Chromosome Instability in Fanconi Anemia: From Breaks to Phenotypic Consequences

Abstract
1. Introduction
2. Double Strand Breaks Are at the Center of Chromosome Aberrations in FA
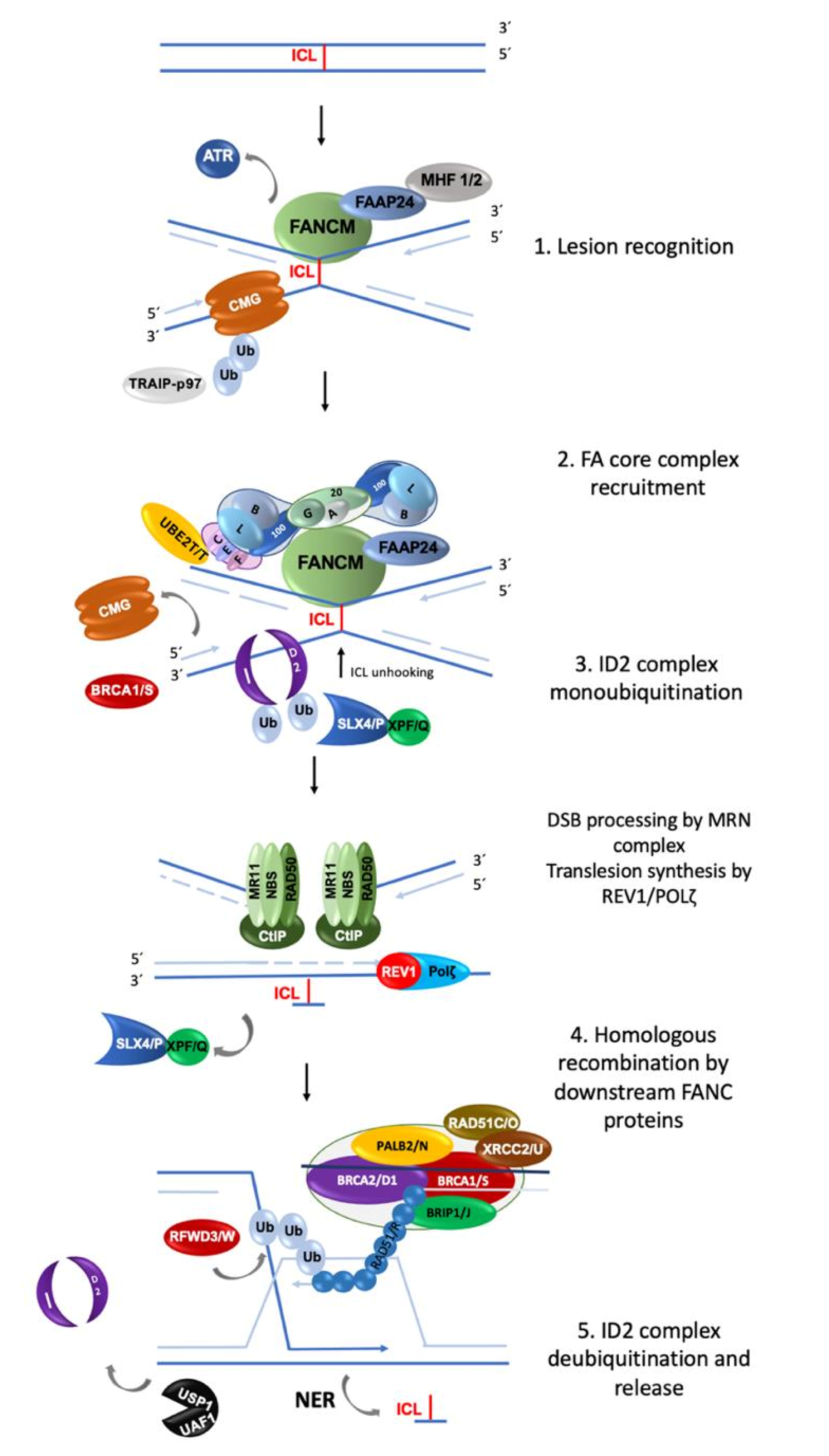
2.1. Involvement of FA/BRCA Pathway in DNA Repair
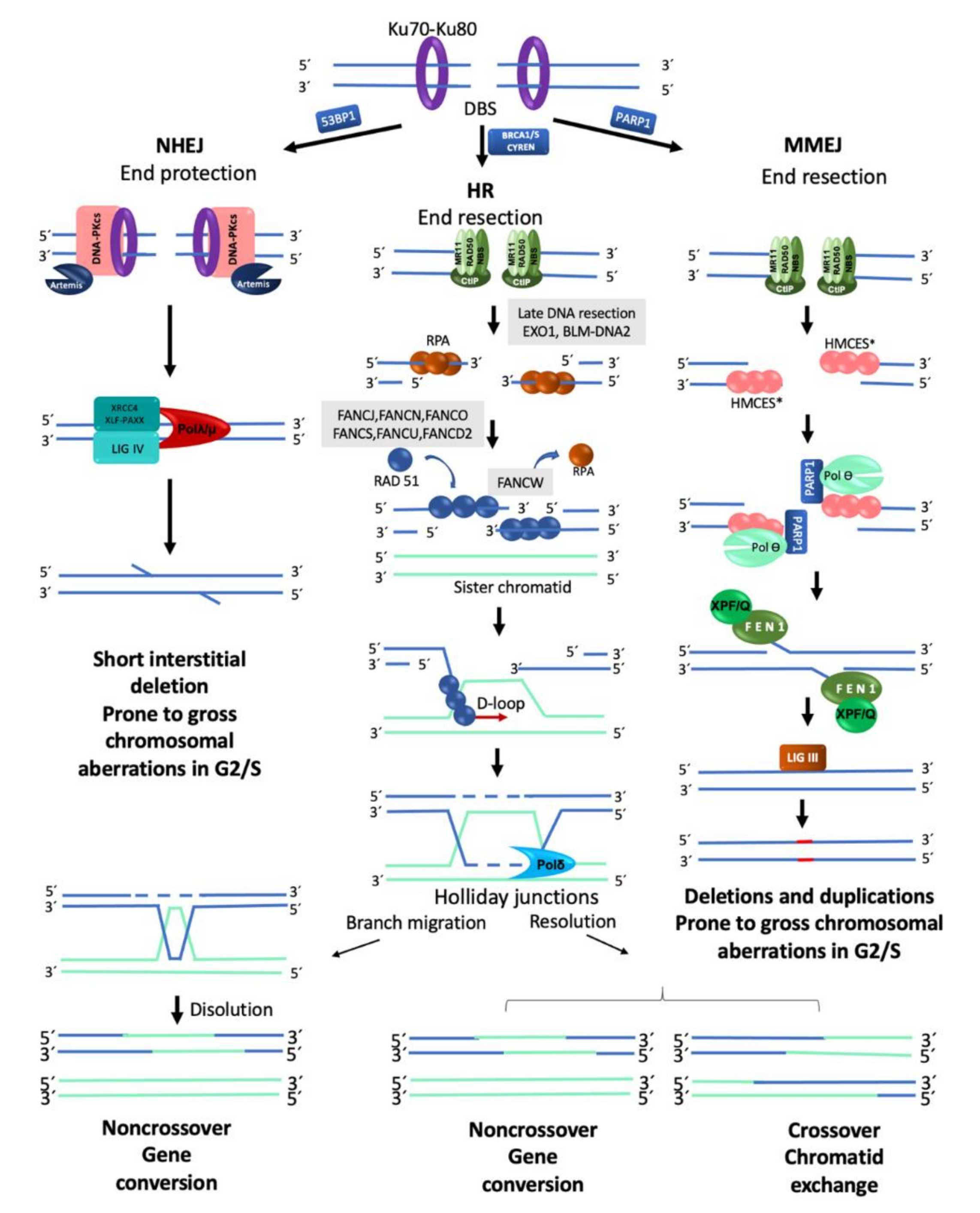
2.2. Repair of Double Strand Breaks
2.2.1. Homologous Recombination
2.2.2. Non-Homologous End Joining
2.2.3. Alternative End Joining (Microhomology Mediated End Joining)
2.2.4. DSB Repair Pathway Choice
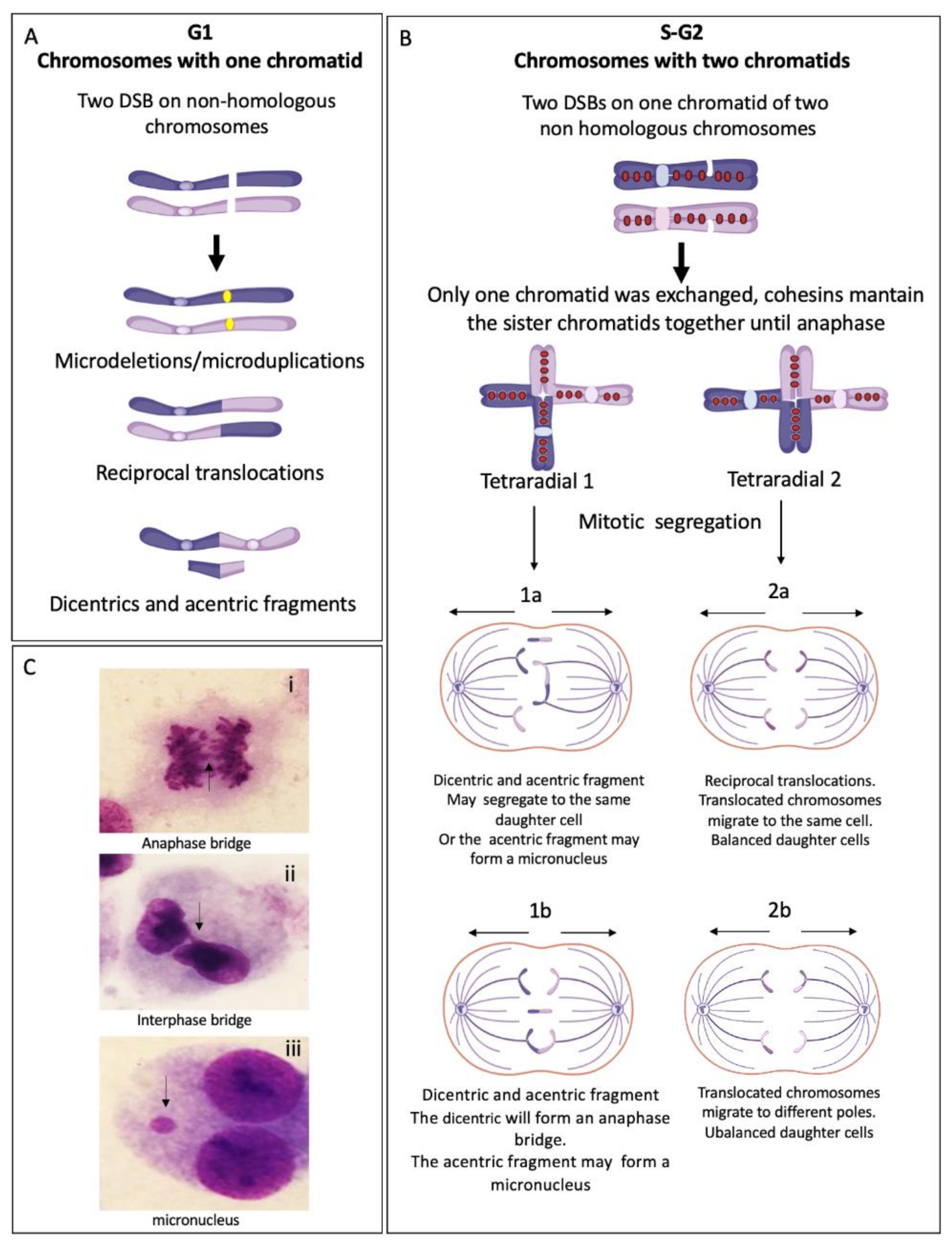
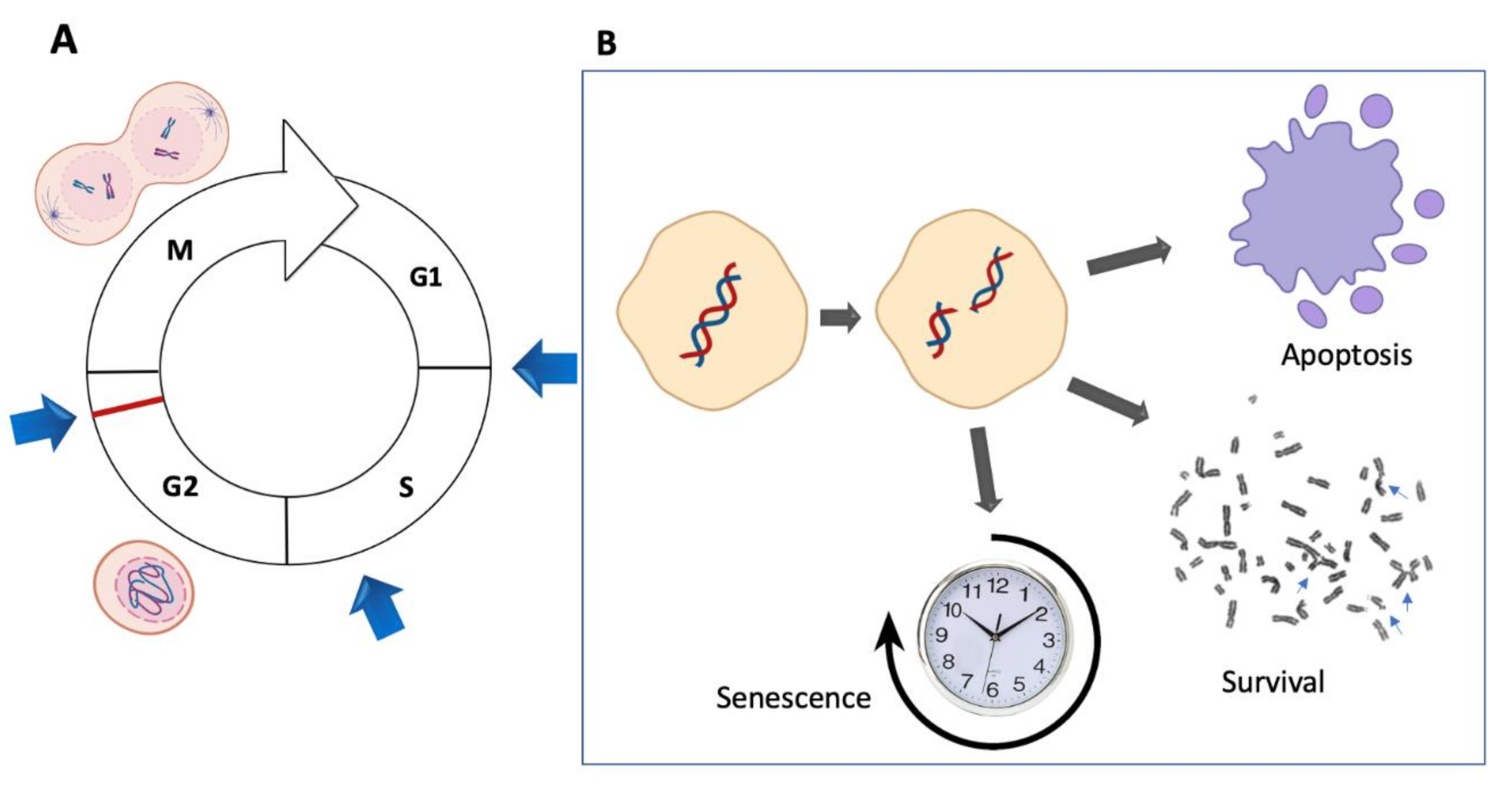
2.3. Double Strand Breaks as the Substrate for Chromosomal Aberrations
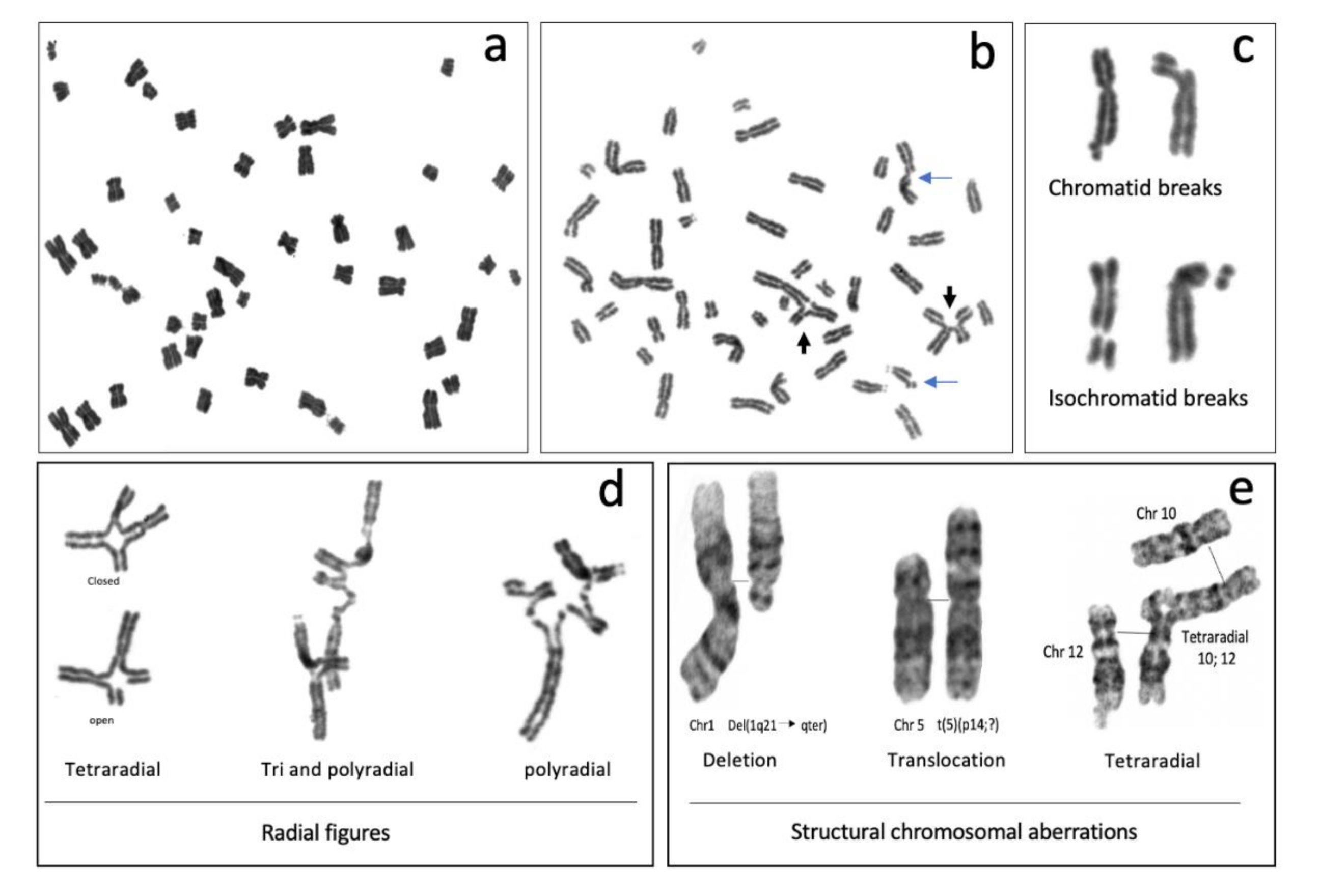
2.3.1. Non-Rejoined Structural Chromosomal Aberrations: Breaks
2.3.2. Rejoined Structural Chromosome Aberrations
2.3.3. Other Chromosome Aberrations
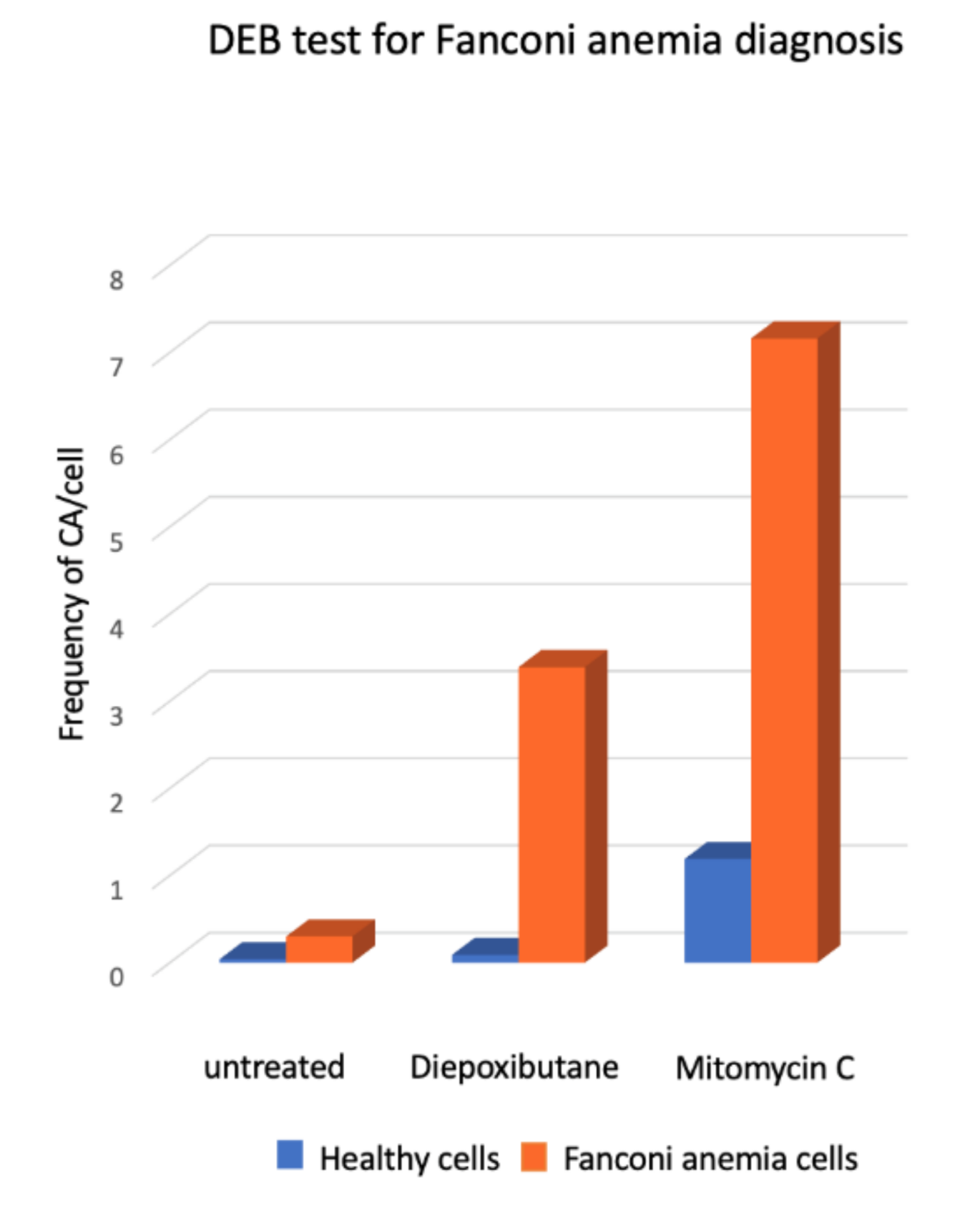
2.4. Chromosome Aberrations for the Diagnosis of Fanconi anemia
3. Fanconi Anemia Proteins beyond ICL Repair
3.1. Fanconi Anemia Proteins Are Involved in Replication Fork Protection
3.1.1. Nucleotide Depletion
3.1.2. Transcription-Replication Collision
3.1.3. Repetitive DNA Sequences
3.1.4. Common Fragile Sites
4. The Control of the Cell Cycle Checkpoints in FA Cells
5. Clinical Consequences of FA/BRCA Pathway Failure
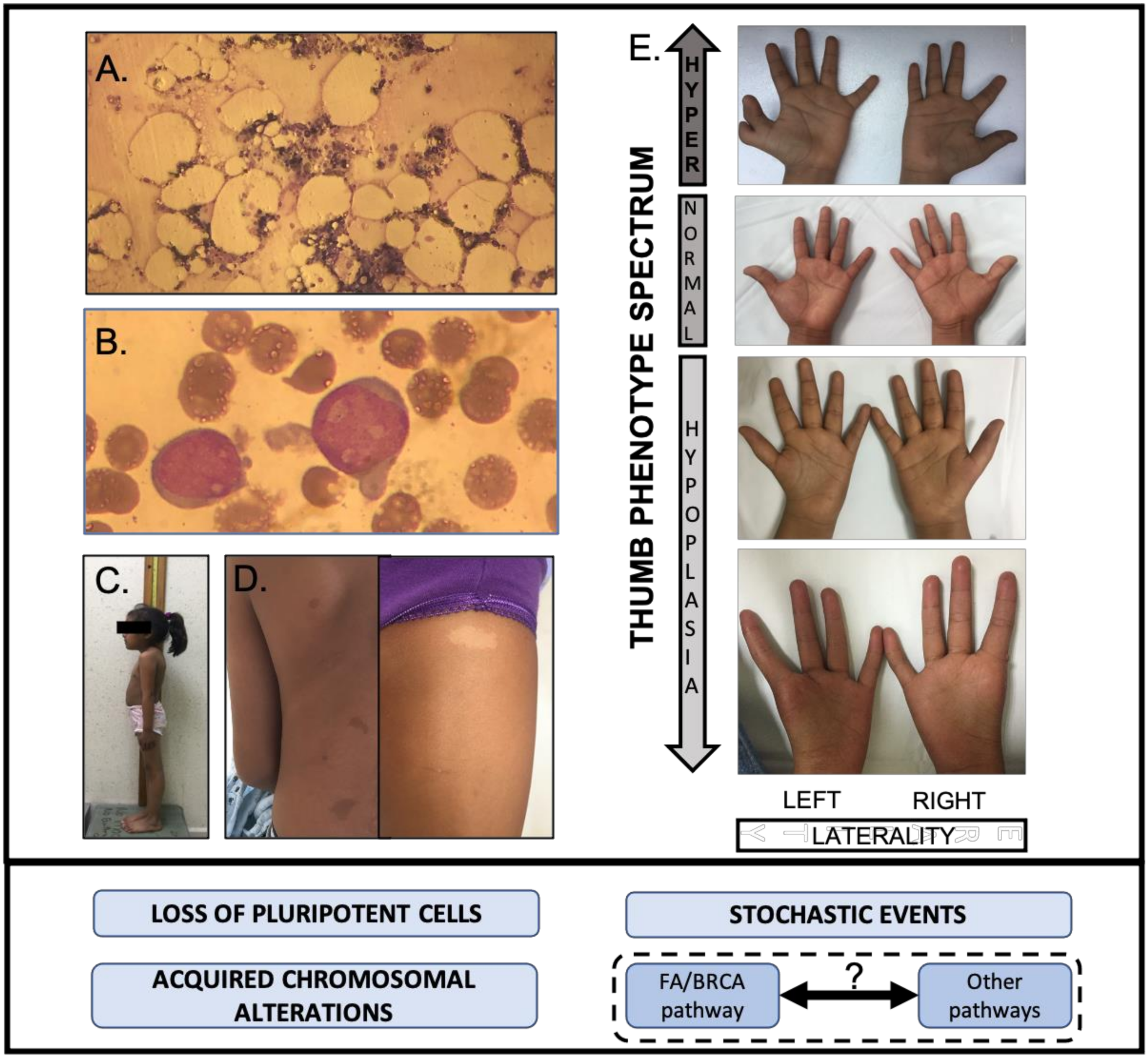
5.1. Development Alterations
5.2. Hematological Manifestations
5.3. Oncologic Susceptibility
5.3.1. Hematologic Neoplasias
MDS
Leukemia
Bone Marrow Abnormalities
5.3.2. Solid Tumors
5.3.3. Skin Cancer
5.3.4. Childhood Solid Cancer
5.3.5. Increased Risk for Heterozygotes
5.3.6. Somatic Mutations in FANC Genes in Sporadic Cancer
5.4. Infertility
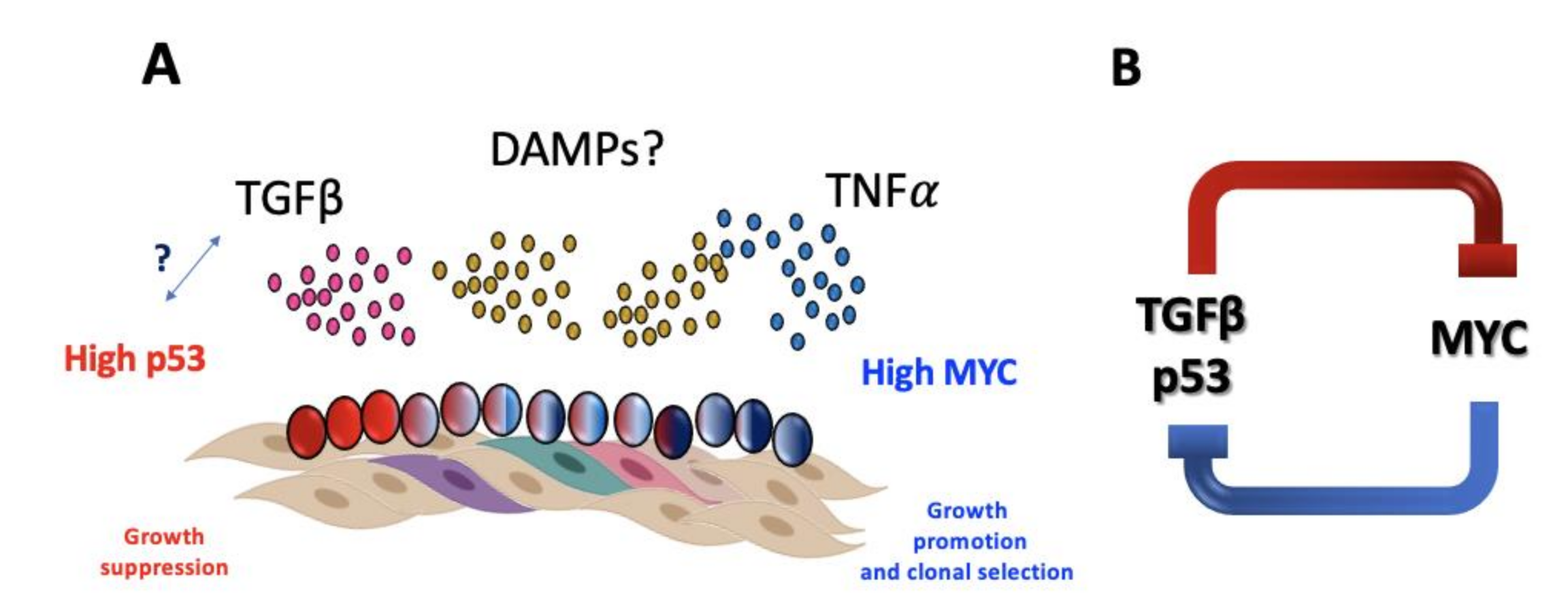
6. The Dichotomy of Aging and Cancer in FA
7. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Wegman-Ostrosky, T.; Savage, S.A. The genomics of inherited bone marrow failure: From mechanism to the clinic. Br. J. Haematol. 2017, 177, 526–542. [Google Scholar] [CrossRef] [PubMed]
- Bogliolo, M.; Surrallés, J. Fanconi anemia: A model disease for studies on human genetics and advanced therapeutics. Curr. Opin. Genet. Dev. 2015, 33, 32–40. [Google Scholar] [CrossRef] [PubMed]
- Rodríguez, A.; D’Andrea, A. Fanconi anemia pathway. Curr. Biol. 2017, 27, R986–R988. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, J.; Nakamura, M. DNA-protein crosslink formation by endogenous aldehydes and AP sites. DNA Repair 2020, 88, 102806. [Google Scholar] [CrossRef] [PubMed]
- Schärer, O.D. DNA interstrand crosslinks: Natural and drug-induced DNA adducts that induce unique cellular responses. ChemBioChem 2005, 6, 27–32. [Google Scholar] [CrossRef]
- Rosado, I.V.; Langevin, F.; Crossan, G.P.; Takata, M.; Patel, K.J. Formaldehyde catabolism is essential in cells deficient for the Fanconi anemia DNA-repair pathway. Nat. Struct. Mol. Biol. 2011, 18, 1432–1434. [Google Scholar] [CrossRef]
- Garaycoechea, J.I.; Crossan, G.P.; Langevin, F.; Mulderrig, L.; Louzada, S.; Yang, F.; Guilbaud, G.; Park, N.; Roerink, S.; Nik-Zainal, S.; et al. Alcohol and endogenous aldehydes damage chromosomes and mutate stem cells. Nature 2018, 553, 171–177. [Google Scholar] [CrossRef]
- Fiesco-Roa, M.O.; Giri, N.; McReynolds, L.J.; Best, A.F.; Alter, B.P. Genotype-phenotype associations in Fanconi anemia: A literature review. Blood Rev. 2019, 37, 100589. [Google Scholar] [CrossRef]
- Ceccaldi, R.; Sarangi, P.; D’Andrea, A.D. The Fanconi anaemia pathway: New players and new functions. Nat. Rev. Mol. Cell Biol. 2016, 17, 337. [Google Scholar] [CrossRef]
- Bogliolo, M.; Bluteau, D.; Lespinasse, J.; Pujol, R.; Vasquez, N.; D’Enghien, C.D.; Stoppa-Lyonnet, D.; Leblanc, T.; Soulier, J.; Surrallés, J. Biallelic truncating FANCM mutations cause early-onset cancer but not Fanconi anemia. Genet. Med. 2018, 20, 458–463. [Google Scholar] [CrossRef]
- Zhang, J.; Dewar, J.M.; Budzowska, M.; Motnenko, A.; Cohn, M.A.; Walter, J.C. DNA interstrand cross-link repair requires replication-fork convergence. Nat. Struct. Mol. Biol. 2015, 22, 242–247. [Google Scholar] [CrossRef] [PubMed]
- Renaudin, X.; Rosselli, F. The FANC/BRCA Pathway Releases Replication Blockades by Eliminating DNA Interstrand Cross-Links. Genes 2020, 11, 585. [Google Scholar] [CrossRef] [PubMed]
- Collis, S.J.; Ciccia, A.; Deans, A.J.; Hořejší, Z.; Martin, J.S.; Maslen, S.L.; Skehel, J.M.; Elledge, S.J.; West, S.C.; Boulton, S.J. FANCM and FAAP24 Function in ATR-Mediated Checkpoint Signaling Independently of the Fanconi Anemia Core Complex. Mol. Cell 2008, 32, 313–324. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.M.; Kee, Y.; Gurtan, A.; D’Andrea, A.D. Cell cycle-dependent chromatin loading of the Fanconi anemia core complex by FANCM/FAAP24. Blood 2008, 111, 5215–5222. [Google Scholar] [CrossRef] [PubMed]
- Tan, W.; van Twest, S.; Murphy, V.J.; Deans, A.J. ATR-Mediated FANCI Phosphorylation Regulates Both Ubiquitination and Deubiquitination of FANCD2. Front. Cell Dev. Biol. 2020, 8, 2. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Walter, J.C. Mechanism and regulation of incisions during DNA interstrand cross-link repair. DNA Repair 2014, 19, 135–142. [Google Scholar] [CrossRef] [PubMed]
