associated FUCA1 Mutation: A Case of a 5-Year-Old Polish Girl with Two Additional Rare Chromosomal Aberrations and Affected DNA Methylation Patterns

 , , , , ,
, , , , ,  and
and 
Abstract
1. Introduction
2. Materials and Methods
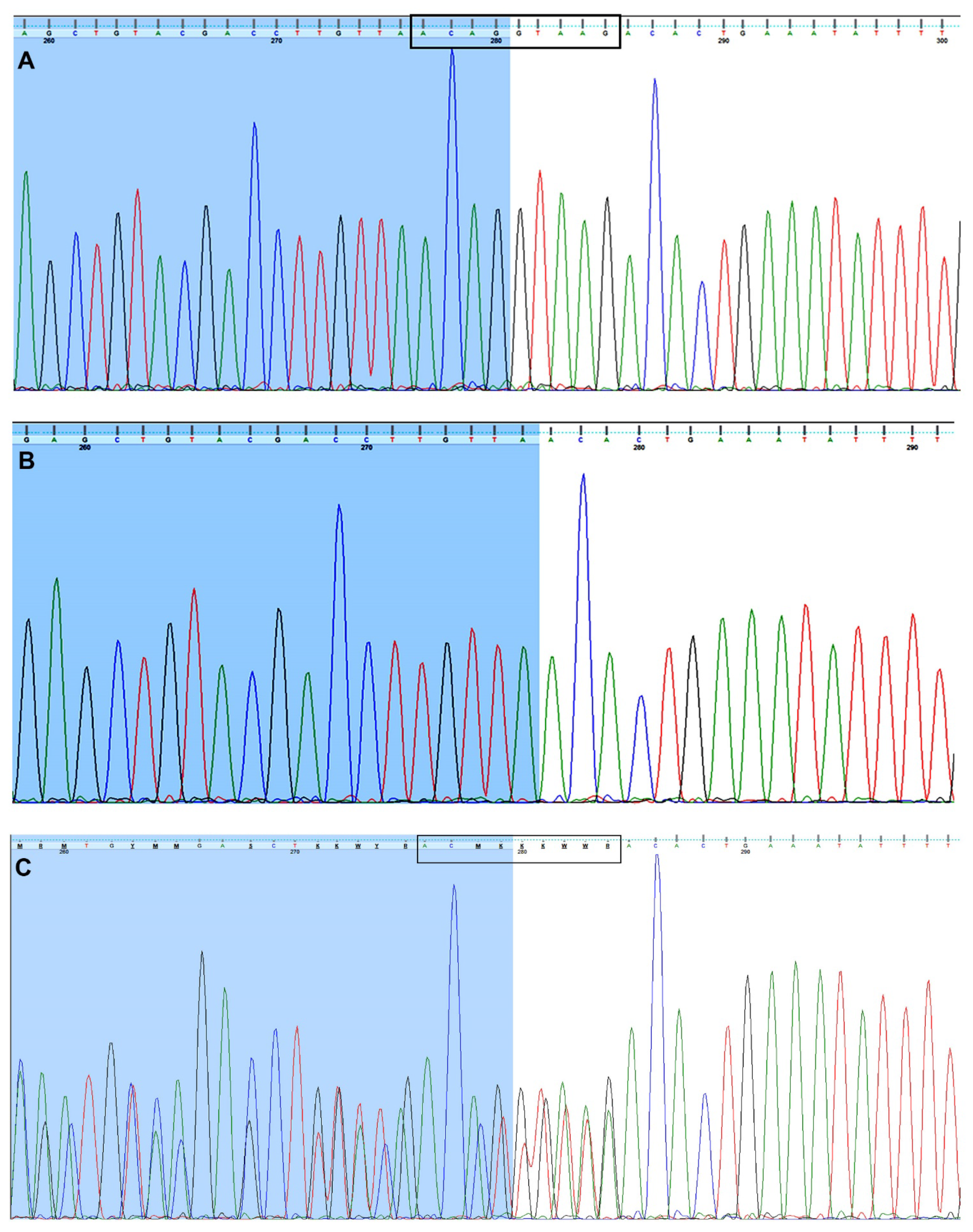
2.1. PCR Amplification and Sanger Sequencing of FUCA1 Exons
2.2. Array-Based Comparative Genomic Hybridization
2.3. Reduced Representation Bisulfite Sequencing
2.4. Raw Data Analysis
2.5. The Variant Calling Procedure
2.6. Kyoto Encyclopedia of Genes and Genomes Pathway Description and Functional Annotation of DMS Associated Genes
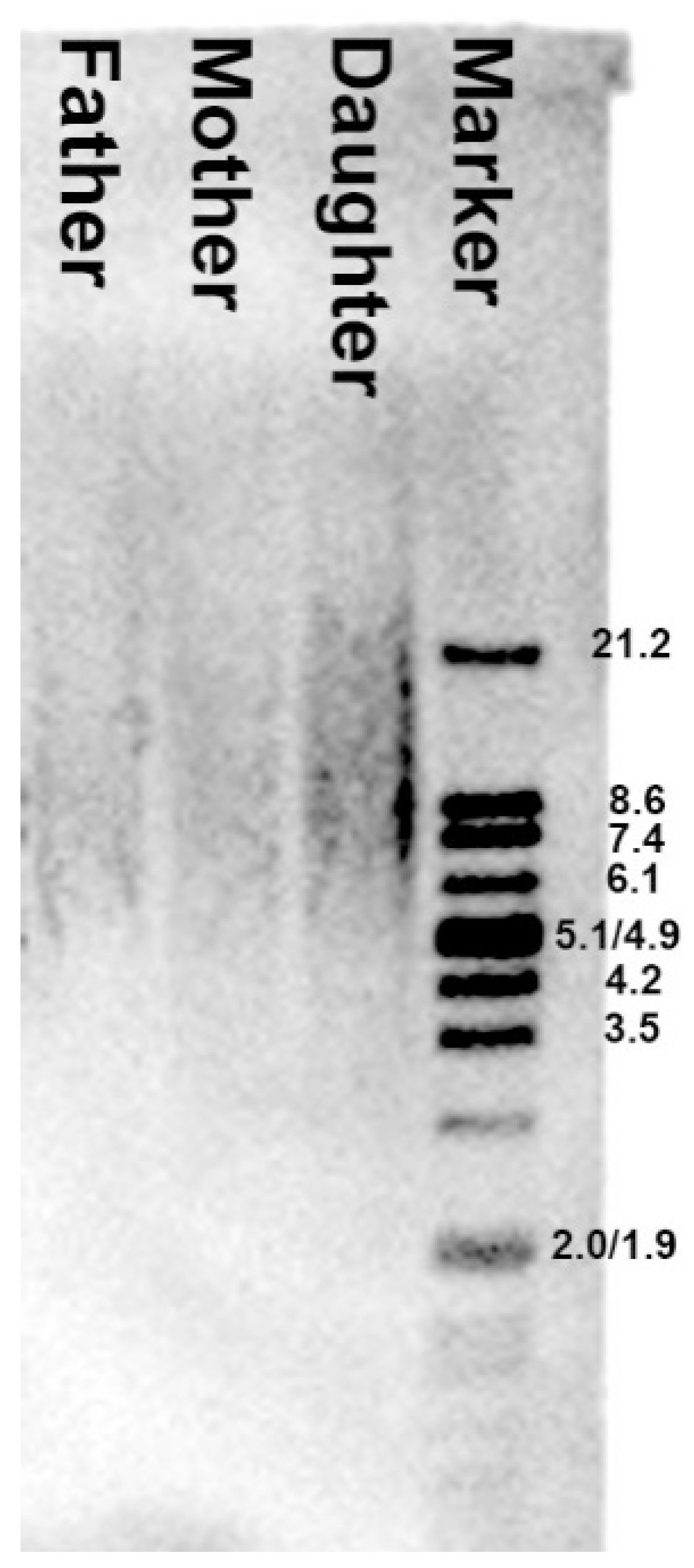
2.7. Telomere Restriction Fragment Length
3. Results
3.1. Case Presentation
3.2. Identification of a Novel Fucosidosis-Related Pathogenic Variant in the FUCA1 Gene
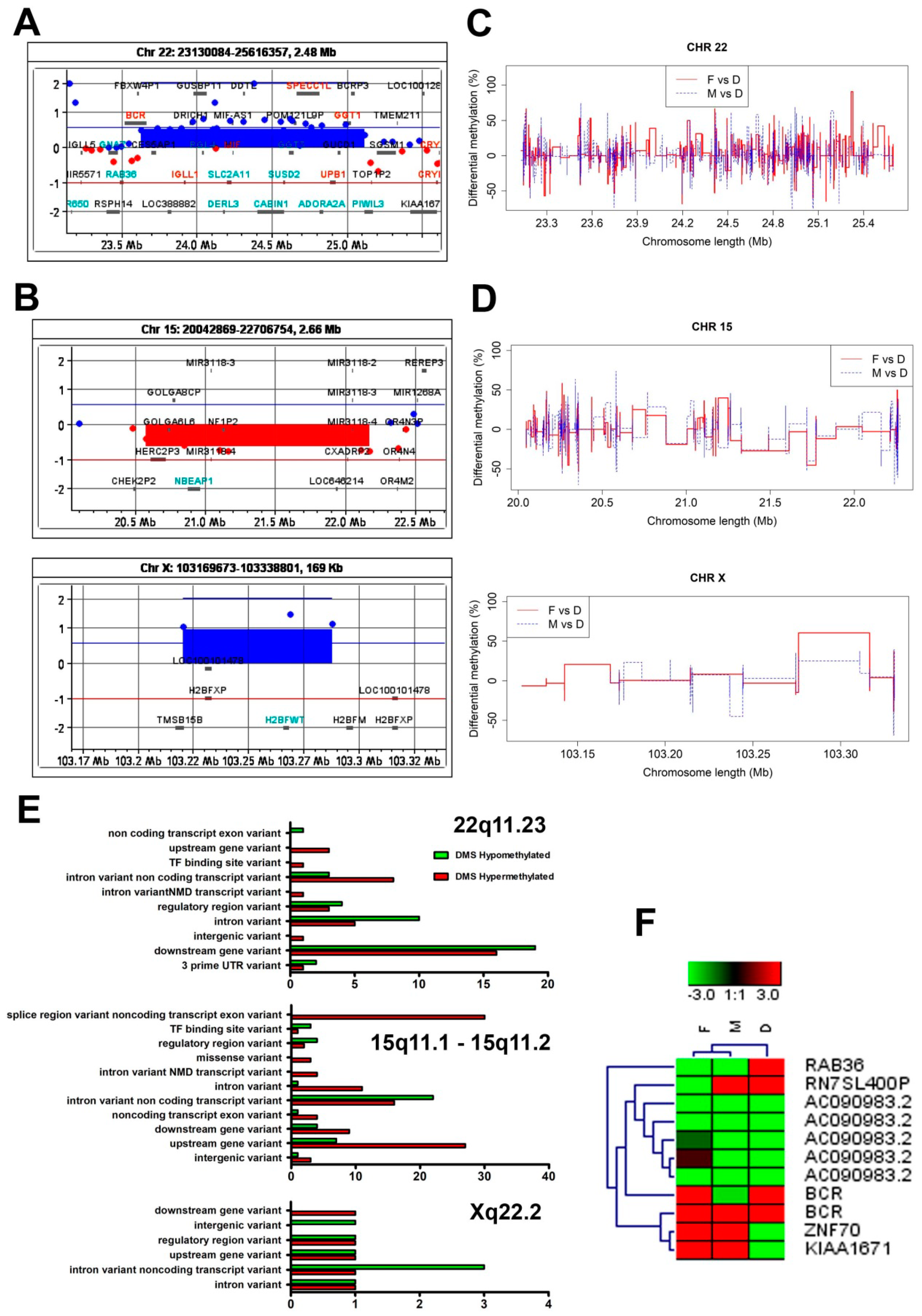
3.3. The Genome-Wide aCGH Analysis Revealed Different Chromosomal Aberrations across the Mother’s and Child’s Genomes
3.4. The Child’s Aberrations of Chromosomes 15 and X Were Associated with Genome-Wide Changes in DNA Methylation Patterns
