Re-Evaluating the Internal Phylogenetic Relationships of Collembola by Means of Mitogenome Data

 ,
, 
 ,
,  and
and 
Abstract
1. Introduction
2. Materials and Methods
2.1. Sample Collection and Mitochondrial DNA Sequencing
2.2. Read Assembly and Mitogenome Annotations
2.3. Phylogenetic Analysis
2.4. Phylogenetic Tree Topology Comparison
2.5. Nucleotide Biases and dN/dS Ratio
3. Results
3.1. New Mitogenomes
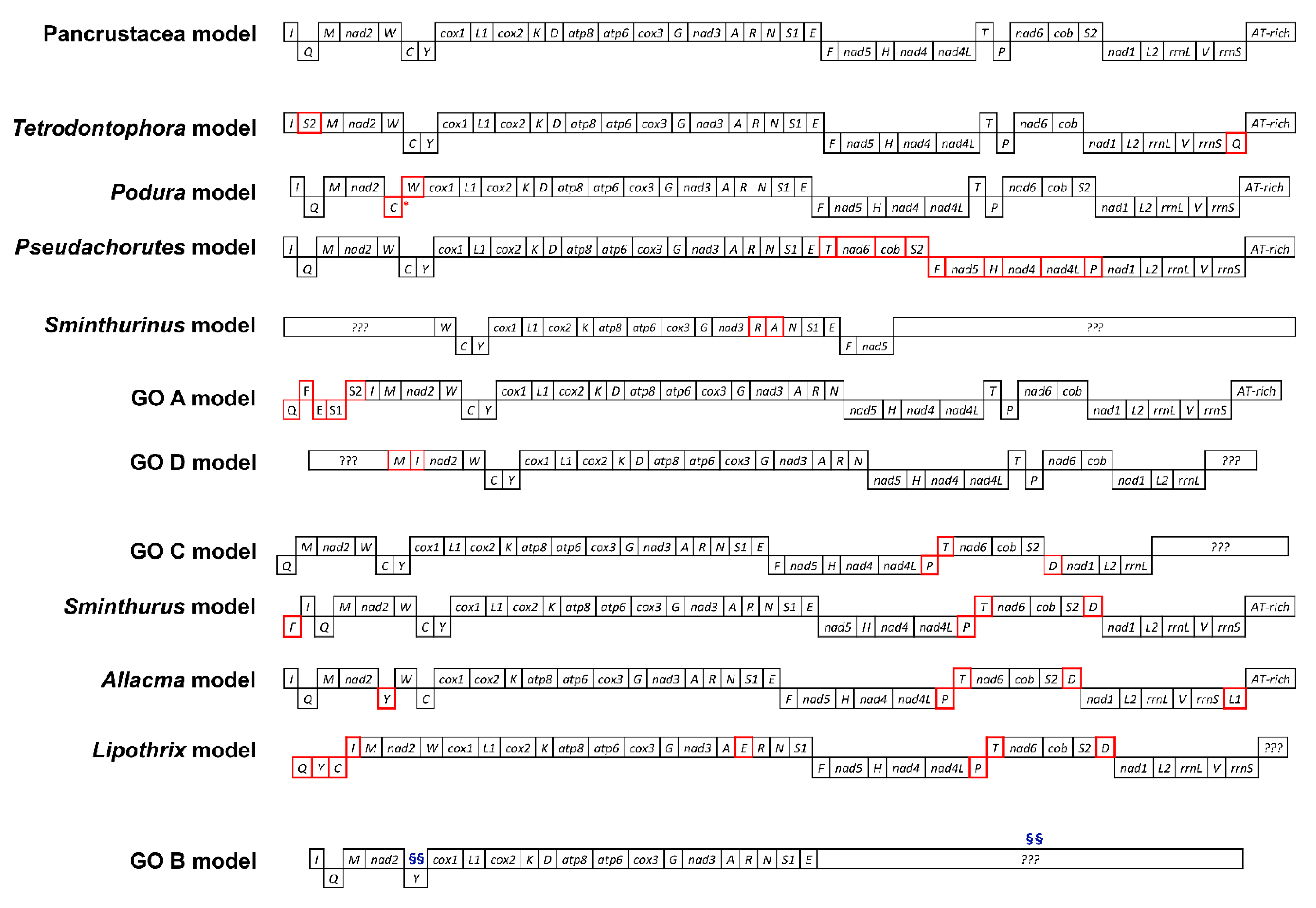
3.2. Dataset Composition and Gene Orders
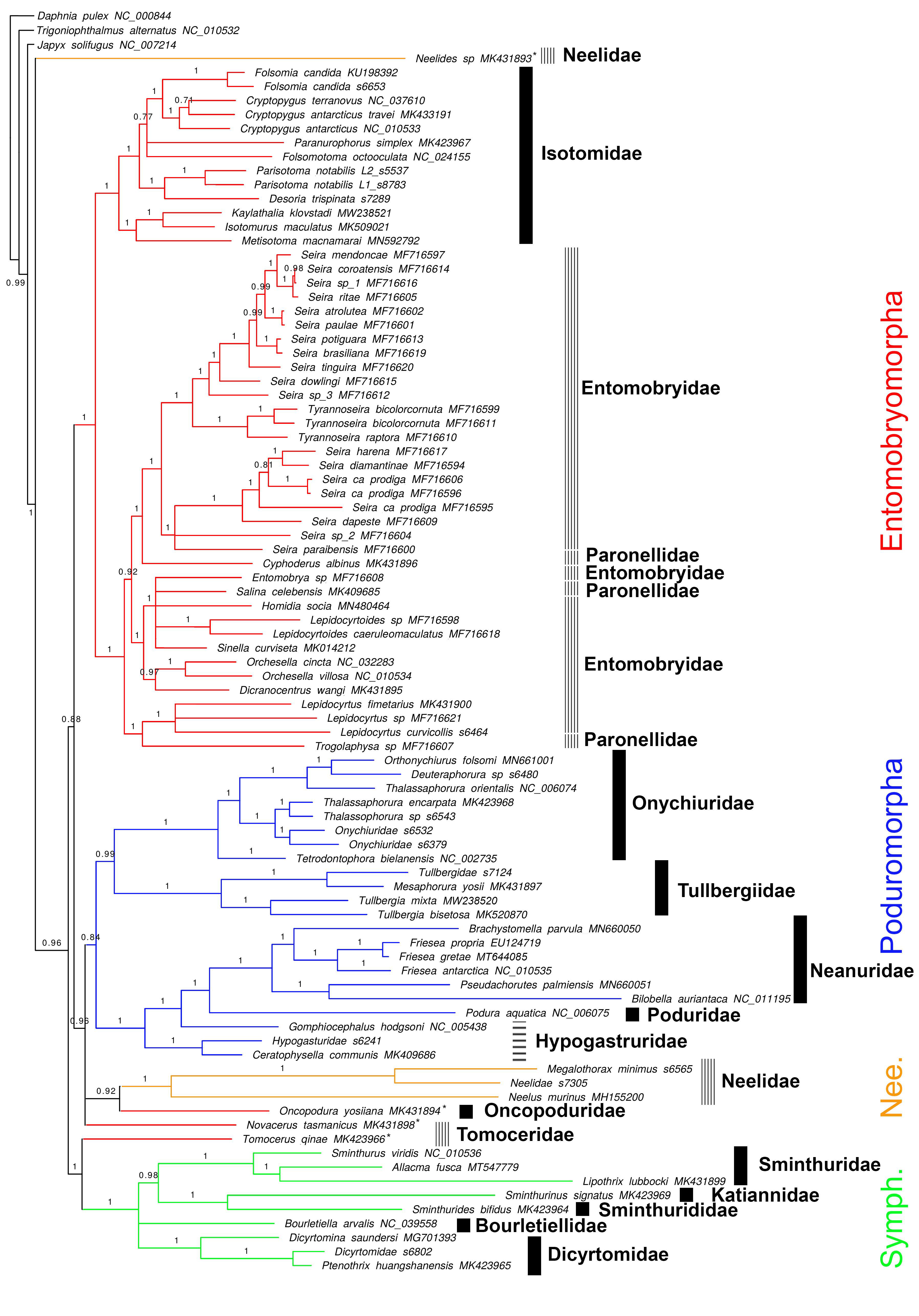
3.3. Phylogenetic Analysis
3.4. Tree Topology Tests
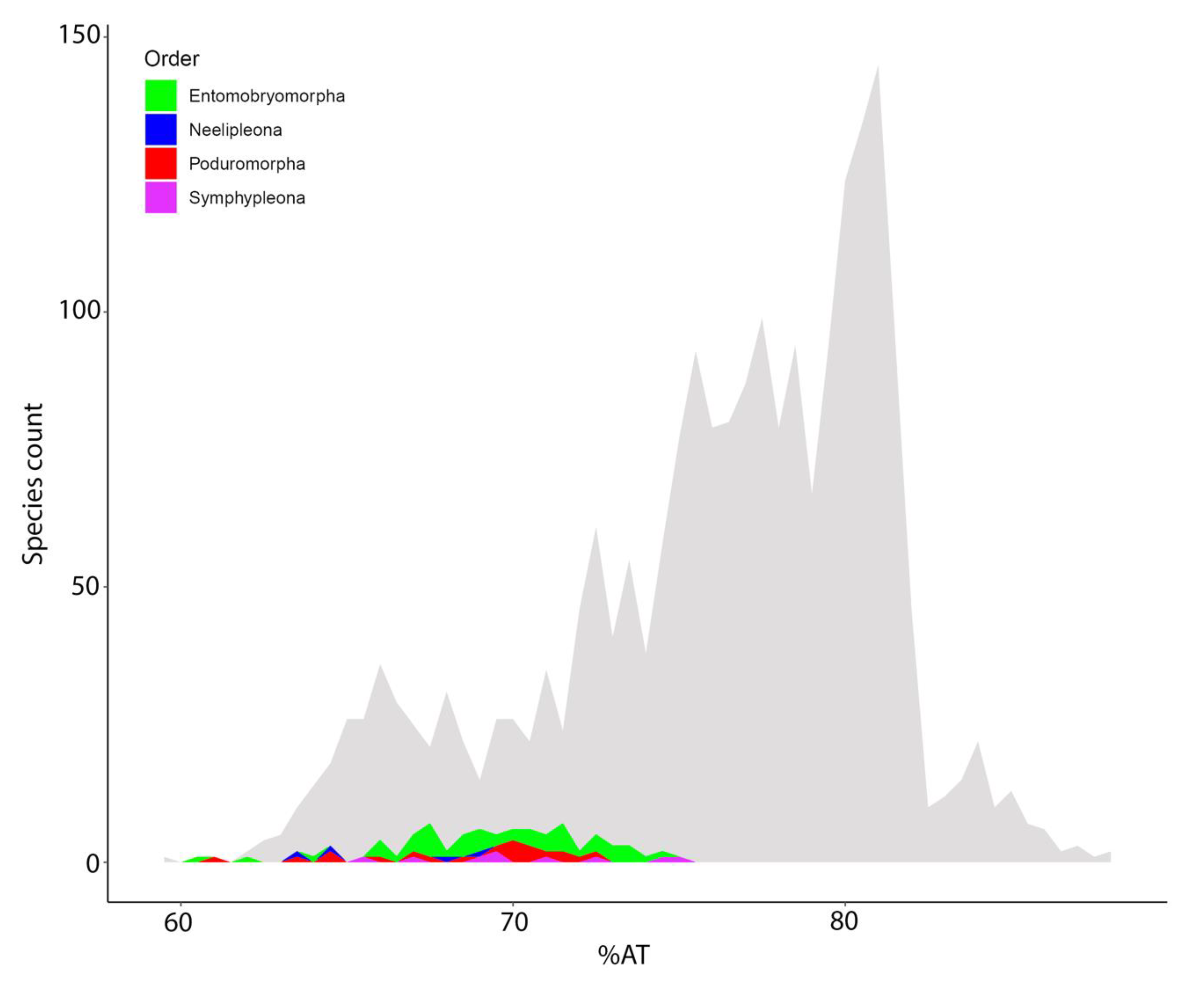
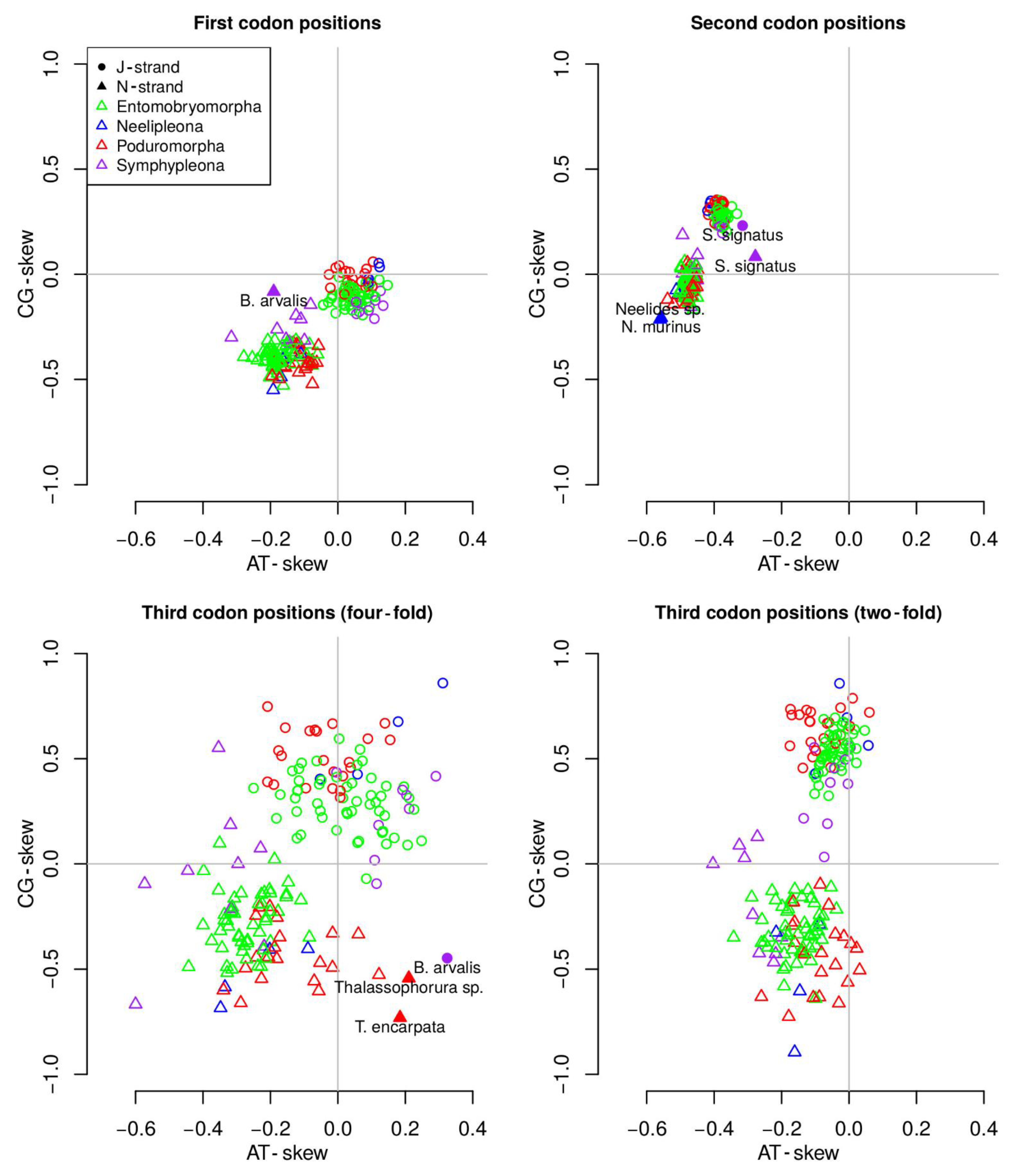
3.5. Nucleotide Biases and dN/dS Ratio
4. Discussion
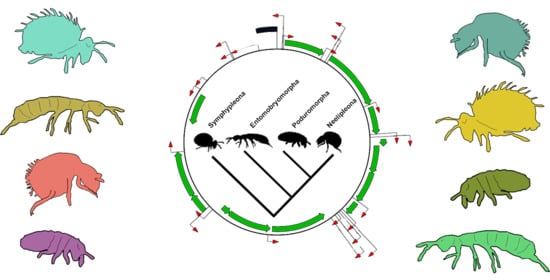
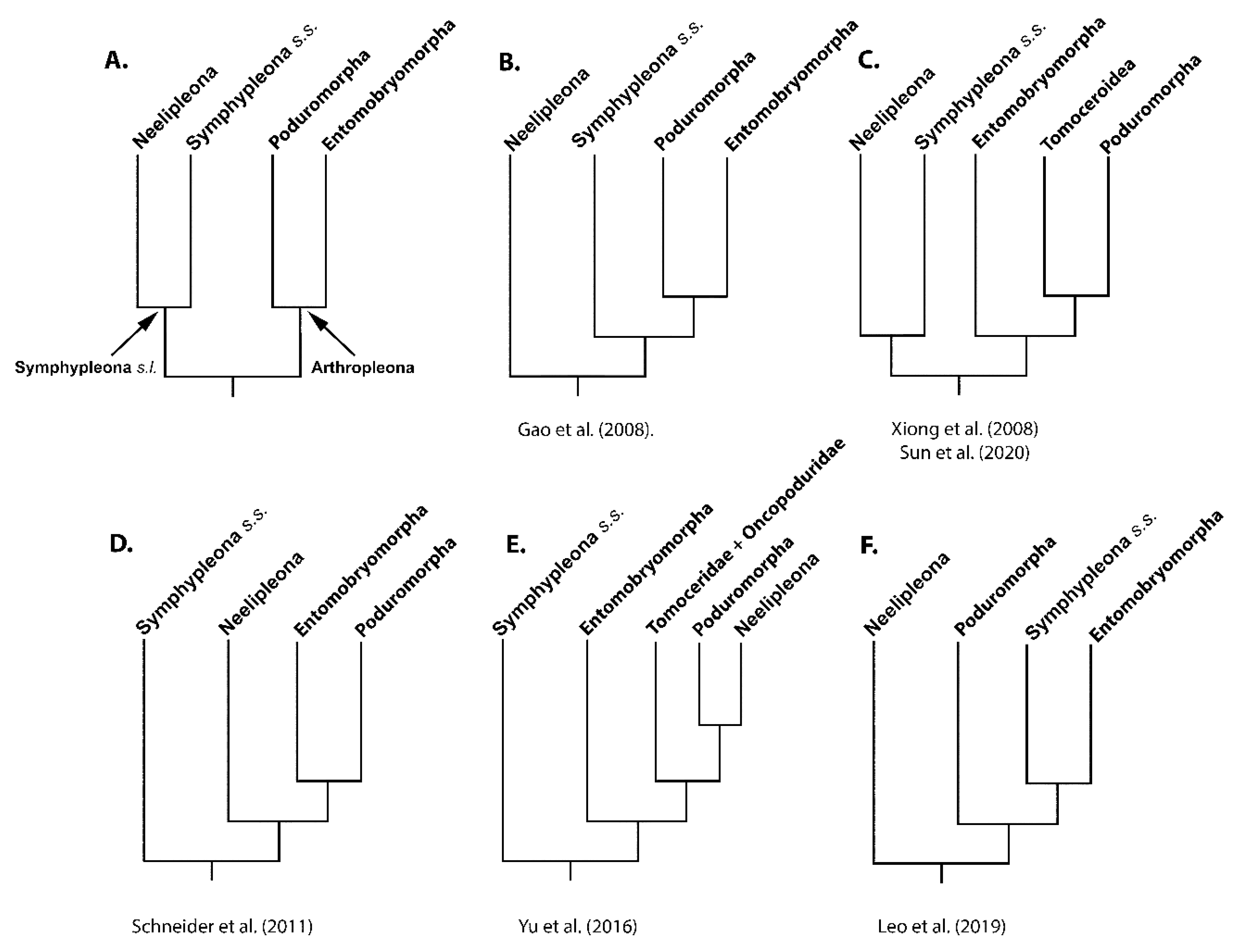
4.1. Deep Collembola Phylogeny
4.2. Collembola Gene Orders
4.3. MtDNA Compositional Biases among Springtails
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Hopkin:, S.P. Biology of the Springtails: (Insecta: Collembola); OUP Oxford: Oxford, UK, 1997; pp. 1–330. ISBN 978-0-19-158925-6. [Google Scholar]
- Leo, C.; Carapelli, A.; Cicconardi, F.; Frati, F.; Nardi, F. Mitochondrial Genome Diversity in Collembola: Phylogeny, Dating and Gene Order. Diversity 2019, 11, 169. [Google Scholar] [CrossRef]
- Nardi, F.; Spinsanti, G.; Boore, J.L.; Carapelli, A.; Dallai, R.; Frati, F. Hexapod origins: Monophyletic or paraphyletic? Science 2003, 299, 1887–1889. [Google Scholar] [CrossRef] [PubMed]
- Luan, Y.X.; Mallatt, J.M.; Xie, R.D.; Yang, Y.M.; Yin, W.Y. The phylogenetic positions of three basal-hexapod groups (Protura, Diplura, and Collembola) based on ribosomal RNA gene sequences. Mol. Biol. Evol. 2005, 22, 1579–1592. [Google Scholar] [CrossRef] [PubMed]
- Carapelli, A.; Liò, P.; Nardi, F.; Van Der Wath, E.; Frati, F. Phylogenetic analysis of mitochondrial protein coding genes confirms the reciprocal paraphyly of Hexapoda and Crustacea. BMC Evol. Biol. 2007, 7, S8. [Google Scholar] [CrossRef] [PubMed]
- D’Haese, C.A. Morphological appraisal of Collembola phylogeny with special emphasis on Poduromorpha and a test of the aquatic origin hypothesis. Zool. Scr. 2003, 32, 563–586. [Google Scholar] [CrossRef]
- Frati, F.; Simon, C.; Sullivan, J.; Swofford, D.L. Evolution of the mitochondrial cytochrome oxidase II gene in Collembola. J. Mol. Evol. 1997, 44, 145–158. [Google Scholar] [CrossRef]