- Budzowska, M.; Graham, T.G.; Sobeck, A.; Waga, S.; Walter, J.C. Regulation of the Rev1–pol ζ complex during bypass of a DNA interstrand cross-link. EMBO J. 2015, 34, 1971–1985. [Google Scholar] [CrossRef]
- Clairmont, C.S.; Sarangi, P.; Ponnienselvan, K.; Galli, L.D.; Csete, I.; Moreau, L.; Adelmant, G.; Chowdhury, D.; Marto, J.A.; D’Andrea, A.D. TRIP13 regulates DNA repair pathway choice through REV7 conformational change. Nat. Cell Biol. 2020, 22, 87–96. [Google Scholar] [CrossRef]
- Marín, M.; Ramírez, M.J.; Carmona, M.A.; Jia, N.; Ogi, T.; Bogliolo, M.; Surrallés, J. Functional comparison of XPF missense mutations associated to multiple DNA repair disorders. Genes 2019, 10, 60. [Google Scholar] [CrossRef]
- Castella, M.; Jacquemont, C.; Thompson, E.L.; Yeo, J.E.; Cheung, R.S.; Huang, J.W.; Sobeck, A.; Hendrickson, E.A.; Taniguchi, T. FANCI Regulates Recruitment of the FA Core Complex at Sites of DNA Damage Independently of FANCD2. PLoS Genet. 2015, 11, e1005563. [Google Scholar] [CrossRef]
- Niraj, J.; Färkkilä, A.; D’Andrea, A.D. The fanconi anemia pathway in cancer. Annu. Rev. Cancer Biol. 2019, 3, 457–478. [Google Scholar] [CrossRef] [PubMed]
- Ranjha, L.; Howard, S.M.; Cejka, P. Main steps in DNA double-strand break repair: An introduction to homologous recombination and related processes. Chromosoma 2018, 127, 187–214. [Google Scholar] [CrossRef] [PubMed]
- Rageul, J.; Kim, H. Fanconi anemia and the underlying causes of genomic instability. Environ. Mol. Mutagen. 2020. [Google Scholar] [CrossRef] [PubMed]
- Shakeel, S.; Rajendra, E.; Alcón, P.; O’Reilly, F.; Chorev, D.S.; Maslen, S.; Degliesposti, G.; Russo, C.J.; He, S.; Hill, C.H.; et al. Structure of the Fanconi anaemia monoubiquitin ligase complex. Nature 2019, 575, 234–237. [Google Scholar] [CrossRef]
- Pannunzio, N.R.; Watanabe, G.; Lieber, M.R. Nonhomologous DNA end-joining for repair of DNA double-strand breaks. J. Biol. Chem. 2018, 293, 10512–10523. [Google Scholar] [CrossRef]
- Scully, R.; Panday, A.; Elango, R.; Willis, N.A. DNA double-strand break repair-pathway choice in somatic mammalian cells. Nat. Rev. Mol. Cell Biol. 2019, 20, 698–714. [Google Scholar] [CrossRef]
- Liu, T.; Huang, J. DNA End Resection: Facts and Mechanisms. Genom. Proteom. Bioinform. 2016, 14, 126–130. [Google Scholar] [CrossRef]
- Shukla, V.; Halabelian, L.; Balagere, S.; Samaniego-Castruita, D.; Feldman, D.E.; Arrowsmith, C.H.; Rao, A.; Aravind, L. HMCES Functions in the Alternative End-Joining Pathway of the DNA DSB Repair during Class Switch Recombination in B Cells. Mol. Cell 2020, 77, 384–394. [Google Scholar] [CrossRef]
- Kim, Y.; Lach, F.P.; Desetty, R.; Hanenberg, H.; Auerbach, A.D.; Smogorzewska, A. Mutations of the SLX4 gene in Fanconi anemia. Nat. Genet. 2011, 43, 142–146. [Google Scholar] [CrossRef]
- Liu, Y.; West, S.C. Happy Hollidays: 40th Anniversary of the Holliday junction. Nat. Rev. Mol. Cell Biol. 2004, 5, 937–944. [Google Scholar] [CrossRef]
- Colavito, S.; Prakash, R.; Sung, P. Promotion and regulation of homologous recombination by DNA helicases. Methods 2010, 51, 329–335. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Cohn, M.A.; Kee, Y.; Haas, W.; Gygi, S.P.; D’Andrea, A.D. UAF1 is a subunit of multiple deubiquitinating enzyme complexes. J. Biol. Chem. 2009, 284, 5343–5351. [Google Scholar] [CrossRef] [PubMed]
- Iliakis, G.; Murmann, T.; Soni, A. Alternative end-joining repair pathways are the ultimate backup for abrogated classical non-homologous end-joining and homologous recombination repair: Implications for the formation of chromosome translocations. Mutat. Res. Genet. Toxicol. Environ. Mutagen. 2015, 793, 166–175. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.; Wu, W.; Wu, W.; Rosidi, B.; Zhang, L.; Wang, H.; Iliakis, G. PARP-1 and Ku compete for repair of DNA double strand breaks by distinct NHEJ pathways. Nucleic Acids Res. 2006, 34, 6170–6182. [Google Scholar] [CrossRef] [PubMed]
- Seol, J.H.; Shim, E.Y.; Lee, S.E. Microhomology-mediated end joining: Good, bad and ugly. Mutat. Res. Fundam. Mol. Mech. Mutagen. 2018, 809, 81–87. [Google Scholar] [CrossRef] [PubMed]
- Chang, H.H.Y.; Pannunzio, N.R.; Adachi, N.; Lieber, M.R. Non-homologous DNA end joining and alternative pathways to double-strand break repair. Nat. Rev. Mol. Cell Biol. 2017, 18, 495–506. [Google Scholar] [CrossRef]
- Löbrich, M.; Jeggo, P.A. The impact of a negligent G2/M checkpoint on genomic instability and cancer induction. Nat. Rev. Cancer 2007, 7, 861–869. [Google Scholar] [CrossRef]
- Yun, M.H.; Hiom, K. CtIP-BRCA1 modulates the choice of DNA double-strand-break repair pathway throughout the cell cycle. Nature 2009, 459, 460–463. [Google Scholar] [CrossRef]
- Fradet-Turcotte, A.; Canny, M.D.; Escribano-Díaz, C.; Orthwein, A.; Leung, C.C.Y.; Huang, H.; Landry, M.C.; Kitevski-Leblanc, J.; Noordermeer, S.M.; Sicheri, F.; et al. 53BP1 is a reader of the DNA-damage-induced H2A Lys 15 ubiquitin mark. Nature 2013, 499, 50–54. [Google Scholar] [CrossRef]