3.5. Descriptive Characteristic of Molecular Pathways Relevant for DMS Associated Genes
3.6. Genes Covering the Differentially Methylated Sites are Involved in a Wide Range of Metabolic and Biochemical Pathways
3.7. Analysis of the Telomere Restriction Fragment Length Showed No Abnormal Telomere Shortening in the Child’s Genome
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Stepien, K.M.; Ciara, E.; Jezela-Stanek, A. Fucosidosis-Clinical Manifestation, Long-Term Outcomes, and Genetic Profile-Review and Case Series. Genes 2020, 11, 1383. [Google Scholar] [CrossRef] [PubMed]
- Van Hoof, F.; Hers, H.G. Mucopolysaccharidosis by absence of α-fucosidase. Lancet 1968, 1, 1198. [Google Scholar] [CrossRef]
- Kretz, K.A.; Cripe, D.; Carson, G.S.; Fukushima, H.; O’Brien, J.S. Structure and sequence of the human α-L-fucosidase gene and pseudogene. Genomics 1992, 12, 276–280. [Google Scholar] [CrossRef]
- Willems, P.J.; Gatti, R.; Darby, J.K.; Romeo, G.; Durand, P.; Dumon, J.E.; O’Brien, J.S. Fucosidosis revisited: A review of 77 patients. Am. J. Med. Genet. 1991, 38, 111–131. [Google Scholar] [CrossRef] [PubMed]
- Willems, P.J.; Darby, J.K.; DiCioccio, R.A.; Nakashima, P.; Eng, C.; Kretz, K.A.; Cavalli-Sforza, L.L.; Shooter, E.M.; O’Brien, J.S. Identification of a mutation in the structural α-L-fucosidase gene in fucosidosis. Am. J. Hum. Genet. 1988, 43, 756–763. [Google Scholar]
- Willems, P.J.; Seo, H.C.; Coucke, P.; Tonlorenzi, R.; O’Brien, J.S. Spectrum of mutations in fucosidosis. Eur. J. Hum. Genet. 1999, 7, 60–67. [Google Scholar] [CrossRef]
- Reuter, M.S.; Tawamie, H.; Buchert, R.; Gebril, O.H.; Froukh, T.; Thiel, C.; Uebe, S.; Ekici, A.B.; Krumbiegel, M.; Zweier, C.; et al. Diagnostic Yield and Novel Candidate Genes by Exome Sequencing in 152 Consanguineous Families with Neurodevelopmental Disorders. JAMA Psychiatry 2017, 74, 293–299. [Google Scholar] [CrossRef]
- HGMD Professional. Available online: http://www.hgmd.cf.ac.uk/ac/index.php (accessed on 8 December 2020).