- D’Haese, C.A. Were the first springtails semi-aquatic? A phylogenetic approach by means of 28S rDNA and optimization alignment. Proc. R. Soc. B Biol. Sci. 2002, 269, 1143–1151. [Google Scholar] [CrossRef]
- Gao, Y.; Bu, Y.; Luan, Y.X. Phylogenetic relationships of basal hexapods reconstructed from nearly complete 18S and 28S rRNA gene sequences. Zool. Sci. 2008, 25, 1139–1145. [Google Scholar] [CrossRef]
- Schneider, C.; Cruaud, C.; D’Haese, C. Unexpected diversity in Neelipleona revealed by molecular phylogeny approach (Hexapoda, Collembola). Soil Org. 2011, 83, 383–398. [Google Scholar]
- Xiong, Y.; Gao, Y.; Yin, W.Y.; Luan, Y.X. Molecular phylogeny of Collembola inferred from ribosomal RNA genes. Mol. Phylogenet. Evol. 2008, 49, 728–735. [Google Scholar] [CrossRef]
- Yu, D.; Zhang, F.; Stevens, M.I.; Yan, Q.; Liu, M.; Hu, F. New insight into the systematics of Tomoceridae (Hexapoda, Collembola) by integrating molecular and morphological evidence. Zool. Scr. 2016, 45, 286–299. [Google Scholar] [CrossRef]
- Misof, B.; Liu, S.; Meusemann, K.; Peters, R.S.; Donath, A.; Mayer, C.; Frandsen, P.B.; Ware, J.; Flouri, T.; Beutel, R.G.; et al. Phylogenomics resolves the timing and pattern of insect evolution. Science 2014, 346, 763–767. [Google Scholar] [CrossRef] [PubMed]
- Sun, X.; Yu, D.; Xie, Z.; Dong, J.; Ding, Y.; Yao, H.; Greenslade, P. Phylomitogenomic analyses on collembolan higher taxa with enhanced taxon sampling and discussion on method selection. PLoS ONE 2020, 15, e0230827. [Google Scholar] [CrossRef] [PubMed]
- Godeiro, N.N.; Pacheco, G.; Liu, S.; Gioia Cipola, N.; Berbel-Filho, W.M.; Zhang, F.; Gilbert, M.T.P.; Bellini, B.C. Phylogeny of Neotropical Seirinae (Collembola, Entomobryidae) based on mitochondrial genomes. Zool. Scr. 2020, 49, 329–339. [Google Scholar] [CrossRef]
- Boore, J.L.; Brown, W.M. Big trees from little genomes: Mitochondrial gene order as a phylogenetic tool. Curr. Opin. Genet. Dev. 1998, 8, 668–674. [Google Scholar] [CrossRef]
- Boore, J.L. Animal mitochondrial genomes. Nucleic Acids Res. 1999, 27, 1767–1780. [Google Scholar] [CrossRef]
- Bernt, M.; Braband, A.; Schierwater, B.; Stadler, P.F. Genetic aspects of mitochondrial genome evolution. Mol. Phylogenetics Evol. 2013, 69, 328–338. [Google Scholar] [CrossRef]
- Carapelli, A.; Cucini, C.; Fanciulli, P.P.; Frati, F.; Convey, P.; Nardi, F. Molecular comparison among three Antarctic endemic springtail species and description of the mitochondrial genome of Friesea gretae (Hexapoda, Collembola). Diversity 2020, 12, 450. [Google Scholar] [CrossRef]