- Arnoult, N.; Correia, A.; Ma, J.; Merlo, A.; Garcia-Gomez, S.; Maric, M.; Tognetti, M.; Benner, C.W.; Boulton, S.J.; Saghatelian, A.; et al. Regulation of DNA Repair pathway choice in S/G2 by the NHEJ inhibitor CYREN Europe PMC Funders Group. Nature 2017. [Google Scholar] [CrossRef]
- Auerbach, A.D. Fanconi anemia and its diagnosis. Mutat. Res. Fundam. Mol. Mech. Mutagen. 2009, 668, 4–10. [Google Scholar] [CrossRef] [PubMed]
- Yu, V.P.C.C.; Koehler, M.; Steinlein, C.; Schmid, M.; Hanakahi, L.A.; Van Gool, A.J.; West, S.C.; Venkitaraman, A.R. Gross chromosomal rearrangements and genetic exchange between nonhomologous chromosomes following BRCA2 inactivation. Genes Dev. 2000, 14, 1400–1406. [Google Scholar] [PubMed]
- Nalepa, G.; Clapp, D.W. Fanconi anaemia and cancer: An intricate relationship. Nat. Rev. Cancer 2018, 18, 168–185. [Google Scholar] [CrossRef] [PubMed]
- Srinivasan, M.; Fumasoni, M.; Petela, N.J.; Murray, A.; Nasmyth, K.A. Cohesion is established during dna replication utilising chromosome associated cohesin rings as well as those loaded de novo onto nascent dnas. Elife 2020, 9. [Google Scholar] [CrossRef] [PubMed]
- Newell, A.E.H.; Akkari, Y.M.N.; Torimaru, Y.; Rosenthal, A.; Reifsteck, C.A.; Cox, B.; Grompe, M.; Olson, S.B. Interstrand crosslink-induced radials form between non-homologous chromosomes, but are absent in sex chromosomes. DNA Repair 2004, 3, 535–542. [Google Scholar] [CrossRef] [PubMed]
- Hanlon Newell, A.E.; Hemphill, A.; Akkari, Y.M.N.; Hejna, J.; Moses, R.E.; Olson, S.B. Loss of homologous recombination or non-homologous end-joining leads to radial formation following DNA interstrand crosslink damage. Cytogenet. Genome Res. 2008, 121, 174–180. [Google Scholar] [CrossRef]
- Kubbies, M.; Schindler, D.; Hoehn, H.; Schinzel, A.; Rabinovitch, P.S. Endogenous blockage and delay of the chromosome cycle despite normal recruitment and growth phase explain poor proliferation and frequent endomitosis in Fanconi anemia cells. Am. J. Hum. Genet. 1985, 37, 1022. [Google Scholar]
- Nicoletti, E.; Rao, G.; Bueren, J.A.; Río, P.; Navarro, S.; Surrallés, J.; Choi, G.; Schwartz, J.D. Mosaicism in Fanconi anemia: Concise review and evaluation of published cases with focus on clinical course of blood count normalization. Ann. Hematol. 2020, 99, 913–924. [Google Scholar] [CrossRef]
- Esmer, C.; Sánchez, S.; Ramos, S.; Molina, B.; Frias, S.; Carnevale, A. DEB Test for Fanconi Anemia Detection in Patients with Atypical Phenotypes. Am. J. Med. Genet. 2003, 124, 35–39. [Google Scholar] [CrossRef]
- Sedlackova, H.; Rask, M.B.; Gupta, R.; Choudhary, C.; Somyajit, K.; Lukas, J. Equilibrium between nascent and parental MCM proteins protects replicating genomes. Nature 2020, 587, 297–302. [Google Scholar] [CrossRef]
- Fragkos, M.; Naim, V. Rescue from replication stress during mitosis. Cell Cycle 2017, 16, 613–633. [Google Scholar] [CrossRef]
- Zeman, M.K.; Cimprich, K.A. Causes and consequences of replication stress. Nat. Cell Biol. 2014, 16, 2–9. [Google Scholar] [CrossRef]
- Primo, L.M.F.; Teixeira, L.K. DNA replication stress: Oncogenes in the spotlight. Genet. Mol. Biol. 2020, 43. [Google Scholar] [CrossRef]
- Schwab, R.A.; Nieminuszczy, J.; Shah, F.; Langton, J.; Lopez Martinez, D.; Liang, C.C.; Cohn, M.A.; Gibbons, R.J.; Deans, A.J.; Niedzwiedz, W. The Fanconi Anemia Pathway Maintains Genome Stability by Coordinating Replication and Transcription. Mol. Cell 2015, 60, 351–361. [Google Scholar] [CrossRef]
- Kolinjivadi, A.M.; Crismani, W.; Ngeow, J. Emerging functions of Fanconi anemia genes in replication fork protection pathways. Hum. Mol. Genet. 2020, 29, R158–R164. [Google Scholar] [CrossRef]
- Panday, A.; Willis, N.A.; Elango, R.; Menghi, F.; Duffey, E.E.; Liu, E.T.; Scully, R. FANCM regulates repair pathway choice at stalled replication forks. bioRxiv 2020. [Google Scholar] [CrossRef]
- Molina, B.; Marchetti, F.; Gómez, L.; Ramos, S.; Torres, L.; Ortiz, R.; Altamirano-Lozano, M.; Carnevale, A.; Frias, S. Hydroxyurea induces chromosomal damage in G2 and enhances the clastogenic effect of mitomycin C in Fanconi anemia cells. Environ. Mol. Mutagen. 2015, 56, 457–467. [Google Scholar] [CrossRef]
- García-Rubio, M.L.; Pérez-Calero, C.; Barroso, S.I.; Tumini, E.; Herrera-Moyano, E.; Rosado, I.V.; Aguilera, A. The Fanconi Anemia Pathway Protects Genome Integrity from R-loops. PLoS Genet. 2015, 11, e1005674. [Google Scholar] [CrossRef]
- Pan, X.; Chen, Y.; Biju, B.; Ahmed, N.; Kong, J.; Goldenberg, M.; Huang, J.; Mohan, N.; Klosek, S.; Parsa, K.; et al. FANCM suppresses DNA replication stress at ALT telomeres by disrupting TERRA R-loops. Sci. Rep. 2019, 9, 1–14. [Google Scholar] [CrossRef]
- Vinciguerra, P.; Godinho, S.A.; Parmar, K.; Pellman, D.; D’Andrea, A.D. Cytokinesis failure occurs in Fanconi anemia pathway-deficient murine and human bone marrow hematopoietic cells. J. Clin. Investig. 2010, 120, 3834–3842. [Google Scholar] [CrossRef]
- Madireddy, A.; Kosiyatrakul, S.T.; Boisvert, R.A.; Herrera-Moyano, E.; García-Rubio, M.L.; Gerhardt, J.; Vuono, E.A.; Owen, N.; Yan, Z.; Olson, S.; et al. FANCD2 Facilitates Replication through Common Fragile Sites. Mol. Cell 2016, 64, 388–404. [Google Scholar] [CrossRef] [PubMed]