- Williamson, M.; Cragg, H.; Grant, J.; Kretz, K.; O’Brien, J.; Willems, P.J.; Young, E.; Winchester, B. A 5’ splice site mutation in fucosidosis. J. Med. Genet. 1993, 30, 218–223. [Google Scholar] [CrossRef][Green Version]
- Wali, G.; Wali, G.M.; Sue, C.M.; Kumar, K.R. A novel homozygous mutation in the FUCA1 gene highlighting fucosidosis as a cause of dystonia: Case report and literature review. Neuropediatrics 2019, 50, 248–252. [Google Scholar] [CrossRef]
- Michalski, J.C.; Klein, A. Glycoprotein lysosomal storage disorders: α- and β-mannosidosis, fucosidosis and α-N-acetylgalactosaminidase deficiency. Biochim. Biophys. Acta 1999, 1455, 69–84. [Google Scholar] [CrossRef]
- Bjornsson, H.T. The Mendelian disorders of the epigenetic machinery. Genome Res. 2015, 25, 1473–1481. [Google Scholar] [CrossRef] [PubMed]
- Zeng, Y.; Chen, T. DNA Methylation Reprogramming during Mammalian Development. Genes 2019, 10, 257. [Google Scholar] [CrossRef] [PubMed]
- Vajen, B.; Thomay, K.; Schlegelberger, B. Induction of Chromosomal Instability via Telomere Dysfunction and Epigenetic Alterations in Myeloid Neoplasia. Cancers 2013, 5, 857–874. [Google Scholar] [CrossRef] [PubMed]
- Koressaar, T.; Remm, M. Enhancements and modifications of primer design program Primer3. Bioinformatics 2007, 23, 1289–1291. [Google Scholar] [CrossRef] [PubMed]
- Untergasser, A.; Cutcutache, I.; Koressaar, T.; Ye, J.; Faircloth, B.C.; Remm, M.; Rozen, S.G. Primer3-new capabilities and interfaces. Nucleic Acids Res. 2012, 40, e115. [Google Scholar] [CrossRef]
- Nicholas, K.; Nicholas, H.B.; Deerfield, D. GeneDoc: Analysis and visualization of genetic variation. EMBnet. News 1997, 4, 14. [Google Scholar]
- Xi, Y.; Li, W. BSMAP: Whole genome bisulfite sequence MAPping program. BMC Bioinform. 2009, 10, 232. [Google Scholar] [CrossRef]
- Akalin, A.; Kormaksson, M.; Li, S.; Garrett-Bakelman, F.E.; Figueroa, M.E.; Melnick, A.; Mason, C.E. MethylKit: A comprehensive R package for the analysis of genome-wide DNA methylation profiles. Genome. Biol. 2012, 13, R87. [Google Scholar] [CrossRef]
- Dodt, M.; Roehr, J.T.; Ahmed, R.; Dieterich, C. FLEXBAR-Flexible Barcode and Adapter Processing for Next-Generation Sequencing Platforms. Biology 2012, 1, 895–905. [Google Scholar] [CrossRef]
- Picard Toolkit. Broad Institute, GitHub Repository. 2019. Available online: https://broadinstitute.github.io/picard/ (accessed on 8 December 2020).