- Cicconardi, F.; Borges, P.A.V.; Strasberg, D.; Oromí, P.; López, H.; Pérez-Delgado, A.J.; Casquet, J.; Caujapé-Castells, J.; Fernández-Palacios, J.M.; Thébaud, C.; et al. MtDNA metagenomics reveals large-scale invasion of belowground arthropod communities by introduced species. Mol. Ecol. 2017, 26, 3104–3115. [Google Scholar] [CrossRef]
- Carapelli, A.; Convey, P.; Nardi, F.; Frati, F. The mitochondrial genome of the Antarctic springtail Folsomotoma octooculata (Hexapoda; Collembola), and an update on the phylogeny of collembolan lineages based on mitogenomic data. Entomologia 2014, 2. [Google Scholar] [CrossRef]
- Cook, C.E.; Yue, Q.; Akam, M. Mitochondrial genomes suggest that hexapods and crustaceans are mutually paraphyletic. Proc. R. Soc. B Biol. Sci. 2005, 272, 1295–1304. [Google Scholar] [CrossRef] [PubMed]
- Dong, J.; Zhang, F.; Wang, X. Complete mitochondrial genome of Pseudachorutes palmiensis (Collembola: Neanuridae). Mitochondrial DNA Part B Resour. 2020, 5, 394–395. [Google Scholar] [CrossRef] [PubMed]
- Nardi, F.; Cucini, C.; Leo, C.; Frati, F.; Fanciulli, P.P.; Carapelli, A. The complete mitochondrial genome of the springtail Allacma fusca, the internal phylogenetic relationships and gene order of Symphypleona. Mitochondrial DNA Part B Resour. 2020, 5, 3121–3123. [Google Scholar] [CrossRef]
- Carpenter, G. Insecta: Aptera-Collembola; British Museum (Natural History): London, UK, 1902; pp. 221–223. [Google Scholar]
- Stevens, M.I.; Fjellberg, A.; Greenslade, P.; Hogg, I.D.; Sunnucks, P. Redescription of the Antarctic springtail Desoria klovstadi using morphological and molecular evidence. Polar Biol. 2006, 29, 820–830. [Google Scholar] [CrossRef]
- Stevens, M.I.; D’Haese, C.A. Morphologically tortured: Taxonomic placement of an Antarctic springtail (Collembola: Isotomidae) misguided by morphology and ecology. Zool. Scr. 2017, 46, 180–187. [Google Scholar] [CrossRef]
- Dierckxsens, N.; Mardulyn, P.; Smits, G. NOVOPlasty: De novo assembly of organelle genomes from whole genome data. Nucleic Acids Res. 2017, 45, e18. [Google Scholar] [CrossRef]
- Li, D.; Liu, C.M.; Luo, R.; Sadakane, K.; Lam, T.W. MEGAHIT: An ultra-fast single-node solution for large and complex metagenomics assembly via succinct de Bruijn graph. Bioinformatics 2015, 31, 1674–1676. [Google Scholar] [CrossRef]