- Schoder, C.; Liehr, T.; Velleuer, E.; Wilhelm, K.; Blaurock, N.; Weise, A.; Mrasek, K. New aspects on chromosomal instability: Chromosomal break-points in Fanconi anemia patients co-localize on the molecular level with fragile sites. Int. J. Oncol. 2009, 36, 307–312. [Google Scholar]
- Milletti, G.; Strocchio, L.; Pagliara, D.; Girardi, K.; Carta, R.; Mastronuzzi, A.; Locatelli, F.; Nazio, F. Canonical and noncanonical roles of fanconi anemia proteins: Implications in cancer predisposition. Cancers 2020, 12, 2684. [Google Scholar] [CrossRef] [PubMed]
- Rodríguez, A.; Jesús Naveja, J.; Torres, L.; De Teresa, B.G.; Juárez-Figueroa, U.; Ayala-Zambrano, C.; Azpeitia, E.; Mendoza, L.; Frías, S. WIP1 contributes to the adaptation of fanconi anemia cells to DNA damage as determined by the regulatory network of the fanconi anemia and checkpoint recovery pathways. Front. Genet. 2019, 10, 411. [Google Scholar] [CrossRef] [PubMed]
- Bartek, J.; Lukas, J. DNA damage checkpoints: From initiation to recovery or adaptation. Curr. Opin. Cell Biol. 2007, 19, 238–245. [Google Scholar] [CrossRef] [PubMed]
- Musacchio, A. The Molecular Biology of Spindle Assembly Checkpoint Signaling Dynamics. Curr. Biol. 2015, 25, R1002–R1018. [Google Scholar] [CrossRef] [PubMed]
- Ceccaldi, R.; Parmar, K.; Mouly, E.; Delord, M.; Kim, J.M.; Regairaz, M.; Pla, M.; Vasquez, N.; Zhang, Q.S.; Pondarre, C.; et al. Bone marrow failure in fanconi anemia is triggered by an exacerbated p53/p21 DNA damage response that impairs hematopoietic stem and progenitor cells. Cell Stem Cell 2012, 11, 36–49. [Google Scholar] [CrossRef]
- Sala-Trepat, M.; Rouillard, D.; Escarceller, M.; Laquerbe, A.; Moustacchi, E.; Papadopoulo, D. Arrest of S-phase progression is impaired in Fanconi anemia cells. Exp. Cell Res. 2000, 260, 208–215. [Google Scholar] [CrossRef]
- Fischer, M. Census and evaluation of p53 target genes. Oncogene 2017, 36, 3943–3956. [Google Scholar] [CrossRef]
- Luo, Q.; Beaver, J.M.; Liu, Y.; Zhang, Z. Dynamics of p53: A master decider of cell fate. Genes 2017, 8, 66. [Google Scholar] [CrossRef]
- Batchelor, E.; Mock, C.S.; Bhan, I.; Loewer, A.; Lahav, G. Recurrent Initiation: A Mechanism for Triggering p53 Pulses in Response to DNA Damage. Mol. Cell 2008, 30, 277–289. [Google Scholar] [CrossRef] [PubMed]
- Wu, M.; Ye, H.; Tang, Z.; Shao, C.; Lu, G.; Chen, B.; Yang, Y.; Wang, G.; Hao, H. P53 Dynamics Orchestrates With Binding Affinity To Target Genes for Cell Fate Decision. Cell Death Dis. 2017, 8, e3130. [Google Scholar] [CrossRef] [PubMed]
- Rodríguez, A.; Torres, L.; Juárez, U.; Sosa, D.; Azpeitia, E.; De Teresa, B.G.; Cortés, E.; Ortíz, R.; Salazar, A.M.; Ostrosky-Wegman, P.; et al. Fanconi anemia cells with unrepaired DNA damage activate components of the checkpoint recovery process. Theor. Biol. Med. Model. 2015, 12, 1–22. [Google Scholar] [CrossRef]
- McHugh, P.J.; Ward, T.A.; Chovanec, M. A prototypical Fanconi anemia pathway in lower eukaryotes? Cell Cycle 2012, 11, 3739–3744. [Google Scholar] [CrossRef]
- Parmar, K.; D’Andrea, A.; Niedernhofer, L.J. Mouse models of Fanconi anemia. Mutat. Res. Fundam. Mol. Mech. Mutagen. 2009, 668, 133–140. [Google Scholar] [CrossRef]
- Juchau, M.R. Chemical teratogenesis in humans: Biochemical and molecular mechanisms. Prog. Drug Res. 1997, 49, 25–92. [Google Scholar]
- Kaminen-Ahola, N. Fetal alcohol spectrum disorders: Genetic and epigenetic mechanisms. Prenat. Diagn. 2020, 40, 1185–1192. [Google Scholar] [CrossRef]
- Van Wassenhove, L.D.; Mochly-Rosen, D.; Weinberg, K.I. Aldehyde dehydrogenase 2 in aplastic anemia, Fanconi anemia and hematopoietic stem cells. Mol. Genet. Metab. 2016, 119, 28–36. [Google Scholar] [CrossRef]
- Hira, A.; Yabe, H.; Yoshida, K.; Okuno, Y.; Shiraishi, Y.; Chiba, K.; Tanaka, H.; Miyano, S.; Nakamura, J.; Kojima, S.; et al. Variant ALDH2 is associated with accelerated progression of bone marrow failure in Japanese Fanconi anemia patients. Blood 2013, 122, 3206–3209. [Google Scholar] [CrossRef]
- Yabe, M.; Yabe, H.; Morimoto, T.; Fukumura, A.; Ohtsubo, K.; Koike, T.; Yoshida, K.; Ogawa, S.; Ito, E.; Okuno, Y.; et al. The phenotype and clinical course of Japanese Fanconi Anaemia infants is influenced by patient, but not maternal ALDH2 genotype. Br. J. Haematol. 2016, 175, 457–461. [Google Scholar] [CrossRef]
- Giampietro, P.F.; Adler-Brecher, B.; Verlander, P.C.; Pavlakis, S.G.; Davis, J.G.; Auerbach, A.D. The need for more accurate and timely diagnosis in Fanconi anemia: A report from the International Fanconi Anemia Registry. Pediatrics 1993, 91, 1116–1120. [Google Scholar]
- Bakker, S.T.; De Winter, J.P.; Te Riele, H. Learning from a paradox: Recent insights into Fanconi anaemia through studying mouse models. DMM Dis. Model. Mech. 2013, 6, 40–47. [Google Scholar] [CrossRef]
- Bianchi, F.T.; Berto, G.E.; Di Cunto, F. Impact of DNA repair and stability defects on cortical development. Cell. Mol. Life Sci. 2018, 75, 3963–3976. [Google Scholar] [CrossRef]
- García-De Teresa, B.; Hernández-Gómez, M.; Frías, S. DNA Damage as a Driver for Growth Delay: Chromosome Instability Syndromes with Intrauterine Growth Retardation. Biomed Res. Int. 2017, 2017. [Google Scholar] [CrossRef]
