Hereditary Optic Neuropathies: Induced Pluripotent Stem Cell-Based 2D/3D Approaches



Abstract
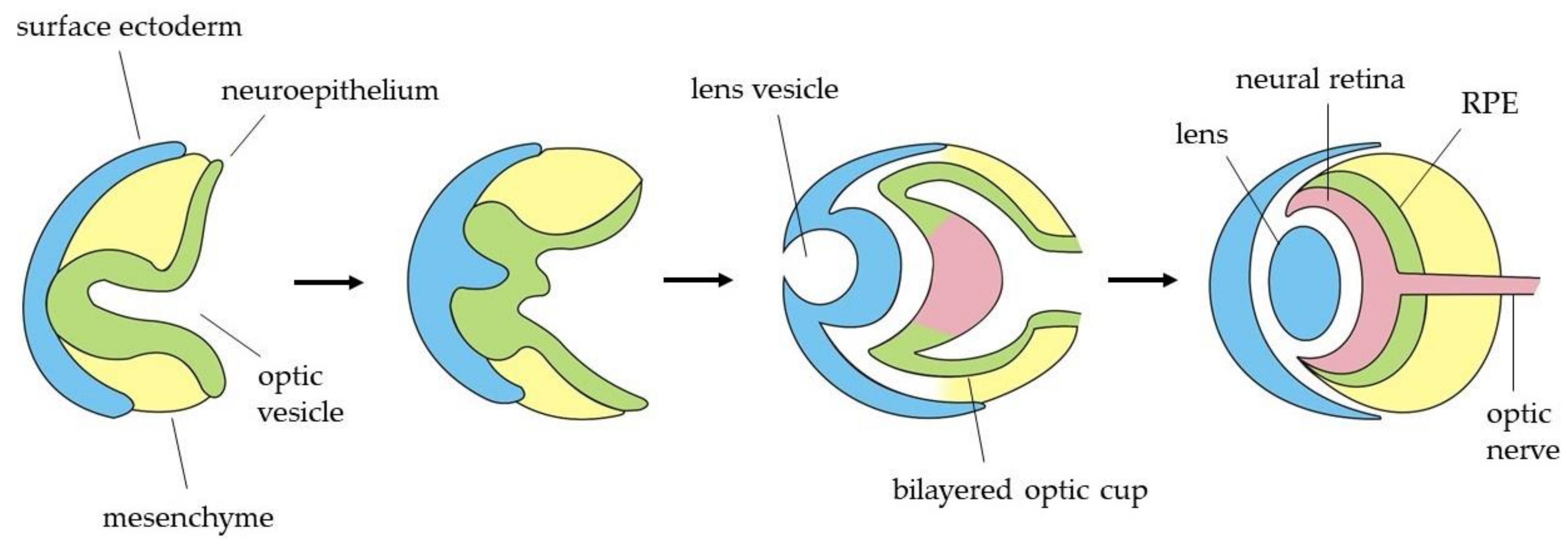
1. Introduction
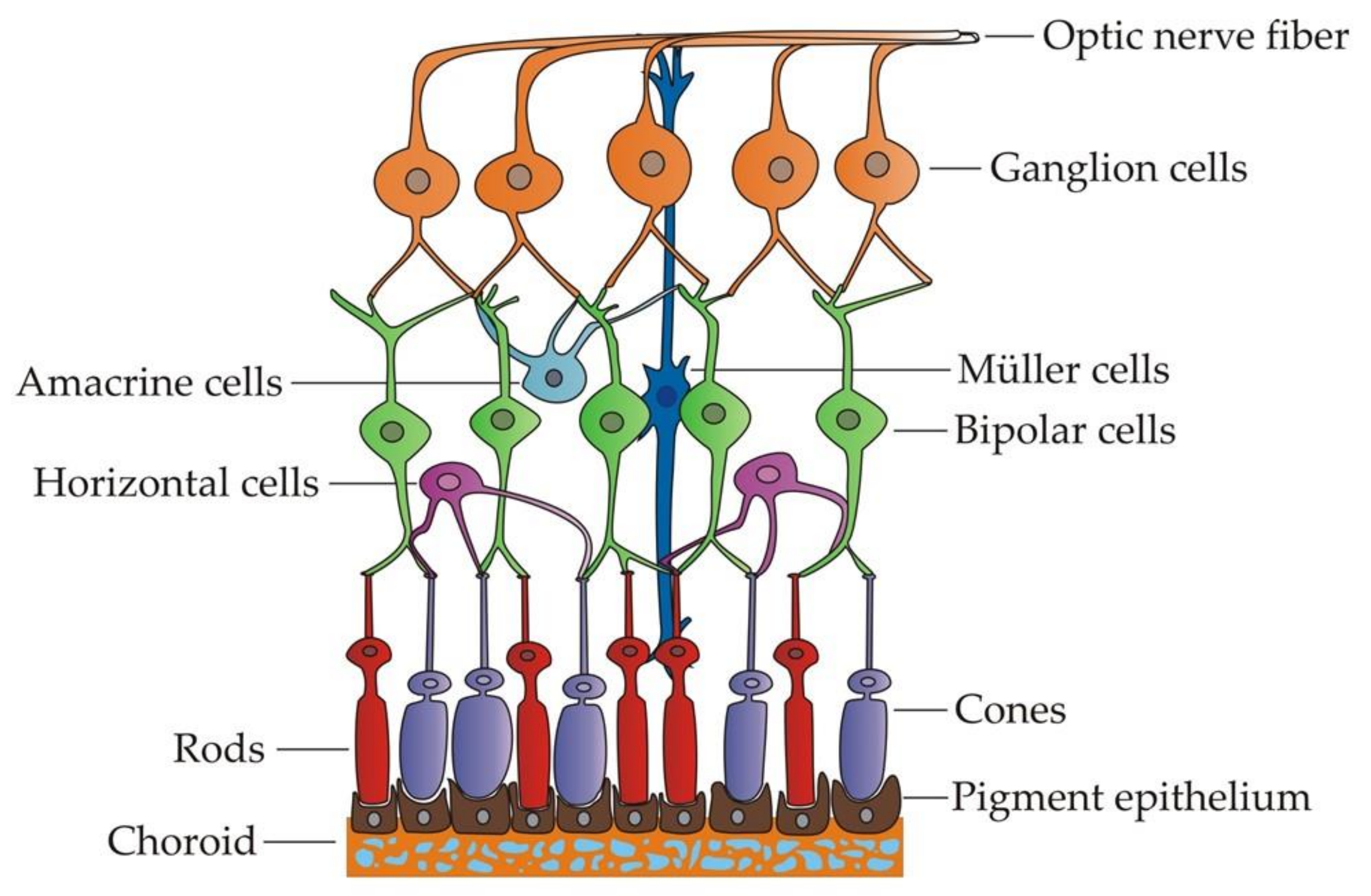
2. Vulnerability of RGCs in Inherited Optic Neuropathies
3. Applications of iPSC-Derived RGCs
3.1. 2D Approaches
3.2. 3D Approaches

{kind=link}
{kind=link}
| Authors | Adapted Protocol | Differentiation Approach | Differentiation Factors | Culture Matrix | Markers for RGC Identification | Isolation Method | Organoids Culture Time (Weeks) | Time for RGC Differentiation (Days) | Reference |
|---|---|---|---|---|---|---|---|---|---|
| Zhong et al., 2014 | Meyer et al., 2011 | EBs, neural rosettes, OV-like structures | N2, B27, FBS, taurine | Matrigel (growth factor reduced) | BRN3, HUC/D, TUJ1 | N/A | 25 | 35 | [116] |
| Reichman et al., 2014 | Original | Neural retina-like structures | N2, bFGF | - | BRN3A, BRN3B | N/A | 16 | 21 | [117] |
| Wiley et al., 2016 | Nakano et al., 2012 | EBs, optic cup-like structures | IWR1e, SAG, CHIR99021, N2 | - | HuC/D | N/A | 11 | 45 | [118] |
| Lowe et al., 2016 | Original | Cysts, cell sheets, retinal organoids | Matrigel, N2, B27, taurine, FBS | - | ISL1/2, TUJ1 | N/A | 22 | 54 | [119] |
| Kobayashi et al., 2018 | Kuwahara et al., 2015 | EBs, retinal organoids | BMP4, N2, CHIR99021, SU5402, FBS | - | ATOH7, BRN3B, ISL1, RBPMS, THY1 | Two-step immunopanning with anti-Thy1 antibodies | 17 | 40 | [121] |
| Mellough et al., 2019 | Original | EBs, retinal organoids | IGF-I, B27, N2 | - | OPN4, Smi32, RBPMS | N/A | 21 | 35 | [124] |
4. Perspectives and Concluding Remarks
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Yu-Wai-Man, P.; Griffiths, P.G.; Chinnery, P.F. Mitochondrial optic neuropathies—Disease mechanisms and therapeutic strategies. Prog. Retin. Eye Res. 2011, 30, 81–114. [Google Scholar] [CrossRef] [PubMed]
- Carelli, V.; Ross-Cisneros, F.N.; Sadun, A.A. Optic nerve degeneration and mitochondrial dysfunction: Genetic and acquired optic neuropathies. Neurochem. Int. 2002, 40, 573–584. [Google Scholar] [CrossRef]
- Bagli, E.; Zikou, A.K.; Agnantis, N.; Kitsos, G. Mitochondrial membrane dynamics and inherited optic neuropathies. In Vivo 2017, 31, 511–525. [Google Scholar] [PubMed]
- Lopez Sanchez, M.I.G.; Crowston, J.G.; Mackey, D.A.; Trounce, I.A. Emerging mitochondrial therapeutic targets in optic neuropathies. Pharmacol. Ther. 2016, 165, 132–152. [Google Scholar] [CrossRef]
- Yu-Wai-Man, P.; Votruba, M.; Moore, A.T.; Chinnery, P.F. Treatment strategies for inherited optic neuropathies: Past, present and future. Eye 2014, 28, 521–537. [Google Scholar] [CrossRef]
- Klopstock, T.; Metz, G.; Yu-Wai-Man, P.; Büchner, B.; Gallenmüller, C.; Bailie, M.; Nwali, N.; Griffiths, P.G.; Von Livonius, B.; Reznicek, L.; et al. Persistence of the treatment effect of idebenone in Leber’s hereditary optic neuropathy. Brain 2013, 136, e230. [Google Scholar] [CrossRef]
- Yu-Wai-Man, P.; Newman, N.J.; Carelli, V.; Moster, M.L.; Biousse, V.; Sadun, A.A.; Klopstock, T.; Vignal-Clermont, C.; Sergott, R.; Rudolph, G.; et al. Bilateral visual improvement with unilateral gene therapy injection for Leber hereditary optic neuropathy. Sci. Transl. Med. 2020, 12, eaaz7423. [Google Scholar] [CrossRef]
- Dahlmann-Noor, A.H.; Vijay, S.; Limb, G.A.; Khaw, P.T. Strategies for optic nerve rescue and regeneration in glaucoma and other optic neuropathies. Drug Discov. Today 2010, 15, 287–299. [Google Scholar] [CrossRef]
- Avior, Y.; Sagi, I.; Benvenisty, N. Pluripotent stem cells in disease modelling and drug discovery. Nat. Rev. Mol. Cell Biol. 2016, 17, 170–182. [Google Scholar] [CrossRef]
- Takahashi, K.; Yamanaka, S.; Zhang, Y.; Li, Y.; Feng, C.; Li, X.; Lin, L.; Guo, L.; Wang, H.; Liu, C.; et al. Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. Cell 2006, 126, 663–676. [Google Scholar] [CrossRef]
- Takahashi, K.; Tanabe, K.; Ohnuki, M.; Narita, M.; Ichisaka, T.; Tomoda, K.; Yamanaka, S. Induction of pluripotent stem cells from adult human fibroblasts by defined factors. Cell 2007, 131, 861–872. [Google Scholar] [CrossRef] [PubMed]
- Yu, J.; Vodyanik, M.A.; Smuga-Otto, K.; Antosiewicz-Bourget, J.; Frane, J.L.; Tian, S.; Nie, J.; Jonsdottir, G.A.; Ruotti, V.; Stewart, R.; et al. Induced pluripotent stem cell lines derived from human somatic cells. Science 2007, 318, 1917–1920. [Google Scholar] [CrossRef] [PubMed]