- Robinson, J.T.; Thorvaldsdóttir, H.; Winckler, W.; Guttman, M.; Lander, E.S.; Getz, G.; Mesirov, J.P. Integrative Genomics Viewer. Nat. Biotechnol. 2011, 29, 24–26. [Google Scholar] [CrossRef]
- Bernt, M.; Donath, A.; Jühling, F.; Externbrink, F.; Florentz, C.; Fritzsch, G.; Pütz, J.; Middendorf, M.; Stadler, P.F. MITOS: Improved de novo metazoan mitochondrial genome annotation. Mol. Phylogenet. Evol. 2013, 69, 313–319. [Google Scholar] [CrossRef]
- Wernersson, R.; Pedersen, A.G. RevTrans: Multiple alignment of coding DNA from aligned amino acid sequences. Nucleic Acids Res. 2003, 31, 3537–3539. [Google Scholar] [CrossRef]
- Katoh, K.; Standley, D.M. MAFFT multiple sequence alignment software version 7: Improvements in performance and usability. Mol. Biol. Evol. 2013, 30, 772–780. [Google Scholar] [CrossRef] [PubMed]
- Castresana, J. Selection of conserved blocks from multiple alignments for their use in phylogenetic analysis. Mol. Biol. Evol. 2000, 17, 540–552. [Google Scholar] [CrossRef] [PubMed]
- Lanfear, R.; Frandsen, P.B.; Wright, A.M.; Senfeld, T.; Calcott, B. Partitionfinder 2: New methods for selecting partitioned models of evolution for molecular and morphological phylogenetic analyses. Mol. Biol. Evol. 2017, 34, 772–773. [Google Scholar] [CrossRef] [PubMed]
- Miller, M.A.; Schwartz, T.; Pickett, B.E.; He, S.; Klem, E.B.; Scheuermann, R.H.; Passarotti, M.; Kaufman, S.; O’Leary, M.A. A RESTful API for access to phylogenetic pools via the CIPRES Science Gateway. Evol. Bioinform. Online 2015, 11, 43–48. [Google Scholar] [CrossRef]
- Ronquist, F.; Teslenko, M.; Van Der Mark, P.; Ayres, D.L.; Darling, A.; Höhna, S.; Larget, B.; Liu, L.; Suchard, M.A.; Huelsenbeck, J.P. Mrbayes 3.2: Efficient bayesian phylogenetic inference and model choice across a large model space. Syst. Biol. 2012, 61, 539–542. [Google Scholar] [CrossRef]
- Rambaut, A.; Drummond, A.J.; Xie, D.; Baele, G.; Suchard, M.A. Posterior summarization in Bayesian phylogenetics using Tracer 1.7. Syst. Biol. 2018, 67, 901–904. [Google Scholar] [CrossRef]
- Nguyen, L.T.; Schmidt, H.A.; Von Haeseler, A.; Minh, B.Q. IQ-TREE: A fast and effective stochastic algorithm for estimating maximum-likelihood phylogenies. Mol. Biol. Evol. 2015, 32, 268–274. [Google Scholar] [CrossRef]
- Suchard, M.A.; Lemey, P.; Baele, G.; Ayres, D.L.; Drummond, A.J.; Rambaut, A. Bayesian phylogenetic and phylodynamic data integration using BEAST 1.10. Virus Evol. 2018, 4, 1–5. [Google Scholar] [CrossRef]