- Webb, M.L.; Rosen, H.; Taghinia, A.; McCarty, E.R.; Cerrato, F.; Upton, J.; Labow, B.I. Incidence of Fanconi anemia in children with congenital thumb anomalies referred for diepoxybutane testing. J. Hand Surg. Am. 2011, 36, 1052–1057. [Google Scholar] [CrossRef]
- Shimamura, A.; Alter, B.P. Pathophysiology and Management of Inherited Bone Marrow Failure Syndromes. Blood Rev. 2010, 24, 101–122. [Google Scholar] [CrossRef]
- Wilks, D.J.; Kay, S.P.J.; Bourke, G. Fanconi’s anaemia and unilateral thumb polydactyly—Don’t miss it. J. Plast. Reconstr. Aesthetic Surg. 2012, 65, 1083–1086. [Google Scholar] [CrossRef]
- Elmakky, A.; Stanghellini, I.; Landi, A.; Percesepe, A. Role of Genetic Factors in the Pathogenesis of Radial Deficiencies in Humans. Curr. Genom. 2015, 16, 264–278. [Google Scholar] [CrossRef]
- Sathyanarayana, V.; Lee, B.; Wright, N.B.; Santos, R.; Bonney, D.; Wynn, R.; Patel, L.; Chandler, K.; Cheesman, E.; Schindler, D.; et al. Patterns and frequency of renal abnormalities in Fanconi anaemia: Implications for long-term management. Pediatr. Nephrol. 2018, 33, 1547–1551. [Google Scholar] [CrossRef]
- Ruggiero, J.L.; Dodds, M.; Freese, R.; Polcari, I.C.; Maguiness, S.; Hook, K.P.; Boull, C. Cutaneous Findings in Fanconi Anemia. J. Am. Acad. Dermatol. 2020. [Google Scholar] [CrossRef]
- Salas-Labadía, C.; Gómez-Carmona, S.; Cruz-Alcívar, R.; Martínez-Anaya, D.; Del Castillo-Ruiz, V.; Durán-Mckinster, C.; Ulloa-Avilés, V.; Yokoyama-Rebollar, E.; Ruiz-Herrera, A.; Navarrete-Meneses, P.; et al. Genetic and clinical characterization of 73 Pigmentary Mosaicism patients: Revealing the genetic basis of clinical manifestations. Orphanet J. Rare Dis. 2019, 14, 259. [Google Scholar] [CrossRef]
- Thomas, I.T.; Frias, J.L.; Cantu, E.S.; Lafer, C.Z.; Flannery, D.B.; Graham, J.G. Association of pigmentary anomalies with chromosomal and genetic mosaicism and chimerism. Am. J. Hum. Genet. 1989, 45, 193–205. [Google Scholar]
- Alter, B.P.; Giri, N.; Savage, S.A.; Rosenberg, P.S. Cancer in the national cancer institute inherited bone marrow failure syndrome cohort after fifteen years of follow-up. Haematologica 2018, 103, 30–39. [Google Scholar] [CrossRef]
- Kutler, D.I.; Singh, B.; Satagopan, J.; Batish, S.D.; Berwick, M.; Giampietro, P.F.; Hanenberg, H.; Auerbach, A.D. A 20-year perspective on the International Fanconi Anemia Registry (IFAR). Blood 2003, 101, 1249–1256. [Google Scholar] [CrossRef] [PubMed]
- Du, W.; Li, X.; Wilson, A.F.; Pang, Q. A small molecule p53 activator attenuates Fanconi anemia leukemic stem cell proliferation. Stem Cell Res. Ther. 2018, 9, 4–7. [Google Scholar] [CrossRef]
- Li, X.; Wilson, A.F.; Du, W.; Pang, Q. Cell-Cycle-Specific Function of p53 in Fanconi Anemia Hematopoietic Stem and Progenitor Cell Proliferation. Stem Cell Reports 2018, 10, 339–346. [Google Scholar] [CrossRef]
- Pitman, J.L.; McNeilly, A.S.; McNeilly, J.R.; Hays, L.E.; Bagby, G.C.; Sawyer, H.R.; McNatty, K.P. The fate of granulosa cells following premature oocyte loss and the development of ovarian cancers. Int. J. Dev. Biol. 2012, 56, 949–958. [Google Scholar] [CrossRef]
- Mechilli, M.; Schinoppi, A.; Kobos, K.; Natarajan, A.T.; Palitti, F. DNA repair deficiency and acetaldehyde-induced chromosomal alterations in CHO cells. Mutagenesis 2008, 23, 51–56. [Google Scholar] [CrossRef]
- Edenberg, H.J. The genetics of alcohol metabolism: Role of alcohol dehydrogenase and aldehyde dehydrogenase variants. Alcohol Res. Health 2007, 30, 5–13. [Google Scholar]
- Garaycoechea, J.I.; Crossan, G.P.; Langevin, F.; Daly, M.; Arends, M.J.; Patel, K.J. Genotoxic consequences of endogenous aldehydes on mouse haematopoietic stem cell function. Nature 2012, 489, 571–575. [Google Scholar] [CrossRef]
- Garaycoechea, J.I.; Patel, K.J. Why does the bone marrow fail in Fanconi anemia? Blood 2014, 123, 26–34. [Google Scholar] [CrossRef] [PubMed]
- Guervilly, J.H.; Macé-Aimé, G.; Rosselli, F. Loss of CHK1 function impedes DNA damage-induced FANCD2 monoubiquitination but normalizes the abnormal G2 arrest in Fanconi anemia. Hum. Mol. Genet. 2008, 17, 679–689. [Google Scholar] [CrossRef] [PubMed]
- Kennedy, R.D.; Chen, C.C.; Stuckert, P.; Archila, E.M.; De La Vega, M.A.; Moreau, L.A.; Shimamura, A.; D’Andrea, A.D. Fanconi anemia pathway-deficient tumor cells are hypersensitive to inhibition of ataxia telangiectasia mutated. J. Clin. Investig. 2007, 117, 1440–1449. [Google Scholar] [CrossRef] [PubMed]
- Ceccaldi, R.; Briot, D.; Larghero, J.; Vasquez, N.; D’Enghien, C.D.; Chamousset, D.; Noguera, M.E.; Waisfisz, Q.; Hermine, O.; Pondarre, C.; et al. Spontaneous abrogation of the G2 DNA damage checkpoint has clinical benefits but promotes leukemogenesis in Fanconi anemia patients. J. Clin. Investig. 2011, 121, 184–194. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Kozono, D.E.; O’Connor, K.W.; Vidal-Cardenas, S.; Rousseau, A.; Hamilton, A.; Moreau, L.; Gaudiano, E.F.; Greenberger, J.; Bagby, G.; et al. TGF-β inhibition rescues hematopoietic stem cell defects and bone marrow failure in Fanconi anemia. Cell Stem Cell 2016, 18, 668–681. [Google Scholar] [CrossRef] [PubMed]