- Volarevic, V.; Markovic, B.S.; Gazdic, M.; Volarevic, A.; Jovicic, N.; Arsenijevic, N.; Armstrong, L.; Djonov, V.; Lako, M.; Stojkovic, M. Ethical and safety issues of stem cell-based therapy. Int. J. Med. Sci. 2018, 15, 36–45. [Google Scholar] [CrossRef] [PubMed]
- Shi, Y.; Inoue, H.; Wu, J.C.; Yamanaka, S. Induced pluripotent stem cell technology: A decade of progress. Nat. Rev. Drug Discov. 2017, 16, 115–130. [Google Scholar] [CrossRef] [PubMed]
- Sayed, N.; Liu, C.; Wu, J.C. Translation of human-induced pluripotent stem cells from clinical trial in a dish to precision medicine. J. Am. Coll. Cardiol. 2016, 67, 2161–2176. [Google Scholar] [CrossRef]
- Cooke, J.A.; Meyer, J.S. Human pluripotent stem cell-derived retinal ganglion cells: Applications for the study and treatment of optic neuropathies. Curr. Ophthalmol. Rep. 2015, 3, 200–206. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Hendriks, W.T.; Warren, C.R.; Cowan, C.A. Genome editing in human pluripotent stem cells: Approaches, pitfalls, and solutions. Cell Stem Cell 2016, 18, 53–65. [Google Scholar] [CrossRef]
- Ortuño-Costela, M.D.C.; Cerrada, V.; García-López, M.; Gallardo, M.E. The challenge of bringing iPSCs to the patient. Int. J. Mol. Sci. 2019, 20, 6305. [Google Scholar] [CrossRef]
- Boutin, M.E.; Hampton, C.; Quinn, R.; Ferrer, M.; Song, M.J. 3D engineering of ocular tissues for disease modeling and drug testing. Adv. Exp. Med. Biol. 2019, 1186, 171–193. [Google Scholar]
- Milea, D.; Amati-Bonneau, P.; Reynier, P.; Bonneau, D. Genetically determined optic neuropathies. Curr. Opin. Neurol. 2010, 23, 24–28. [Google Scholar] [CrossRef]
- Yu-Wai-Man, P.; Chinnery, P.F. Dominant optic atrophy: Novel OPA1 mutations and revised prevalence estimates. Ophthalmology 2013, 120, 1–2. [Google Scholar] [CrossRef] [PubMed]
- Gorman, G.S.; Schaefer, A.M.; Ng, Y.; Gomez, N.; Blakely, E.L.; Alston, C.L.; Feeney, C.; Horvath, R.; Yu-Wai-Man, P.; Chinnery, P.F.; et al. Prevalence of nuclear and mitochondrial DNA mutations related to adult mitochondrial disease. Ann. Neurol. 2015, 77, 753–759. [Google Scholar] [CrossRef] [PubMed]
- Fraser, J.A.; Biousse, V.; Newman, N.J. The neuro-ophthalmology of mitochondrial disease. Surv. Ophthalmol. 2010, 55, 299–334. [Google Scholar] [CrossRef]
- Carelli, V.; Ross-Cisneros, F.N.; Sadun, A.A. Mitochondrial dysfunction as a cause of optic neuropathies. Prog. Retin. Eye Res. 2004, 23, 53–89. [Google Scholar] [CrossRef] [PubMed]
- Yu-Wai-Man, P.; Griffiths, P.G.; Hudson, G.; Chinnery, P.F. Inherited mitochondrial optic neuropathies. J. Med. Genet. 2009, 46, 145–158. [Google Scholar] [CrossRef]
- Lin, C.S.; Sharpley, M.S.; Fan, W.; Waymire, K.G.; Sadun, A.A.; Carelli, V.; Ross-Cisneros, F.N.; Baciu, P.; Sung, E.; McManus, M.J.; et al. Mouse mtDNA mutant model of Leber hereditary optic neuropathy. Proc. Natl. Acad. Sci. USA 2012, 109, 20065–20070. [Google Scholar] [CrossRef]
- Caporali, L.; Maresca, A.; Capristo, M.; Del Dotto, V.; Tagliavini, F.; Valentino, M.L.; La Morgia, C.; Carelli, V. Incomplete penetrance in mitochondrial optic neuropathies. Mitochondrion 2017, 36, 130–137. [Google Scholar] [CrossRef]
- Hudson, G.; Keers, S.; Man, P.Y.W.; Griffiths, P.; Huoponen, K.; Savontaus, M.L.; Nikoskelainen, E.; Zeviani, M.; Carrara, F.; Horvath, R.; et al. Identification of an X-chromosomal locus and haplotype modulating the phenotype of a mitochondrial DNA disorder. Am. J. Hum. Genet. 2005, 77, 1086–1091. [Google Scholar] [CrossRef]
- Yang, T.-C.; Yarmishyn, A.A.; Yang, Y.-P.; Lu, P.-C.; Chou, S.-J.; Wang, M.-L.; Lin, T.-C.; Hwang, D.-K.; Chou, Y.-B.; Chen, S.-J.; et al. Mitochondrial transport mediates survival of retinal ganglion cells in affected LHON patients. Hum. Mol. Genet. 2020, 29, 1454–1464. [Google Scholar] [CrossRef]
- Istikharah, R.; Tun, A.W.; Kaewsutthi, S.; Aryal, P.; Kunhapan, B.; Katanyoo, W.; Chuenkongkaew, W.; Lertrit, P. Identification of the variants in PARL, the nuclear modifier gene, responsible for the expression of LHON patients in Thailand. Exp. Eye Res. 2013, 116, 55–57. [Google Scholar] [CrossRef]