- Kishino, H.; Miyata, T.; Hasegawa, M. Maximum likelihood inference of protein phylogeny and the origin of chloroplasts. J. Mol. Evol. 1990, 31, 151–160. [Google Scholar] [CrossRef]
- Hassanin, A.; Léger, N.; Deutsch, J. Evidence for multiple reversals of asymmetric mutational constraints during the evolution of the mitochondrial genome of Metazoa, and consequences for phylogenetic inferences. Syst. Biol. 2005, 54, 277–298. [Google Scholar] [CrossRef]
- Gao, F.; Chen, C.; Arab, D.A.; Du, Z.; He, Y.; Ho, S.Y. EasyCodeML: A visual tool for analysis of selection using CodeML. Ecol. Evol. 2019, 9, 3891–3898. [Google Scholar] [CrossRef] [PubMed]
- Zhang, F.; Sun, D.D.; Yu, D.Y.; Wang, B.X. Molecular phylogeny supports S-chaetae as a key character better than jumping organs and body scales in classification of Entomobryoidea (Collembola). Sci. Rep. 2015, 5, 12471. [Google Scholar] [CrossRef] [PubMed]
- Carapelli, A.; Fanciulli, P.P.P.; Frati, F.; Leo, C. Mitogenomic data to study the taxonomy of Antarctic springtail species (Hexapoda: Collembola) and their adaptation to extreme environments. Polar Biol. 2019, 42, 715–732. [Google Scholar] [CrossRef]
- Prada, C.F.; Boore, J.L. Gene annotation errors are common in the mammalian mitochondrial genomes database. BMC Genomics 2019, 20, 73. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
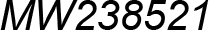{kind=link}
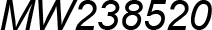{kind=link}
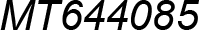{kind=link}
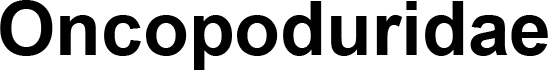{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| AN | Order | Family | Species | Gene Order |
|---|---|---|---|---|
| NC_010533 | Entomobr. | Isotomidae | Cryptopygus antarcticus | Pancrustacea model |
| MK433191 | Entomobr. | Isotomidae | Cryptopygus a. travei | Pancrustacea model |
| KX863671 | Entomobr. | Isotomidae | Cryptopygus terranovus | Pancrustacea model |
| MK431896 | Entomobr. | Paronellidae | Cyphoderus albinus | Pancrustacea model |
| s7289 | Entomobr. | Isotomidae | Desoria trispinata | Pancrustacea model § |
| MK431895 | Entomobr. | Entomobryidae | Dicranocentrus wangi | Pancrustacea model |
| MF716608 | Entomobr. | Entomobryidae | Entomobrya sp. | Pancrustacea model * |
| KU198392 | Entomobr. | Isotomidae | Folsomia candida | Pancrustacea model |
| s6653 | Entomobr. | Isotomidae | Folsomia candida | Pancrustacea model § |
| NC_024155 | Entomobr. | Isotomidae | Folsomotoma octooculata | Pancrustacea model |