- Rodríguez, A.; Zhang, K.; Färkkilä, A.; Filiatrault, J.; Yang, C.; Velázquez, M.; Furutani, E.; Goldman, D.C.; García de Teresa, B.; Garza-Mayén, G.; et al. MYC Promotes Bone Marrow Stem Cell Dysfunction in Fanconi Anemia. Cell Stem Cell 2020. [Google Scholar] [CrossRef]
- Alter, B.P. Fanconi anemia and the development of leukemia. Best Pract. Res. Clin. Haematol. 2014, 27, 214–221. [Google Scholar] [CrossRef]
- Cioc, A.M.; Wagner, J.E.; MacMillan, M.L.; DeFor, T.; Hirsch, B. Diagnosis of myelodysplastic syndrome among a cohort of 119 patients with fanconi anemia: Morphologic and cytogenetic characteristics. Am. J. Clin. Pathol. 2010, 133, 92–100. [Google Scholar] [CrossRef]
- Alter, B.P.; Greene, M.H.; Velazquez, I.; Rosenberg, P.S. Cancer in Fanconi anemia. Blood 2003, 101, 2072–2073. [Google Scholar] [CrossRef]
- De Latour, R.P.; Soulier, J. How I treat MDS and AML in Fanconi anemia. Blood 2016, 127, 2971–2979. [Google Scholar] [CrossRef]
- Alter, B.P. Cancer in Fanconi anemia, 1927–2001. Cancer 2003, 97, 425–440. [Google Scholar] [CrossRef] [PubMed]
- Auerbach, A.D.; Allen, R.G. Leukemia and preleukemia in Fanconi anemia patients. A review of the literature and report of the International Fanconi Anemia Registry. Cancer Genet. Cytogenet. 1991, 51, 1–12. [Google Scholar] [CrossRef]
- Mitchell, R.; Wagner, J.E.; Hirsch, B.; Defor, T.E.; Zierhut, H.; Macmillan, M.L. Haematopoietic cell transplantation for acute leukaemia and advanced myelodysplastic syndrome in Fanconi anaemia. Br. J. Haematol. 2013, 164, 384–395. [Google Scholar] [CrossRef] [PubMed]
- Bagby, G.C.; Fleischman, A. The stem cell fitness landscape and pathways of molecular leukemogenesis. Front. Biosci. 2011, S3, 487–500. [Google Scholar] [CrossRef][Green Version]
- Rosenberg, P.S.; Greene, M.H.; Alter, B.P. Cancer incidence in persons with Fanconi anemia. Blood 2003, 101, 822–826. [Google Scholar] [CrossRef]
- Quentin, S.; Cuccuini, W.; Ceccaldi, R.; Nibourel, O.; Pondarre, C.; Pagès, M.P.; Vasquez, N.; D’Enghien, C.D.; Larghero, J.; De Latour, R.P.; et al. Myelodysplasia and leukemia of fanconi anemia are associated with a specific pattern of genomic abnormalities that includes cryptic RUNX1/AML1 lesions. Blood 2011, 117, e161–e170. [Google Scholar] [CrossRef]
- Ye, C.J.; Sharpe, Z.; Heng, H.H. Origins and consequences of chromosomal instability: From cellular adaptation to genome chaos-mediated system survival. Genes 2020, 11, 1162. [Google Scholar] [CrossRef]
- Chao, M.M.; Thomay, K.; Goehring, G.; Wlodarski, M.; Pastor, V.; Schlegelberger, B.; Schindler, D.; Kratz, C.P.; Niemeyer, C. Mutational Spectrum of Fanconi Anemia Associated Myeloid Neoplasms. Klin. Padiatr. 2017, 229, 329–334. [Google Scholar] [CrossRef]
- Van Zeeburg, H.J.T.; Snijders, P.J.F.; Wu, T.; Gluckman, E.; Soulier, J.; Surralles, J.; Castella, M.; Van Der Wal, J.E.; Wennerberg, J.; Califano, J.; et al. Clinical and molecular characteristics of squamous cell carcinomas from Fanconi anemia patients. J. Natl. Cancer Inst. 2008, 100, 1649–1653. [Google Scholar] [CrossRef]
- Ramírez, M.J.; Minguillón, J.; Loveless, S.; Lake, K.; Carrasco, E.; Stjepanovic, N.; Balmaña, J.; Català, A.; Mehta, P.A.; Surrallés, J. Chromosome fragility in the buccal epithelium in patients with Fanconi anemia. Cancer Lett. 2020, 472, 1–7. [Google Scholar] [CrossRef]
- Velleuer, E.; Dietrich, R.; Pomjanski, N.; de Santana Almeida Araujo, I.K.; Silva de Araujo, B.E.; Sroka, I.; Biesterfeld, S.; Böcking, A.; Schramm, M. Diagnostic accuracy of brush biopsy–based cytology for the early detection of oral cancer and precursors in Fanconi anemia. Cancer Cytopathol. 2020, 128, 403–413. [Google Scholar] [CrossRef] [PubMed]
- Kao, W.H.; Riker, A.I.; Kushwaha, D.S.; Ng, K.; Enkemann, S.A.; Jove, R.; Buettner, R.; Zinn, P.O.; Sánchez, N.P.; Villa, J.L.; et al. Upregulation of fanconi anemia DNA repair genes in melanoma compared with non-melanoma skin cancer. J. Investig. Dermatol. 2011, 131, 2139–2142. [Google Scholar] [CrossRef] [PubMed]
- Malric, A.; Defachelles, A.-S.; Leblanc, T.; Lescoeur, B.; Lacour, B.; Peuchmaur, M.; Maurage, C.-A.; Pierron, G.; Guillemont, D.; Dubois d’Enghien, C.; et al. Fanconi Anemia and Solid Malignancies in Childhood: A National Retrospective Study. Pediatr. Blood Cancer 2015, 62, 463–470. [Google Scholar] [CrossRef] [PubMed]
- Meyer, S.; Tischkowitz, M.; Chandler, K.; Gillespie, A.; Birch, J.M.; Evans, D.G. Fanconi anaemia, BRCA2 mutations and childhood cancer: A developmental perspective from clinical and epidemiological observations with implications for genetic counselling. J. Med. Genet. 2014, 51, 71–75. [Google Scholar] [CrossRef] [PubMed]
- Howlett, N.G.; Taniguchi, T.; Olson, S.; Cox, B.; Waisfisz, Q.; De Die-Smulders, C.; Persky, N.; Grompe, M.; Joenje, H.; Pals, G.; et al. Biallelic inactivation of BRCA2 in Fanconi anemia. Science 2002, 297, 606–609. [Google Scholar] [CrossRef]
- Sklavos, M.M.; Giri, N.; Stratton, P.; Alter, B.P.; Pinto, L.A. Anti-müllerian hormone deficiency in females with Fanconi Anemia. J. Clin. Endocrinol. Metab. 2014, 99, 1608–1614. [Google Scholar] [CrossRef]