- Jiang, P.; Jin, X.; Peng, Y.; Wang, M.; Liu, H.; Liu, X.; Zhang, Z.; Ji, Y.; Zhang, J.; Liang, M.; et al. The exome sequencing identified the mutation in YARS2 encoding the mitochondrial tyrosyl-tRNA synthetase as a nuclear modifier for the phenotypic manifestation of Leber’s hereditary optic neuropathy-associated mitochondrial DNA mutation. Hum. Mol. Genet. 2016, 25, 584–596. [Google Scholar] [CrossRef] [PubMed]
- Newman, N.J. Treatment of hereditary optic neuropathies. Nat. Rev. Neurol. 2012, 8, 545–556. [Google Scholar] [CrossRef] [PubMed]
- Pfeffer, G.; Burke, A.; Yu-Wai-Man, P.; Compston, A.; Chinnery, P.F. Clinical features of MS associated with Leber hereditary optic neuropathy mtDNA mutations. Neurology 2013, 81, 2073–2081. [Google Scholar] [CrossRef] [PubMed]
- Delettre, C.; Lenaers, G.; Pelloquin, L.; Belenguer, P.; Hamel, C.P. OPA1 (Kjer type) dominant optic atrophy: A novel mitochondrial disease. Mol. Genet. Metab. 2002, 75, 97–107. [Google Scholar] [CrossRef]
- Chun, B.Y.; Rizzo, J.F. Dominant optic atrophy and Leber’s hereditary optic neuropathy: Update on clinical features and current therapeutic approaches. Semin. Pediatr. Neurol. 2017, 24, 129–134. [Google Scholar] [CrossRef]
- Alexander, C.; Votruba, M.; Pesch, U.E.A.; Thiselton, D.L.; Mayer, S.; Moore, A.; Rodriguez, M.; Kellner, U.; Leo-Kottler, B.; Auburger, G.; et al. OPA1, encoding a dynamin-related GTPase, is mutated in autosomal dominant optic atrophy linked to chromosome 3q28. Nat. Genet. 2000, 26, 211–215. [Google Scholar] [CrossRef]
- Delettre, C.; Lenaers, G.; Griffoin, J.M.; Gigarel, N.; Lorenzo, C.; Belenguer, P.; Pelloquin, L.; Grosgeorge, J.; Turc-Carel, C.; Perret, E.; et al. Nuclear gene OPA1, encoding a mitochondrial dynamin-related protein, is mutated in dominant optic atrophy. Nat. Genet. 2000, 26, 207–210. [Google Scholar] [CrossRef]
- Chun, B.Y.; Rizzo, J.F. Dominant optic atrophy: Updates on the pathophysiology and clinical manifestations of the optic atrophy 1 mutation. Curr. Opin. Ophthalmol. 2016, 27, 475–480. [Google Scholar] [CrossRef]
- Le Roux, B.; Lenaers, G.; Zanlonghi, X.; Amati-Bonneau, P.; Chabrun, F.; Foulonneau, T.; Caignard, A.; Leruez, S.; Gohier, P.; Procaccio, V.; et al. OPA1: 516 unique variants and 831 patients registered in an updated centralized Variome database. Orphanet J. Rare Dis. 2019, 14, 214. [Google Scholar] [CrossRef]
- Lenaers, G.; Hamel, C.; Delettre, C.; Amati-Bonneau, P.; Procaccio, V.; Bonneau, D.; Reynier, P.; Milea, D. Dominant optic atrophy. Orphanet J. Rare Dis. 2012, 7, 46. [Google Scholar] [CrossRef]
- Hanein, S.; Perrault, I.; Roche, O.; Gerber, S.; Khadom, N.; Rio, M.; Boddaert, N.; Jean-Pierre, M.; Brahimi, N.; Serre, V.; et al. TMEM126A, encoding a mitochondrial protein, is mutated in autosomal-recessive nonsyndromic optic atrophy. Am. J. Hum. Genet. 2009, 84, 493–498. [Google Scholar] [CrossRef] [PubMed]
- Yu-Wai-Man, P.; Griffiths, P.G.; Gorman, G.S.; Lourenco, C.M.; Wright, A.F.; Auer-Grumbach, M.; Toscano, A.; Musumeci, O.; Valentino, M.L.; Caporali, L.; et al. Multi-system neurological disease is common in patients with OPA1 mutations. Brain 2010, 133, 771–786. [Google Scholar] [CrossRef] [PubMed]
- Bonneau, D.; Colin, E.; Oca, F.; Ferre, M.; Chevrollier, A.; Guéguen, N.; Desquiret-Dumas, V.; N’Guyen, S.; Barth, M.; Zanlonghi, X.; et al. Early-onset Behr syndrome due to compound heterozygous mutations in OPA1. Brain 2014, 137, e301. [Google Scholar] [CrossRef] [PubMed]
- Yahalom, G.; Anikster, Y.; Huna-Baron, R.; Hoffmann, C.; Blumkin, L.; Lev, D.; Tsabari, R.; Nitsan, Z.; Lerman, S.F.; Ben-Zeev, B.; et al. Costeff syndrome: Clinical features and natural history. J. Neurol. 2014, 261, 2275–2282. [Google Scholar] [CrossRef] [PubMed]
- Carelli, V.; Schimpf, S.; Fuhrmann, N.; Valentino, M.L.; Zanna, C.; Iommarini, L.; Papke, M.; Schaich, S.; Tippmann, S.; Baumann, B.; et al. A clinically complex form of dominant optic atrophy (OPA8) maps on chromosome 16. Hum. Mol. Genet. 2011, 20, 1893–1905. [Google Scholar] [CrossRef] [PubMed]
- Rigoli, L.; Bramanti, P.; Di Bella, C.; De Luca, F. Genetic and clinical aspects of Wolfram syndrome 1, a severe neurodegenerative disease. Pediatr. Res. 2018, 83, 921–929. [Google Scholar] [CrossRef]