- Liu, Y.; Siegmund, K.D.; Laird, P.W.; Berman, B.P. Bis-SNP: Combined DNA methylation and SNP calling for Bisulfite-seq data. Genome. Biol. 2012, 13, R61. [Google Scholar] [CrossRef]
- Danecek, P.; Auton, A.; Abecasis, G.; Albers, C.A.; Banks, E.; DePristo, M.A.; Handsaker, R.; Lunter, G.; Marth, G.; Sherry, S.T.; et al. The variant call format and VCF tools. Bioinformatics 2011, 27, 2156–2158. [Google Scholar] [CrossRef] [PubMed]
- Dennis, G., Jr.; Sherman, B.T.; Hosack, D.A.; Yang, J.; Gao, W.; Lane, H.C.; Lempicki, R.A. DAVID: Database for Annotation, Visualization, and Integrated Discovery. Genome Biol. 2003, 4, P3. [Google Scholar] [CrossRef] [PubMed]
- Sturn, A.; Quackenbush, J.; Trajanoski, Z. Genesis: Cluster analysis of microarray data. Bioinformatics 2002, 18, 207–208. [Google Scholar] [CrossRef] [PubMed]
- Wnuk, M.; Lewinska, A.; Gurgul, A.; Zabek, T.; Potocki, L.; Oklejewicz, B.; Bugno-Poniewierska, M.; Wegrzyn, M.; Slota, E. Changes in DNA methylation patterns and repetitive sequences in blood lymphocytes of aged horses. Age 2014, 36, 31–48. [Google Scholar] [CrossRef] [PubMed]
- Kehler, J.; Tolkunova, E.; Koschorz, B.; Pesce, M.; Gentile, L.; Boiani, M.; Lomelí, H.; Nagy, A.; McLaughlin, K.J.; Schöler, H.R.; et al. Oct 4 is required for primordial germ cell survival. EMBO Rep. 2004, 5, 1078–1083. [Google Scholar] [CrossRef] [PubMed]
- Kalimuthu, S.N.; Chetty, R. Gene of the month: SMARCB1. J. Clin. Pathol. 2016, 69, 484–489. [Google Scholar] [CrossRef]
- Choi, S.K.; Kim, M.J.; You, J.S. SMARCB1 Acts as a Quiescent Gatekeeper for Cell Cycle and Immune Response in Human Cells. Int. J. Mol. Sci. 2020, 21, 3969. [Google Scholar] [CrossRef]
- Tan, M.; van Tol, H.T.; Rosenkranz, D.; Roovers, E.F.; Damen, M.J.; Stout, T.A.; Wu, W.; Roelen, B.A. PIWIL3 Forms a Complex with TDRKH in Mammalian Oocytes. Cells 2020, 9, 1356. [Google Scholar] [CrossRef]
- Chatterjee, A.; Rodger, E.J.; Stockwell, P.A.; Weeks, R.J.; Morison, I.M. Technical considerations for reduced representation bisulfite sequencing with multiplexed libraries. J. Biomed. Biotechnol. 2012, 2012, 741542. [Google Scholar] [CrossRef]
- Baylin, S.B.; Esteller, M.; Rountree, M.R.; Bachman, K.E.; Schuebel, K.; Herman, J.G. Aberrant patterns of DNA methylation, chromatin formation and gene expression in cancer. Hum. Mol. Genet. 2001, 10, 687–692. [Google Scholar] [CrossRef]
- Wang, L.; Yang, M.; Hong, S.; Tang, T.; Zhuang, J.; Huang, H. Fucosidosis in a Chinese boy: A case report and literature review. J. Int. Med. Res. 2020, 48, 300060520911269. [Google Scholar] [CrossRef] [PubMed]
- Tüttelmann, F.; Simoni, M.; Kliesch, S.; Ledig, S.; Dworniczak, B.; Wieacker, P.; Röpke, A. Copy number variants in patients with severe oligozoospermia and Sertoli-cell-only syndrome. PLoS ONE 2011, 6, e19426. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.P.; Wang, K.G.; Huang, H.K.; Peng, C.R.; Chern, S.R.; Wu, P.S.; Chen, Y.N.; Chen, S.W.; Lee, C.C.; Wang, W. Detection of mosaic 15q11.1-q11.2 deletion encompassing NBEAP1 and POTEB in a fetus with diffuse lymphangiomatosis. Taiwan J. Obs. Gynecol. 2017, 56, 230–233. [Google Scholar] [CrossRef] [PubMed]
- Prajapati, P.; Sripada, L.; Singh, K.; Roy, M.; Bhatelia, K.; Dalwadi, P.; Singh, R. Systemic Analysis of miRNAs in PD Stress Condition: miR-5701 Modulates Mitochondrial-Lysosomal Cross Talk to Regulate Neuronal Death. Mol. Neurobiol. 2018, 55, 4689–4701. [Google Scholar] [PubMed]
- Boulard, M.; Gautier, T.; Mbele, G.O.; Gerson, V.; Hamiche, A.; Angelov, D.; Bouvet, P.; Dimitrov, S. The NH2 tail of the novel histone variant H2BFWT exhibits properties distinct from conventional H2B with respect to the assembly of mitotic chromosomes. Mol. Cell Biol. 2006, 26, 1518–1526. [Google Scholar] [CrossRef]