| MN480464 | Entomobr. | Entomobryidae | Homidia socia | Pancrustacea model |
| MK509021 | Entomobr. | Isotomidae | Isotomurus maculatus | Pancrustacea model |
| MW238521 | Entomobr. | Isotomidae | Kaylathalia klovstadi | Pancrustacea model |
| MF716618 | Entomobr. | Entomobryidae | Lepidocyrtoides caeruleomaculatus | Pancrustacea model |
| MF716598 | Entomobr. | Entomobryidae | Lepidocyrtoides sp. | Pancrustacea model |
| s6464 | Entomobr. | Lepidocyrtinae | Lepidocyrtus curvicollis | Pancrustacea model § |
| MK431900 | Entomobr. | Entomobryidae | Lepidocyrtus fimetarius | Pancrustacea model |
| MF716621 | Entomobr. | Lepidocyrtinae | Lepidocyrtus sp. | Not analysed |
| MN592792 | Entomobr. | Isotomidae | Metisotoma macnamarai | Pancrustacea model |
| MK431898 | Entomobr. | Tomoceridae | Novacerus tasmanicus | Pancrustacea model |
| MK431894 | Entomobr. | Oncopoduridae | Oncopodura yosiiana | Pancrustacea model |
| KT985987 | Entomobr. | Entomobryidae | Orchesella cincta | Pancrustacea model |
| EU016195 | Entomobr. | Entomobryidae | Orchesella villosa | Pancrustacea model |
| MK423967 | Entomobr. | Isotomidae | Paranurophorus simplex | Pancrustacea model * |
| s8783 | Entomobr. | Isotomidae | Parisotoma notabilis L1 | Pancrustacea model § |
| s5537 | Entomobr. | Isotomidae | Parisotoma notabilis L2 | Pancrustacea model § |
| MK409685 | Entomobr. | Paronellidae | Salina celebensis | Pancrustacea model |
| MF716602 | Entomobr. | Entomobryidae | Seira atrolutea | Pancrustacea model |
| MF716619 | Entomobr. | Entomobryidae | Seira brasiliana | Pancrustacea model |
| MF716595 | Entomobr. | Entomobryidae | Seira ca. prodiga 1 | Pancrustacea model |
| MF716596 | Entomobr. | Entomobryidae | Seira ca. prodiga 2 | Pancrustacea model * |
| MF716606 | Entomobr. | Entomobryidae | Seira ca. prodiga 3 | Pancrustacea model |
| MF716614 | Entomobr. | Entomobryidae | Seira coroatensis | Pancrustacea model |
| MF716609 | Entomobr. | Entomobryidae | Seira dapeste | Pancrustacea model |
| MF716594 | Entomobr. | Entomobryidae | Seira diamantinae | Pancrustacea model |
| MF716615 | Entomobr. | Entomobryidae | Seira dowlingi | Pancrustacea model * |
| MF716617 | Entomobr. | Entomobryidae | Seira harena | Pancrustacea model |
| MF716597 | Entomobr. | Entomobryidae | Seira mendoncae | Pancrustacea model |
| MF716600 | Entomobr. | Entomobryidae | Seira paraibensis | Seira prarabiensis model |