- Metodiev, M.D.; Gerber, S.; Hubert, L.; Delahodde, A.; Chretien, D.; Gérard, X.; Amati-Bonneau, P.; Giacomotto, M.C.; Boddaert, N.; Kaminska, A.; et al. Mutations in the tricarboxylic acid cycle enzyme, aconitase 2, cause either isolated or syndromic optic neuropathy with encephalopathy and cerebellar atrophy. J. Med. Genet. 2014, 51, 834–838. [Google Scholar] [CrossRef]
- Neuhann, T.; Rautenstrauss, B. Genetic and phenotypic variability of optic neuropathies. Expert Rev. Neurother. 2013, 13, 357–367. [Google Scholar] [CrossRef]
- Weinreb, R.N.; Aung, T.; Medeiros, F.A. The pathophysiology and treatment of glaucoma: A review. J. Am. Med. Assoc. 2014, 311, 1901–1911. [Google Scholar] [CrossRef]
- Blumberg, D.; Skaat, A.; Liebmann, J.M. Emerging risk factors for glaucoma onset and progression. Prog. Brain Res. 2015, 221, 81–101. [Google Scholar]
- Wang, R.; Wiggs, J.L. Common and rare genetic risk factors for glaucoma. Cold Spring Harb. Perspect. Med. 2014, 4, a017244. [Google Scholar] [CrossRef] [PubMed]
- Zhou, T.; Souzeau, E.; Siggs, O.M.; Landers, J.; Mills, R.; Goldberg, I.; Healey, P.R.; Graham, S.; Hewitt, A.W.; Mackey, D.A.; et al. Contribution of mutations in known mendelian glaucoma genes to advanced early-onset primary open-angle glaucoma. Investig. Ophthalmol. Vis. Sci. 2017, 58, 1537–1544. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.W.; Sun, P.; Chen, Y.; Jiang, L.P.; Wu, H.P.; Zhang, W.; Gao, F. Research progress on human genes involved in the pathogenesis of glaucoma. Mol. Med. Rep. 2018, 18, 656–674. [Google Scholar] [CrossRef] [PubMed]
- Fuse, N. Genetic bases for glaucoma. J. Exp. Med. 2010, 221, 1–10. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Yu, D.Y.; Cringle, S.J.; Balaratnasingam, C.; Morgan, W.H.; Yu, P.K.; Su, E.N. Retinal ganglion cells: Energetics, compartmentation, axonal transport, cytoskeletons and vulnerability. Prog. Retin. Eye Res. 2013, 36, 217–246. [Google Scholar] [CrossRef] [PubMed]
- Chamberlain, K.A.; Sheng, Z.H. Mechanisms for the maintenance and regulation of axonal energy supply. J. Neurosci. Res. 2019, 97, 897–913. [Google Scholar] [CrossRef]
- Chhetri, J.; Gueven, N. Targeting mitochondrial function to protect against vision loss. Expert Opin. Ther. Targets 2016, 20, 721–736. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Dong, J.; Cull, G.; Fortune, B.; Cioffi, G.A. Varicosities of intraretinal ganglion cell axons in human and nonhuman primates. Investig. Ophthalmol. Vis. Sci. 2003, 44, 2–9. [Google Scholar] [CrossRef]
- Man, C.Y.W.; Chinnery, P.F.; Griffiths, P.G. Optic neuropathies—Importance of spatial distribution of mitochondria as well as function. Med. Hypotheses 2005, 65, 1038–1042. [Google Scholar] [CrossRef]
- Yu-Wai-Man, P.; Votruba, M.; Burté, F.; La Morgia, C.; Barboni, P.; Carelli, V. A neurodegenerative perspective on mitochondrial optic neuropathies. Acta Neuropathol. 2016, 132, 789–806. [Google Scholar] [CrossRef]
- Tezel, G.; Yang, X.; Cai, J. Proteomic identification of oxidatively modified retinal proteins in a chronic pressure-induced rat model of glaucoma. Investig. Ophthalmol. Vis. Sci. 2005, 46, 3177–3187. [Google Scholar] [CrossRef] [PubMed]
- Yu-Wai-Man, P.; Newman, N.J. Inherited eye-related disorders due to mitochondrial dysfunction. Hum. Mol. Genet. 2017, 26, 12–20. [Google Scholar] [CrossRef] [PubMed]
- Mandal, A.; Drerup, C.M. Axonal transport and mitochondrial function in neurons. Front. Cell. Neurosci. 2019, 13, 373. [Google Scholar] [CrossRef] [PubMed]
- Ito, Y.A.; Di Polo, A. Mitochondrial dynamics, transport, and quality control: A bottleneck for retinal ganglion cell viability in optic neuropathies. Mitochondrion 2017, 36, 186–192. [Google Scholar] [CrossRef]
- Lu, A.Q.; Barnstable, C.J. Pluripotent stem cells as models of retina development. Mol. Neurobiol. 2019, 56, 6056–6070. [Google Scholar] [CrossRef]
- Sinn, R.; Wittbrodt, J. An eye on eye development. Mech. Dev. 2013, 130, 347–358. [Google Scholar] [CrossRef]
- Giger, F.A.; Houart, C. The birth of the eye vesicle: When fate decision equals morphogenesis. Front. Neurosci. 2018, 12, 87. [Google Scholar] [CrossRef]
- Heavner, W.; Pevny, L. Eye development and retinogenesis. Cold Spring Harb. Perspect. Biol. 2012, 4, a008391. [Google Scholar] [CrossRef]