- Yang, X.; Zhang, X.; Jiao, J.; Zhang, F.; Pan, Y.; Wang, Q.; Chen, Q.; Cai, B.; Tang, S.; Zhou, Z.; et al. Rare variants in FANCA induce premature ovarian insufficiency. Hum. Genet. 2019, 138, 1227–1236. [Google Scholar] [CrossRef]
- Krausz, C.; Riera-Escamilla, A.; Chianese, C.; Moreno-Mendoza, D.; Ars, E.; Rajmil, O.; Pujol, R.; Bogliolo, M.; Blanco, I.; Rodríguez, I.; et al. From exome analysis in idiopathic azoospermia to the identification of a high-risk subgroup for occult Fanconi anemia. Genet. Med. 2019, 21, 189–194. [Google Scholar] [CrossRef]
- Fu, C.; Begum, K.; Jordan, P.W.; He, Y.; Overbeek, P.A. Dearth and delayed maturation of testicular germ cells in Fanconi anemia E mutant male mice. PLoS ONE 2016, 11, e0159800. [Google Scholar] [CrossRef]
- Fu, C.; Begum, K.; Overbeek, P.A. Primary ovarian insufficiency induced by fanconi anemia e mutation in a mouse model. PLoS ONE 2016, 11, e0144285. [Google Scholar] [CrossRef]
- Simhadri, S.; Peterson, S.; Patel, D.S.; Huo, Y.; Cai, H.; Bowman-Colin, C.; Miller, S.; Ludwig, T.; Ganesan, S.; Bhaumik, M.; et al. Male fertility defect associated with disrupted BRCA1-PALB2 interaction in mice. J. Biol. Chem. 2014, 289, 24617–24629. [Google Scholar] [CrossRef]
- Kato, Y.; Alavattam, K.G.; Sin, H.S.; Meetei, A.R.; Pang, Q.; Andreassen, P.R.; Namekawa, S.H. FANCB is essential in the male germline and regulates H3K9 methylation on the sex chromosomes during meiosis. Hum. Mol. Genet. 2015, 24, 5234–5249. [Google Scholar] [CrossRef] [PubMed]
- Tsui, V.; Crismani, W. The Fanconi Anemia Pathway and Fertility. Trends Genet. 2019, 35, 199–214. [Google Scholar] [CrossRef] [PubMed]
- Brosh, R.M.; Bellani, M.; Liu, Y.; Seidman, M.M. Fanconi Anemia: A DNA repair disorder characterized by accelerated decline of the hematopoietic stem cell compartment and other features of aging. Ageing Res. Rev. 2017, 33, 67–75. [Google Scholar] [CrossRef] [PubMed]
- Savage, S.A.; Walsh, M.F. Myelodysplastic Syndrome, Acute Myeloid Leukemia, and Cancer Surveillance in Fanconi Anemia. Hematol. Oncol. Clin. North Am. 2018, 32, 657–668. [Google Scholar] [CrossRef] [PubMed]
- Giri, N.; Batista, D.L.; Alter, B.P.; Stratakis, C.A. Endocrine abnormalities in patients with fanconi anemia. J. Clin. Endocrinol. Metab. 2007, 92, 2624–2631. [Google Scholar] [CrossRef]
- López-Otín, C.; Blasco, M.A.; Partridge, L.; Serrano, M.; Kroemer, G. The hallmarks of aging. Cell 2013, 153, 1194–1217. [Google Scholar] [CrossRef]
- Alter, B.P.; Giri, N.; Savage, S.A.; Rosenberg, P.S. Telomere length in inherited bone marrow failure syndromes. Haematologica 2015, 100, 49–54. [Google Scholar] [CrossRef]
- Kumari, U.; Ya Jun, W.; Huat Bay, B.; Lyakhovich, A. Evidence of mitochondrial dysfunction and impaired ROS detoxifying machinery in Fanconi Anemia cells. Oncogene 2014, 33, 165–172. [Google Scholar] [CrossRef]
- Helbling-Leclerc, A.; Dessarps-Freichey, F.; Evrard, C.; Rosselli, F. Fanconi anemia proteins counteract the implementation of the oncogene-induced senescence program. Sci. Rep. 2019, 9, 1–11. [Google Scholar] [CrossRef]
- Chen, G.Y.; Nuñez, G. Sterile inflammation: Sensing and reacting to damage. Nat. Rev. Immunol. 2010, 10, 826–837. [Google Scholar] [CrossRef] [PubMed]
- Zinger, A.; Cho, W.C.; Ben-Yehuda, A. Cancer and aging—The inflammatory connection. Aging Dis. 2017, 8, 611–627. [Google Scholar] [CrossRef] [PubMed]
- Gong, T.; Liu, L.; Jiang, W.; Zhou, R. DAMP-sensing receptors in sterile inflammation and inflammatory diseases. Nat. Rev. Immunol. 2020, 20, 95–112. [Google Scholar] [CrossRef] [PubMed]
- Garaycoechea, J.I.; Crossan, G.P.; Langevin, F.F.; Mulderrig, L.; Louzada, S.; Yang, F.; Guilbaud, G.; Park, N.; Roerink, S.; Nik-Zainal, S.; et al. Main steps in DNA double-strand break repair: An introduction to homologous recombination and related processes. Nature 2018, 668, 51–56. [Google Scholar]
- Dufour, C.; Corcione, A.; Svahn, J.; Haupt, R.; Poggi, V.; Béka’ssy, A.N.; Scimè, R.; Pistorio, A.; Pistoia, V. TNF-α and IFN-γ are overexpressed in the bone marrow of Fanconi anemia patients and TNF-α suppresses erythropoiesis in vitro. Blood 2003, 102, 2053–2059. [Google Scholar] [CrossRef] [PubMed]
- Rosselli, F.; Sanceau, J.; Gluckman, E.; Wietzerbin, J.; Moustacchi, E. Abnormal lymphokine production: A novel feature of the genetic disease Fanconi anemia. II. In vitro and in vivo spontaneous overproduction of tumor necrosis factor α. Blood 1994, 83, 1216–1225. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Sejas, D.P.; Qiu, Y.; Williams, D.A.; Pang, Q. Inflammatory ROS promote and cooperate with the Fanconi anemia mutation for hematopoietic senescence. J. Cell Sci. 2007, 120, 1572–1583. [Google Scholar] [CrossRef]
- Hsu, J.I.; Dayaram, T.; Tovy, A.; De Braekeleer, E.; Jeong, M.; Wang, F.; Zhang, J.; Heffernan, T.P.; Gera, S.; Kovacs, J.J.; et al. PPM1D Mutations Drive Clonal Hematopoiesis in Response to Cytotoxic Chemotherapy. Cell Stem Cell 2018, 23, 700–713. [Google Scholar] [CrossRef]
- McNerney, M.E.; Le Beau, M.M. The Harmful Consequences of Increased Fitness in Hematopoietic Stem Cells. Cell Stem Cell 2018, 23, 634–635. [Google Scholar] [CrossRef]