| MF716601 | Entomobr. | Entomobryidae | Seira paulae | Pancrustacea model |
| MF716613 | Entomobr. | Entomobryidae | Seira potiguara | Pancrustacea model |
| MF716605 | Entomobr. | Entomobryidae | Seira ritae | Pancrustacea model * |
| MF716616 | Entomobr. | Entomobryidae | Seira sp. 1 | Pancrustacea model |
| MF716604 | Entomobr. | Entomobryidae | Seira sp. 2 | Pancrustacea model |
| MF716612 | Entomobr. | Entomobryidae | Seira sp. 3 | Seira sp. 3 model |
| MF716620 | Entomobr. | Entomobryidae | Seira tinguira | Pancrustacea model * |
| MK014212 | Entomobr. | Entomobryidae | Sinella curviseta | Pancrustacea model |
| MK423966 | Entomobr. | Tomoceridae | Tomocerus qinae | Pancrustacea model |
| MF716607 | Entomobr. | Paronellidae | Trogolaphysa sp. | Trogolaphysa model |
| MF716599 | Entomobr. | Entomobryidae | Tyrannoseira bicolorcornuta | Pancrustacea model |
| MF716611 | Entomobr. | Entomobryidae | Tyrannoseira bicolorcornuta | Pancrustacea model * |
| MF716610 | Entomobr. | Entomobryidae | Tyrannoseira raptora | Pancrustacea model |
| s7305 | Neeliple. | Neelidae | gen. sp. | Pancrustacea model § |
| s6565 | Neeliple. | Neelidae | Megalothorax minimus | Pancrustacea model § |
| MK431893 | Neeliple. | Neelidae | Neelides sp. | Pancrustacea model * |
| MH155200 | Neeliple. | Neelidae | Neelus murinus | Pancrustacea model |
| EU084034 | Podurom. | Neanuridae | Bilobella aurantiaca | Pancrustacea model |
| MN660050 | Podurom. | Brachystomellidae | Brachystomella parvula | Pancrustacea model * |
| MK409686 | Podurom. | Hypogastruridae | Ceratophysella communis | Pancrustacea model |
| s6480 | Podurom. | Onychiuridae | Deuteraphorura sp. | Tetrodontophora model § |
| NC010535 | Podurom. | Neanuridae | Friesea antarctica | Pancrustacea model |
| MT644085 | Podurom. | Neanuridae | Friesea gretae | Pancrustacea model |
| EU124719 | Podurom. | Neanuridae | Friesea propria | Pancrustacea model |
| s6241 | Podurom. | Hypogastruridae | gen. sp. | Pancrustacea model § |
| s6379 | Podurom. | Onychiuridae | gen. sp. | Tetrodontophora model § |
| s6532 | Podurom. | Onychiuridae | gen. sp. | Tetrodontophora model§ |
| s7124 | Podurom. | Tullbergiidae | gen. sp. | Pancrustacea model *§ |
| AY191995 | Podurom. | Hypogastruridae | Gomphiocephalus hodgsoni | Pancrustacea model |
| MK431897 | Podurom. | Tullbergiidae | Mesaphorura yosii | Pancrustacea model |
| MN661001 | Podurom. | Onychiuridae | Orthonychiurus folsomi | Tetrodontophora model |