- Lupo, G.; Novorol, C.; Smith, J.R.; Vallier, L.; Miranda, E.; Alexander, M.; Biagioni, S.; Pedersen, R.A.; Harris, W.A. Multiple roles of Activin/Nodal, bone morphogenetic protein, fibroblast growth factor and Wnt/β-catenin signalling in the anterior neural patterning of adherent human embryonic stem cell cultures. Open Biol. 2013, 3, 120167. [Google Scholar] [CrossRef]
- Fuhrmann, S. Current Topics in Developmental Biology; Elsevier Inc.: Salt Lake City, UT, USA, 2010; pp. 61–84. [Google Scholar]
- Horsford, D.J.; Nguyen, M.T.T.; Sellar, G.C.; Kothary, R.; Arnheiter, H.; McInnes, R.R. Chx10 repression of Mitf is required for the maintenance of mammalian neuroretinal identity. Development 2005, 132, 177–187. [Google Scholar] [CrossRef]
- Miesfeld, J.B.; Brown, N.L. Current Topics in Developmental Biology, 1st ed.; Elsevier Inc.: Salt Lake City, UT, USA, 2019; pp. 351–393. [Google Scholar]
- Stenkamp, D.L. Progress in Molecular Biology and Translational Science, 1st ed.; Elsevier Inc.: Salt Lake City, UT, USA, 2015; pp. 397–414. [Google Scholar]
- Wang, S.W.; Kim, B.S.; Ding, K.; Wang, H.; Sun, D.; Johnson, R.L.; Klein, W.H.; Gan, L. Requirement for math5 in the development of retinal ganglion cells. Genes Dev. 2001, 15, 24–29. [Google Scholar] [CrossRef] [PubMed]
- Pan, L.; Deng, M.; Xie, X.; Gan, L. ISL1 and BRN3B co-regulate the differentiation of murine retinal ganglion cells. Development 2008, 135, 1981–1990. [Google Scholar] [CrossRef] [PubMed]
- Nelson, B.R.; Gumuscu, B.; Hartman, B.H.; Reh, T.A. Notch activity is downregulated just prior to retinal ganglion cell differentiation. Dev. Neurosci. 2006, 28, 128–141. [Google Scholar] [CrossRef] [PubMed]
- Guo, N.-N.; Liu, L.-P.; Zheng, Y.-W.; Li, Y.-M. Inducing human induced pluripotent stem cell differentiation through embryoid bodies: A practical and stable approach. World J. Stem Cells 2020, 12, 25–34. [Google Scholar] [CrossRef]
- Elkabetz, Y.; Panagiotakos, G.; Al Shamy, G.; Socci, N.D.; Tabar, V.; Studer, L. Human ES cell-derived neural rosettes reveal a functionally distinct early neural stem cell stage. Genes Dev. 2008, 22, 152–165. [Google Scholar] [CrossRef]
- Wilson, P.G.; Stice, S.S. Development and differentiation of neural rosettes derived from human embryonic stem cells. Stem Cell Rev. 2006, 2, 67–77. [Google Scholar] [CrossRef]
- Jensen, J.B.; Parmar, M. Strengths and limitations of the neurosphere culture system. Mol. Neurobiol. 2006, 34, 153–154. [Google Scholar] [CrossRef]
- Lamba, D.A.; Karl, M.O.; Ware, C.B.; Reh, T.A. Efficient generation of retinal progenitor cells from human embryonic stem cells. Proc. Natl. Acad. Sci. USA 2006, 103, 12769–12774. [Google Scholar] [CrossRef]
- Tucker, B.A. Use of a synthetic xeno-free culture substrate for induced pluripotent stem cell induction and retinal differentiation. Stem Cells 2013, 2, 16–24. [Google Scholar] [CrossRef]
- Riazifar, H.; Jia, Y.; Chen, J.; Lynch, G.; Huang, T. Chemically induced specification of retinal ganglion cells from human embryonic and induced pluripotent stem cells. Stem Cells Transl. Med. 2014, 3, 424–432. [Google Scholar] [CrossRef]
- Tanaka, T.; Yokoi, T.; Tamalu, F.; Watanabe, S.-I.; Nishina, S.; Azuma, N. Generation of retinal ganglion cells with functional axons from human induced pluripotent stem cells. Sci. Rep. 2015, 5, 8344. [Google Scholar] [CrossRef] [PubMed]
- Nakano, T.; Ando, S.; Takata, N.; Kawada, M.; Muguruma, K.; Sekiguchi, K.; Saito, K.; Yonemura, S.; Eiraku, M.; Sasai, Y. Self-formation of optic cups and storable stratified neural retina from human ESCs. Cell Stem Cell 2012, 10, 771–785. [Google Scholar] [CrossRef] [PubMed]
- Ohlemacher, S.K.; Sridhar, A.; Xiao, Y.; Hochstetler, A.E.; Sarfarazi, M.; Cummins, T.R.; Meyer, J.S. Stepwise differentiation of retinal ganglion cells from human pluripotent stem cells enables analysis of glaucomatous neurodegeneration. Stem Cells 2016, 34, 1553–1562. [Google Scholar] [CrossRef] [PubMed]
- Teotia, P.; Chopra, D.A.; Dravid, S.M.; Van Hook, M.J.; Qiu, F.; Morrison, J.; Rizzino, A.; Ahmad, I. Generation of functional human retinal ganglion cells with target specificity from pluripotent stem cells by chemically defined recapitulation of developmental mechanism. Stem Cells 2017, 35, 572–585. [Google Scholar] [CrossRef]