- McSwiggin, H.M.; O’Doherty, A.M. Epigenetic reprogramming during spermatogenesis and male factor infertility. Reproduction 2018, 156, R9–R21. [Google Scholar] [CrossRef] [PubMed]
- Churikov, D.; Siino, J.; Svetlova, M.; Zhang, K.L.; Gineitis, A.; Bradbury, E.M.; Zalensky, A. Novel human testis-specific histone H2B encoded by the interrupted gene on the X chromosome. Genomics 2004, 84, 745–756. [Google Scholar] [CrossRef]
- Saadeh, H.; Schulz, R. Protection of CpG islands against de novo DNA methylation during oogenesis is associated with the recognition site of E2f1 and E2f2. Epigenetics Chromatin. 2014, 7, 26. [Google Scholar]
- Sambuughin, N.; Goldfarb, L.G.; Sivtseva, T.M.; Davydova, T.K.; Vladimirtsev, V.A.; Osakovskiy, V.L.; Danilova, A.P.; Nikitina, R.S.; Ylakhova, A.N.; Diachkovskaya, M.P.; et al. Adult-onset autosomal dominant spastic paraplegia linked to a GTPase-effector domain mutation of dynamin 2. BMC Neurol. 2015, 15, 223. [Google Scholar] [CrossRef]
- Zúñiga-Ramírez, C.; Kramis-Hollands, M.; Mercado-Pimentel, R.; González-Usigli, H.A.; Sáenz-Farret, M.; Soto-Escageda, A.; Fasano, A. Generalized Dystonia and Paroxysmal Dystonic Attacks due to a Novel ATP1A3 Variant. Tremor Other Hyperkinet Mov. 2019, 13, 9. [Google Scholar] [CrossRef]
- Erro, R.; Sheerin, U.M.; Bhatia, K.P. Paroxysmal dyskinesias revisited: 500 genetically proven cases and a new classification. Mov. Disord. 2014, 29, 1108–1116. [Google Scholar] [CrossRef] [PubMed]
- Erro, R.; Bhatia, K.P.; Espay, A.J.; Striano, P. The epileptic and nonepileptic spectrum of paroxysmal dyskinesias: Channelopathies, synaptopathies and transportopathies. Mov. Disord. 2017, 32, 310–318. [Google Scholar] [CrossRef] [PubMed]
- Erro, R.; Bhatia, K.P. Unravelling of the paroxysmal dyskinesias. J. Neurol. Neurosurg. Psychiatry 2019, 90, 227–234. [Google Scholar] [CrossRef] [PubMed]
- Giffin, N.J.; Benton, S.; Goadsby, P.J. Benign paroxysmal torticollis of infancy: Four new cases and linkage to CACNA1A mutation. Dev. Med. Child. Neurol. 2002, 44, 490–493. [Google Scholar] [CrossRef]
- Gu, J.; Kumar, K.R.; Sue, C.M. CACNA1A mutation presenting with focal dystonia. J. Neurol. Neurosurg. Psychiatry. 2017, 88, e1. [Google Scholar] [CrossRef]
- Sun, X.; Qian, L.L.; Li, Y.; Pfiefer, T.M.; Wang, X.L.; Lee, H.C.; Lu, T. Regulation of KCNMA1 transcription by Nrf2 in coronary arterial smooth muscle cells. J. Mol. Cell. Cardiol. 2020, 140, 68–76. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| FUCA1 | Genomic Position [bp] of PCR Amplicon | PCR Length [bp] | Ta [°C] | 5′-3′ Forward | 5′-3′ Reverse |
|---|---|---|---|---|---|
| Exon 1 | Chr1:23867835-23868368 | 533 | 61 | AGCCCCACCTCCTGTTTG | GGGCCAATCGTTAGTCAGAG |
| Exon 2 | Chr1:23865244-23865980 | 736 | 60 | CTGTAACGGTGTCCCCTTTG | CTCCTTCAGGGCAAAGAATG |
| Exon 3 | Chr1:23862935-23863410 | 475 | 61 | GCAATATGGCCACATGACAG | GGTTTTGGATCCCATTCCTC |
| Exon 4 | Chr1:23859621-23860301 | 680 | 59 | AGCCAGGGAAGATTAAAGGAG | TGCTGTGTTTGTTTTAAATGGTG |
| Exon 5 | Chr1:23854026-23854815 | 789 | TDPCR | TCCCCCTCTGTGAGAAACAC | GCTGTCCTGTGCATTGTAGG |
| Exon 6 | Chr1:23848438-23848888 | 450 | 60 | TCACCAAACCCACTCTCTCC | CCAGGCTTGGCATTCATAG |
| Exon 7 | Chr1:23845925-23846344 | 419 | 59 | GGTAGGAAACAGCCACGC | AGCCAACATCACACCATTGC |
| Exon 8 | Chr1:23845237-23846005 | 768 | 61 | CAGCCTGGTAAGCCTTTTCC | CTTCCCTGCCAGGTTTCTC |
| Exon 8 | Chr1:23844873-23845347 | 474 | 59 | TCTAACAAAGAGGCTGAACTGG | AGGATTTGGCAAGCTCAACC |
| Mother’s 22q11.23 Gain | Daughter’s 15q11.1-q11.2 Microdeletion | Daughter’s X.22.2 Gain |
|---|---|---|