| NC_006075 | Podurom. | Poduridae | Podura aquatica | Podura model * |
| MN660051 | Podurom. | Neanuridae | Pseudachorutes palmiensis | Pseudachorutes model |
| NC_002735 | Podurom. | Onychiuridae | Tetrodontophora bielanensis | Tetrodontophora model |
| MK423968 | Podurom. | Onychiuridae | Thalassaphorura encarpata | Tetrodontophora model |
| NC_006074 | Podurom. | Onychiuridae | Thalassaphorura orientalis | Tetrodontophora model * |
| s6543 | Podurom. | Onychiuridae | Thalassophorura sp. | Tetrodontophora model § |
| MK520870 | Podurom. | Tullbergiidae | Tullbergia bisetosa | Pancrustacea model |
| MW238520 | Podurom. | Tullbergiidae | Tullbergia mixta | Pancrustacea model * |
| MT547779 | Symphy. | Sminthuridae | Allacma fusca | Allacma model |
| KY618680 | Symphy. | Bourletiellidae | Bourletiella arvalis | Pancrustacea model |
| MG701393 | Symphy. | Dicyrtomidae | Dicyrtomina saundersi | Pancrustacea model |
| s6802 | Symphy. | Dicyrtomidae | gen. sp. | Pancrustacea model § |
| MK431899 | Symphy. | Sminthuridae | Lipothrix lubbocki | Lipothirx model |
| MK423965 | Symphy. | Dicyrtomidae | Ptenothrix huangshanensis | Pancrustacea model |
| MK423964 | Symphy. | Sminthurididae | Sminthurides bifidus | Pancrustacea model * |
| MK423969 | Symphy. | Katiannidae | Sminthurinus signatus | Sminthurinus model * |
| NC_010536 | Symphy. | Sminthuridae | Sminthurus viridis | Sminthurus model |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cucini, C.; Fanciulli, P.P.; Frati, F.; Convey, P.; Nardi, F.; Carapelli, A. Re-Evaluating the Internal Phylogenetic Relationships of Collembola by Means of Mitogenome Data. Genes 2021, 12, 44. https://doi.org/10.3390/genes12010044
Cucini C, Fanciulli PP, Frati F, Convey P, Nardi F, Carapelli A. Re-Evaluating the Internal Phylogenetic Relationships of Collembola by Means of Mitogenome Data. Genes. 2021; 12(1):44. https://doi.org/10.3390/genes12010044
Chicago/Turabian StyleCucini, Claudio, Pietro P. Fanciulli, Francesco Frati, Peter Convey, Francesco Nardi, and Antonio Carapelli. 2021. "Re-Evaluating the Internal Phylogenetic Relationships of Collembola by Means of Mitogenome Data" Genes 12, no. 1: 44. https://doi.org/10.3390/genes12010044
APA StyleCucini, C., Fanciulli, P. P., Frati, F., Convey, P., Nardi, F., & Carapelli, A. (2021). Re-Evaluating the Internal Phylogenetic Relationships of Collembola by Means of Mitogenome Data. Genes, 12(1), 44. https://doi.org/10.3390/genes12010044