- Lee, J.; Choi, S.; Kim, Y.; Jun, I.; Sung, J.J.; Dongjin, R.; Kim, Y.I.; Cho, M.S.; Byeon, S.H.; Kim, D.; et al. Defined conditions for differentiation of functional retinal ganglion cells from human pluripotent stem cells. Investig. Ophthalmol. Vis. Sci. 2018, 59, 3531–3542. [Google Scholar] [CrossRef]
- Chavali, V.R.M.; Haider, N.; Rathi, S.; Vrathasha, V.; Alapati, T.; He, J.; Gill, K.; Nikonov, R.; Duong, T.T.; McDougald, D.S.; et al. Dual SMAD inhibition and Wnt inhibition enable efficient and reproducible differentiations of induced pluripotent stem cells into retinal ganglion cells. Sci. Rep. 2020, 10, 11828. [Google Scholar] [CrossRef]
- Gill, K.P.; Hung, S.S.C.; Sharov, A.; Lo, C.Y.; Needham, K.; Lidgerwood, G.E.; Jackson, S.; Crombie, D.E.; Nayagam, B.A.; Cook, A.L.; et al. Enriched retinal ganglion cells derived from human embryonic stem cells. Sci. Rep. 2016, 6, 30552. [Google Scholar] [CrossRef]
- Sluch, V.M.; Davis, C.H.O.; Ranganathan, V.; Kerr, J.M.; Krick, K.; Martin, R.; Berlinicke, C.A.; Marsh-Armstrong, N.; Diamond, J.S.; Mao, H.Q.; et al. Differentiation of human ESCs to retinal ganglion cells using a CRISPR engineered reporter cell line. Sci. Rep. 2015, 5, 16595. [Google Scholar] [CrossRef]
- Hsu, C.C.; Chien, K.H.; Yarmishyn, A.A.; Buddhakosai, W.; Wu, W.J.; Lin, T.C.; Chiou, S.H.; Chen, J.T.; Peng, C.H.; Hwang, D.K.; et al. Modulation of osmotic stress-induced TRPV1 expression rescues human iPSC-derived retinal ganglion cells through PKA. Stem Cell Res. Ther. 2019, 10, 284. [Google Scholar] [CrossRef]
- Lidgerwood, G.E.; Hewitt, A.W.; Pébay, A. Human pluripotent stem cells for the modelling of diseases of the retina and optic nerve: Toward a retina in a dish. Curr. Opin. Pharmacol. 2019, 48, 114–119. [Google Scholar] [CrossRef]
- Khan, S.; Hung, S.S.C.; Wong, R.C.B. The use of induced pluripotent stem cells for studying and treating optic neuropathies. Curr. Opin. Organ Transplant. 2016, 21, 484–489. [Google Scholar] [CrossRef] [PubMed]
- Tucker, B.A.; Solivan-timpe, F.; Roos, B.R.; Anfinson, K.R.; Robin, L.; Wiley, L.A.; Mullins, R.F.; Fingert, J.H. Duplication of TBK1 stimulates autophagy in iPSC-derived retinal cells from a patient with normal tension glaucoma. J. Stem Cell Res. Ther. 2014, 3, 161. [Google Scholar] [CrossRef] [PubMed]
- Galera-Monge, T.; Zurita-Díaz, F.; Moreno-Izquierdo, A.; Fraga, M.F.; Fernández, A.F.; Ayuso, C.; Garesse, R.; Gallardo, M.E. Generation of a human iPSC line from a patient with an optic atrophy “plus” phenotype due to a mutation in the OPA1 gene. Stem Cell Res. 2016, 16, 673–676. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Zurita-Díaz, F.; Galera-Monge, T.; Moreno-Izquierdo, A.; Corton, M.; Ayuso, C.; Garesse, R.; Gallardo, M.E. Establishment of a human DOA “plus” iPSC line, IISHDOi003-A, with the mutation in the OPA1 gene: C.1635C>A; p.Ser545Arg. Stem Cell Res. 2017, 24, 81–84. [Google Scholar] [CrossRef]
- Chen, J.; Riazifar, H.; Guan, M.X.; Huang, T. Modeling autosomal dominant optic atrophy using induced pluripotent stem cells and identifying potential therapeutic targets. Stem Cell Res. Ther. 2016, 7, 2. [Google Scholar] [CrossRef]
- Cerrada, V.; García-López, M.; Moreno-Izquierdo, A.; Villaverde, C.; Zurita, O.; Martin-Merida, M.I.; Arenas, J.; Ayuso, C.; Gallardo, M.E. Derivation of a human DOA iPSC line, IISHDOi006-A, with a mutation in the ACO2 gene: C.1999G>A; p.Glu667Lys. Stem Cell Res. 2019, 40, 101566. [Google Scholar] [CrossRef]
- Peron, C.; Mauceri, R.; Cabassi, T.; Segnali, A.; Maresca, A.; Iannielli, A.; Rizzo, A.; Sciacca, F.L.; Broccoli, V.; Carelli, V.; et al. Generation of a human iPSC line, FINCBi001-A, carrying a homoplasmic m.G3460A mutation in MT-ND1 associated with Leber ’s Hereditary optic Neuropathy (LHON). Stem Cell Res. 2020, 48, 101939. [Google Scholar] [CrossRef]
- Hung, S.S.C.; Van Bergen, N.J.; Jackson, S.; Liang, H.; Mackey, D.A.; Hernández, D.; Lim, S.Y.; Hewitt, A.W.; Trounce, I.; Pébay, A.; et al. Study of mitochondrial respiratory defects on reprogramming to human induced pluripotent stem cells. Aging 2016, 8, 945–957. [Google Scholar] [CrossRef]