- Bowman, R.L.; Busque, L.; Levine, R.L. Clonal Hematopoiesis and Evolution to Hematopoietic Malignancies. Cell Stem Cell 2018, 22, 157–170. [Google Scholar] [CrossRef]
- Elias, H.K.; Bryder, D.; Park, C.Y. Molecular mechanisms underlying lineage bias in aging hematopoiesis. Semin. Hematol. 2017, 54, 4–11. [Google Scholar] [CrossRef] [PubMed]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| FANC Gene/Alias | Cytogenetic Location | Function of the FANC Protein |
|---|---|---|
| FANCA | 16q24.3 | FA core complex |
| FANCB | Xp22.2 | FA core complex |
| FANCC | 9q22.32 | FA core complex |
| FANCD1/BRCA2 | 13q13.1 | Homologous recombination. Enable RAD51 to displace RPA from ssDNA. |
| FANCD2 | 3p25.3 | Monoubiquitinated ID complex recruits the downstream repair proteins and facilitates repair of DNA ICLs |
| FANCE | 6p21.31 | FA core complex; bridge between the FA core complex and FANCD2 |
| FANCF | 11p14.3 | FA core complex |
| FANCG/XRCC9 | 9p13.3 | FA core complex |
| FANCI | 15q26.1 | Monoubiquitinated ID complex recruits the downstream repair proteins and facilitates repair of DNA ICLs |
| FANCJ/BRIP1 | 17q23.2 | FA core complex |
| FANCL | 2p16.1 | E3 ubiquitin-protein ligase, monoubiquitination of FANCD2 |
| 2FANCM | 14q21.2 | FA core complex. Acts by sensing stalled fork by ICLs and recruiting the core complex proteins to the site of ICL |
| FANCN/PALB2 | 16q12.2 | Homologous recombination |
| 2FANCO/RAD51C | 17q22 | Resolution of D-loop structures through Holliday Junction Intermediates and Homologous DNA Pairing and Strand Exchange. |
| FANCP/SLX4 | 16p13.3 | Cooperate with FANCQ-XPF to generate endonucleolytic incisions to unhook the ICL. |
| FANCQ/XPF | 16p13.12 | DNA endonuclease, involved in homologous recombination; responsible for 5′ incision to remove ICLs |
| 2FANCR/RAD51 | 15q15.1 | Interact with the ssDNA-binding protein RPA, RAD52 homologous pairing and strand transfer of DNA |
| 2FANCS/BRCA1 | 17q21.31 | Homologous recombination |
| FANCT/UBE2T | 1q32.1 | E2 ubiquitin-conjugating enzyme, associates with FA core complex, catalyzes monoubiquitination of FANCD2 in association with FANCL |
| FANCU/XRCC2 | 7q36.1 | Homologous recombination |
| FANCV/REV7 | 1p36.22 | Translesion DNA synthesis |
| FANCW/RFWD3 | 16q23.1 | RING-Type E3 Ubiquitin Transferase |
| Non-Homologous End-Joining | Microhomology Mediated End-Joining | Homologous Recombination | |
|---|---|---|---|
| Timing | Fast | Fast | Slow |
| Template dependence | Independent | Independent | Dependent |
| Homology usage | 0–4 bp | 2–20 bp | >100 bp |
| End resection | no | yes | yes |
| Cell cycle phase | G1, S, G2 | G1, S, G2 | S/G2 |
| Accuracy of repair | Mostly accurate, error prone | Frequently error prone | Highly accurate |
| Causes of Replication Stress | DNA Lesion/Configuration | Proteins Involved in Replication Stress Resolution 1 | Outcomes of Unsolved Replication Stress | References |
|---|---|---|---|---|
| Exogenous Sources | ||||
| Aphidicolin | Late replication/recombination intermediates | FANCD2, FANCI, BLM, PICH | Chromosomal aberrations, UFB in CFS and telomeres, telomere fragility, cytokinesis failure, binucleated cells | [55,60] |
| Anticancer drugs (Hydroxyurea) | Nucleotide depletion induced reversed forks “chicken foots” | BRCA2/FANCD1, FANCD2, BRCA1/FANCS, RAD51/FANCR | ||
| Endogenous Sources | ||||
| Complex/repetitive DNA sequences: | ||||
| Transcription-replication collision | R-Loops | FANCM, FA pathway, FANCD2, BLM | Chromosomal aberrations, micronucleus | [53,54,58,59] |
| Centromere | Catenated DNA Stretched DNA Chromosome entanglements | FANCM, FANCD2, BLM, PICH, TOPOIIa, TOPBP1, WRN, TRF1, TRF2 | Centromeric UFB, Incomplete chromatid disjunction. Cytokinesis failure, Chromosome breakage, micronuclei, binucleated cells, polyploidy | [3,53] |
| Telomere | Unresolved R-Loops within ALT telomeres | FANCM-BLM, BRCA2/FANCD1, BRCA1/FANCS, RAD51/FANCR | Telomeric UFB, Cytokinesis failure, Chromosome breakage | [59,63] |
| GC-rich DNA | G-quadruplexes, stem loops | FANCJ | Chromosome breakage | [53,63] |
| Common Fragile sites | Large replicons, scarcity of replication origins | MUS81-EME1, ERCC1-XPF/FANCQ, SLX4/FANCP, FANCD2, BLM-RMI1-RMI2-TOPOIII; | Fragile sites UFB, Chromosomal aberrations, cytokinesis failure, binucleated cells, chromosome mis-segregation, cell death | [21,55,60,61] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
García-de-Teresa, B.; Rodríguez, A.; Frias, S. Chromosome Instability in Fanconi Anemia: From Breaks to Phenotypic Consequences. Genes 2020, 11, 1528. https://doi.org/10.3390/genes11121528
García-de-Teresa B, Rodríguez A, Frias S. Chromosome Instability in Fanconi Anemia: From Breaks to Phenotypic Consequences. Genes. 2020; 11(12):1528. https://doi.org/10.3390/genes11121528
Chicago/Turabian StyleGarcía-de-Teresa, Benilde, Alfredo Rodríguez, and Sara Frias. 2020. "Chromosome Instability in Fanconi Anemia: From Breaks to Phenotypic Consequences" Genes 11, no. 12: 1528. https://doi.org/10.3390/genes11121528
APA StyleGarcía-de-Teresa, B., Rodríguez, A., & Frias, S. (2020). Chromosome Instability in Fanconi Anemia: From Breaks to Phenotypic Consequences. Genes, 11(12), 1528. https://doi.org/10.3390/genes11121528