| BCR (Breakpoint Cluster Region) | HERC2P3 (Hect Domain And RLD 2 Pseudogene 3) | TMSB15B (Thymosin β 15B) |
| CES5AP1 (Carboxylesterase 5A Pseudogene 1) | GOLGA6L6 (Golgin A6 Family Like 6) | H2BFXP (H2B.W Histone 4, Pseudogene) |
| ZDHHC8P1 (Zinc Finger DHHC-Type Containing 8 Pseudogene 1) | GOLGA8CP (Golgin A8 Family Member C, Pseudogene) | LOC100101478 |
| LOC101929374 | NBEAP1 (Neurobeachin Pseudogene 1) | H2BFWT (H2B Histone Family Member W, Testis Specific) |
| LOC388882 | MIR3118-4 (MicroRNA 3118-4) | |
| IGLL1 (Immunoglobulin Lambda Like Polypeptide 1) | MIR3118-3 (MicroRNA 3118-3) | |
| DRICH1 (Aspartate Rich 1) | MIR3118-2 (MicroRNA 3118-2) | |
| GUSBP11 (Glucuronidase, β/Immunoglobulin Lambda-Like Polypeptide 1 Pseudogene) | POTEB3 (POTE Ankyrin Domain Family Member B3) | |
| RGL4 (Ral Guanine Nucleotide Dissociation Stimulator Like 4) | POTEB (POTE Ankyrin Domain Family Member B) | |
| ZNF70 (Zinc Finger Protein 70) | POTEB2 (POTE Ankyrin Domain Family Member B2) | |
| VPREB3 (V-Set Pre-B Cell Surrogate Light Chain 3) | NF1P2 (Neurofibromin 1 Pseudogene 2) | |
| C22orf15 (Chromosome 22 Open Reading Frame 15) | MIR5701-3 (MicroRNA 5701-3) | |
| CHCHD10 (Coiled-Coil-Helix-Coiled-Coil-Helix Domain Containing 10) | MIR5701-1 (MicroRNA 5701-1) | |
| MMP11 (Matrix Metallopeptidase 11) SMARCB1 (SWI/SNF Related, Matrix Associated, Actin Dependent Regulator Of Chromatin, Subfamily B, Member 1) | MIR5701-2 (MicroRNA 5701-2) | |
| DERL3 (Derlin 3) | LINC01193 (Long Intergenic Non-Protein Coding RNA 1193) | |
| SLC2A11 (Solute Carrier Family 2 Member 11) | LOC646214 (P21 (RAC1) Activated Kinase 2 Pseudogene) | |
| MIF-AS1 (MIF Antisense RNA 1) | CXADRP2 (Coxsackie Virus and Adenovirus Receptor Pseudogene 2) | |
| MIF (Macrophage Migration Inhibitory Factor) | ||
| GSTT2B (Glutathione S-Transferase Theta 2B) | ||
| GSTT2 (Glutathione S-Transferase Theta 2, Gene/Pseudogene) | ||
| DDTL (D-Dopachrome Tautomerase Like) DDT (D-Dopachrome Tautomerase) | ||
| GSTTP1 (Glutathione S-Transferase Theta 4) | ||
| LOC391322 (D-Dopachrome Tautomerase-Like) | ||
| GSTT1-AS1 (GSTT1 Antisense RNA 1) | ||
| GSTT1 (Glutathione S-Transferase Theta 1) | ||
| GSTTP2 (Glutathione S-Transferase Theta Pseudogene 2) | ||
| CABIN1 (Calcineurin Binding Protein 1) | ||
| SUSD2 (Sushi Domain Containing 2) | ||
| GGT5 (γ-Glutamyltransferase 5) | ||
| POM121L9P (POM121 Transmembrane Nucleoporin Like 9, Pseudogene) | ||
| SPECC1L (Sperm Antigen With Calponin Homology And Coiled-Coil Domains 1 Like) | ||
| SPECC1L (Sperm Antigen With Calponin Homology And Coiled-Coil Domains 1 Like) | ||
| ADORA2A (Adenosine A2a Receptor) | ||
| ADORA2A-AS1 (ADORA2A Antisense RNA 1) | ||
| UPB1 (β-Ureidopropionase 1) | ||
| GUCD1 (Guanylyl Cyclase Domain Containing 1) | ||
| SNRPD3 (Small Nuclear Ribonucleoprotein D3 Polypeptide) | ||
| GGT1 (γ-Glutamyltransferase 1) | ||
| LRRC75B (Leucine Rich Repeat Containing 75B) | ||
| BCRP3 (Breakpoint Cluster Region Pseudogene 3) | ||
| POM121L10P (POM121 Transmembrane Nucleoporin Like 10, Pseudogene) | ||
| PIWIL3 (Piwi Like RNA-Mediated Gene Silencing 3) |
| RRBS Library | Total Reads (After Filtering) | Aligned Reads | Uniquely Aligned Reads |
|---|---|---|---|
| Father | 16,442,608 | 13,169,725 (80.1%) | 9,822,103 (59.7%) |
| Mother | 18,778,091 | 15,678,863 (83.5%) | 11,785,228 (62.8%) |
| Daughter | 24,380,417 | 19,220,521 (78.8%) | 14,438,686 (59.2%) |
| Type of Pairwise Comparison | Number of DMS | |||
|---|---|---|---|---|
| Chromosome 15 | Chromosome 22 | Chromosome X | All Chromosomes | |
| Hypomethylated DMS | ||||
| F vs. D | 245 (11.78%) | 871 (41.93%) | 25 (1.20%) | 2077 |
| M vs. D | 318 (16.13%) | 1042 (52.87%) | 34 (1.72%) | 1971 |
| Hypermethylated DMS | ||||