- Wong, R.C.B.; Lim, S.Y.; Hung, S.S.C.; Jackson, S.; Khan, S.; Van Bergen, N.J.; De Smit, E.; Liang, H.H.; Kearns, L.S.; Clarke, L.; et al. Mitochondrial replacement in an iPSC model of Leber’s hereditary optic neuropathy. Aging 2017, 9, 1341–1350. [Google Scholar] [CrossRef]
- Wu, Y.; Wang, A.; Chen, Y.; Yarmishyn, A.A. Bioactivity and gene expression profiles of hiPSC-generated retinal ganglion cells in MT-ND4 mutated Leber’s hereditary optic neuropathy. Exp. Cell Res. 2018, 363, 299–309. [Google Scholar] [CrossRef]
- Lu, H.; Yang, Y.; Chen, Y.; Wu, Y.; Wang, C.; Tsai, F.; Hwang, D.; Lin, T.; Chen, S.; Wang, A.; et al. Generation of patient-specific induced pluripotent stem cells from Leber’s hereditary optic neuropathy. Stem Cell Res. 2018, 28, 56–60. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Nguyen, P.; Nguyen, N.; Lin, T.; Yarmishyn, A.A. Glutamate stimulation dysregulates AMPA receptors-induced signal transduction pathway in Leber’s inherited optic neuropathy patient-specific hiPSC-derived retinal ganglion cells. Cells 2019, 8, 625. [Google Scholar] [CrossRef] [PubMed]
- Hockemeyer, D.; Jaenisch, R. Induced pluripotent stem cells meet genome editing. Cell Stem Cell 2016, 18, 573–586. [Google Scholar] [CrossRef] [PubMed]
- Jehuda, R.B.; Shemer, Y.; Binah, O. Genome editing in induced pluripotent stem cells using CRISPR/Cas9. Stem Cell Rev. Rep. 2018, 14, 323–336. [Google Scholar] [CrossRef]
- Oswald, J.; Baranov, P. Regenerative medicine in the retina: From stem cells to cell replacement therapy. Ther. Adv. Ophthalmol. 2018, 10, 1–21. [Google Scholar] [CrossRef]
- Satarian, L.; Javan, M.; Kiani, S.; Hajikaram, M.; Mirnajafi-zadeh, J. Engrafted human induced pluripotent stem cell-derived anterior specified neural progenitors protect the rat crushed optic nerve. PLoS ONE 2013, 8, e71855. [Google Scholar] [CrossRef]
- Parameswaran, S. Continuous non-cell autonomous reprogramming to generate retinal ganglion cells for glaucomatous neuropathy. Stem Cells 2015, 33, 1743–1758. [Google Scholar] [CrossRef][Green Version]
- Crair, M.C.; Mason, X.C.A. Reconnecting eye to brain. Dev. Dyn. 2016, 36, 118–128. [Google Scholar] [CrossRef]
- Fligor, C.M.; Langer, K.B.; Sridhar, A.; Ren, Y.; Shields, P.K.; Edler, M.C.; Ohlemacher, S.K.; Sluch, V.M.; Zack, D.J.; Zhang, C.; et al. Three-Dimensional retinal organoids facilitate the investigation of retinal ganglion cell development, organization and neurite outgrowth from human pluripotent stem cells. Sci. Rep. 2018, 8, 14520. [Google Scholar] [CrossRef]
- Eglen, R.M.; Reisine, T. Human iPS Cell-derived patient tissues and 3D Cell culture part 2: Spheroids, organoids, and disease modeling. SLAS Technol. 2019, 24, 18–27. [Google Scholar] [CrossRef]
- Meyer, J.S.; Howden, S.E.; Wallace, K.A.; Verhoeven, A.D.; Wright, L.S.; Capowski, E.E.; Pinilla, I.; Martin, J.M.; Tian, S.; Stewart, R.; et al. Optic vesicle-like structures derived from human pluripotent stem cells facilitate a customized approach to retinal disease treatment. Stem Cells 2011, 29, 1206–1218. [Google Scholar] [CrossRef] [PubMed]
- Ohlemacher, S.K.; Iglesias, C.L.; Sridhar, A.; Gamm, D.M.; Meyer, J.S. Generation of highly enriched populations of optic vesicle-like retinal cells from human pluripotent stem cells. Curr. Protoc. Stem Cell Biol. 2015, 32, 1H8.1–1H8.20. [Google Scholar] [CrossRef] [PubMed]
- Zhong, X.; Gutierrez, C.; Xue, T.; Hampton, C.; Vergara, M.N.; Cao, L.H.; Peters, A.; Park, T.S.; Zambidis, E.T.; Meyer, J.S.; et al. Generation of three-dimensional retinal tissue with functional photoreceptors from human iPSCs. Nat. Commun. 2014, 5, 4047. [Google Scholar] [CrossRef]
- Reichman, S.; Terray, A.; Slembrouck, A.; Nanteau, C.; Orieux, G.; Habeler, W.; Nandrot, E.F.; Sahel, J.-A.; Monville, C.; Goureau, O. From confluent human iPS cells to self-forming neural retina and retinal pigmented epithelium. Proc. Natl. Acad. Sci. USA 2014, 111, 8518–8523. [Google Scholar] [CrossRef] [PubMed]
- Wiley, L.A.; Burnight, E.R.; Deluca, A.P.; Anfinson, K.R.; Cranston, C.M.; Kaalberg, E.E.; Penticoff, J.A.; Affatigato, L.M.; Mullins, R.F.; Stone, E.M.; et al. cGMP production of patient-specific iPSCs and photoreceptor precursor cells to treat retinal degenerative blindness. Sci. Rep. 2016, 6, 30742. [Google Scholar] [CrossRef]