| F vs. D | 168 (5.58%) | 896 (28.85%) | 26 (0.86%) | 3012 |
| M vs. D | 268 (8.40%) | 1134 (35.55%) | 47 (1.47%) | 3190 |
| Hypomethylated DMS | Hypermethylated DMS |
|---|---|
| Metabolic processes | |
| Metabolic pathways | Metabolic pathways |
| Adipocytokine signaling | - |
| Sphingolipid signaling | Sphingolipid signaling |
| Propanoate metabolism | - |
| Nonalcoholic fatty liver disease (NAFLD) | - |
| Neutrophin signaling | - |
| Central carbon metabolism in cancer | Central carbon metabolism in cancer |
| Carbon metabolism | - |
| Carbohydrate digestion and absorption | - |
| - | Choline metabolism in cancer |
| Cellular signaling pathways | |
| Pathways in cancer | - |
| cAMP signaling pathway | - |
| - | Rap1 signaling |
| - | PI3K-Akt signaling |
| - | MAPK signaling, calcium signaling |
| Biological processes | |
| Regulation of actin cytoskeleton, | Regulation of actin cytoskeleton |
| endocytosis | - |
| - | Proteoglycans in cancer |
| - | Focal adhesion |
| - | Adherens junction |
| Neurological processes | |
| Alzheimer’s disease | - |
| Cholinergic synapse | Cholinergic synapse |
| - | Morphine addiction |
| - | Serotonergic synapse |
| - | Hippo signaling |
| - | Glutamatergic signaling |
| - | Axon guidance |
| - | Long-term potentiation |
| - | Long-term depression |
| Cancerogenic processes | |
| Small cell lung cancer | - |
| - | Melanoma |
| - | Non-small cell lung cancer |
| - | Glioma |
| - | Acute myeloid leukemia |
| Chronic myeloid leukemia | - |
| Physiological processes | |
| Renal cell carcinoma | Renal cell carcinoma |
| - | Vascular smooth muscle contraction |
| - | Progesterone-mediated oocyte maturation |
| - | Oocyte meiosis |
| Cell death and autophagy processes | |
| Apoptosis | Apoptosis |
| - | Regulation of autophagy |
| - | Phagosome |
| Developmental processes | |
| Osteoclast differentiation | - |
| Type II diabetes mellitus | - |
| - | Signaling pathways regulating pluripotency of stem cells |
| - | Osteoclast differentiation |
| RNA metabolism | |
| RNA degradation | Spliceosome |
| - | MicroRNA in cancers |
| DNA repair processes | |
| - | Nucleotide excision repair and mismatch repair |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Domin, A.; Zabek, T.; Kwiatkowska, A.; Szmatola, T.; Deregowska, A.; Lewinska, A.; Mazur, A.; Wnuk, M. The Identification of a Novel Fucosidosis-Associated FUCA1 Mutation: A Case of a 5-Year-Old Polish Girl with Two Additional Rare Chromosomal Aberrations and Affected DNA Methylation Patterns. Genes 2021, 12, 74. https://doi.org/10.3390/genes12010074
Domin A, Zabek T, Kwiatkowska A, Szmatola T, Deregowska A, Lewinska A, Mazur A, Wnuk M. The Identification of a Novel Fucosidosis-Associated FUCA1 Mutation: A Case of a 5-Year-Old Polish Girl with Two Additional Rare Chromosomal Aberrations and Affected DNA Methylation Patterns. Genes. 2021; 12(1):74. https://doi.org/10.3390/genes12010074
Chicago/Turabian StyleDomin, Agnieszka, Tomasz Zabek, Aleksandra Kwiatkowska, Tomasz Szmatola, Anna Deregowska, Anna Lewinska, Artur Mazur, and Maciej Wnuk. 2021. "The Identification of a Novel Fucosidosis-Associated FUCA1 Mutation: A Case of a 5-Year-Old Polish Girl with Two Additional Rare Chromosomal Aberrations and Affected DNA Methylation Patterns" Genes 12, no. 1: 74. https://doi.org/10.3390/genes12010074
APA StyleDomin, A., Zabek, T., Kwiatkowska, A., Szmatola, T., Deregowska, A., Lewinska, A., Mazur, A., & Wnuk, M. (2021). The Identification of a Novel Fucosidosis-Associated FUCA1 Mutation: A Case of a 5-Year-Old Polish Girl with Two Additional Rare Chromosomal Aberrations and Affected DNA Methylation Patterns. Genes, 12(1), 74. https://doi.org/10.3390/genes12010074