- Lowe, A.; Harris, R.; Bhansali, P.; Cvekl, A.; Liu, W. Intercellular adhesion-dependent cell survival and rock-regulated actomyosin-driven forces mediate self-formation of a retinal organoid. Stem Cell Rep. 2016, 6, 743–756. [Google Scholar] [CrossRef]
- Kuwahara, A.; Ozone, C.; Nakano, T.; Saito, K.; Eiraku, M.; Sasai, Y. Generation of a ciliary margin-like stem cell niche from self-organizing human retinal tissue. Nat. Commun. 2015, 6, 6286. [Google Scholar] [CrossRef]
- Kobayashi, W.; Onishi, A.; Tu, H.-Y.; Takihara, Y.; Matsumura, M.; Tsujimoto, K.; Inatani, M.; Nakazawa, T.; Takahashi, M. Culture systems of dissociated mouse and human pluripotent stem cell–derived retinal ganglion cells purified by two-step immunopanning. Investig. Ophthalmol. Vis. Sci. 2018, 59, 776–787. [Google Scholar] [CrossRef]
- Deng, W.L.; Gao, M.L.; Lei, X.L.; Lv, J.N.; Zhao, H.; He, K.W.; Xia, X.X.; Li, L.Y.; Chen, Y.C.; Li, Y.P.; et al. Gene correction reverses ciliopathy and photoreceptor loss in iPSC-derived retinal organoids from retinitis pigmentosa patients. Stem Cell Rep. 2018, 10, 1267–1281. [Google Scholar] [CrossRef]
- Hallam, D.; Hilgen, G.; Dorgau, B.; Zhu, L.; Yu, M.; Bojic, S.; Hewitt, P.; Schmitt, M.; Uteng, M.; Kustermann, S.; et al. Human-induced pluripotent stem cells generate light responsive retinal organoids with variable and nutrient-dependent efficiency. Stem Cells 2018, 36, 1535–1551. [Google Scholar] [CrossRef]
- Mellough, C.B.; Collin, J.; Queen, R.; Hilgen, G.; Dorgau, B.; Zerti, D.; Felemban, M.; White, K.; Sernagor, E.; Lako, M. Systematic comparison of retinal organoid differentiation from human pluripotent stem cells reveals stage specific, cell line, and methodological differences. Stem Cells Transl. Med. 2019, 8, 694–706. [Google Scholar] [CrossRef] [PubMed]
- Chichagova, V.; Dorgau, B.; Felemban, M.; Georgiou, M.; Armstrong, L.; Lako, M. Differentiation of retinal organoids from human pluripotent stem cells. Curr. Protoc. Stem Cell Biol. 2019, 50, e95. [Google Scholar] [CrossRef] [PubMed]
- Artero Castro, A.; Rodríguez Jimenez, F.J.; Jendelova, P.; Erceg, S. Deciphering retinal diseases through the generation of three dimensional stem cell-derived organoids: Concise Review. Stem Cells 2019, 37, 1496–1504. [Google Scholar] [CrossRef] [PubMed]
- Capowski, E.E.; Samimi, K.; Mayerl, S.J.; Phillips, M.J.; Pinilla, I.; Howden, S.E.; Saha, J.; Jansen, A.D.; Edwards, K.L.; Jager, L.D.; et al. Reproducibility and staging of 3D human retinal organoids across multiple pluripotent stem cell lines. Development 2019, 146, dev171686. [Google Scholar] [CrossRef] [PubMed]
- Ovando-Roche, P.; West, E.L.; Branch, M.J.; Sampson, R.D.; Fernando, M.; Munro, P.; Georgiadis, A.; Rizzi, M.; Kloc, M.; Naeem, A.; et al. Use of bioreactors for culturing human retinal organoids improves photoreceptor yields. Stem Cell Res. Ther. 2018, 9, 156. [Google Scholar] [CrossRef]
- Fang, Y.; Eglen, R.M. Three-dimensional cell cultures in drug discovery and development. SLAS Discov. 2017, 22, 456–472. [Google Scholar]
- Achberger, K.; Haderspeck, J.C.; Kleger, A.; Liebau, S. Stem cell-based retina models. Adv. Drug Deliv. Rev. 2019, 140, 33–50. [Google Scholar] [CrossRef]
- DiStefano, T.; Chen, H.Y.; Panebianco, C.; Kaya, K.D.; Brooks, M.J.; Gieser, L.; Morgan, N.Y.; Pohida, T.; Swaroop, A. Accelerated and improved differentiation of retinal organoids from pluripotent stem cells in rotating-wall vessel bioreactors. Stem Cell Rep. 2018, 10, 300–313. [Google Scholar] [CrossRef]
- Kador, K.E.; Alsehli, H.S.; Zindell, A.N.; Lau, L.W.; Andreopoulos, F.M.; Watson, B.D.; Goldberg, J.L. Retinal ganglion cell polarization using immobilized guidance cues on a tissue-engineered scaffold. Acta Biomater. 2014, 10, 4939–4946. [Google Scholar] [CrossRef]
- Kador, K.E.; Montero, R.B.; Venugopalan, P.; Hertz, J.; Zindell, A.N.; Valenzuela, D.A.; Udding, M.S.; Lavik, E.B.; Muller, K.J.; Andropoulos, F.M.; et al. Tissue engineering the retinal ganglion cell nerve fiber layer. Biomaterials 2014, 34, 4242–4250. [Google Scholar] [CrossRef]
- Kundu, J.; Michaelson, A.; Baranov, P.; Young, M.J.; Carrier, R.L. Approaches to cell delivery: Substrates and scaffolds for cell therapy. Cell Based Ther. Retin. Degener. Dis. 2014, 53, 143–154. [Google Scholar]
- Li, K.; Zhong, X.; Yang, S.; Luo, Z.; Li, K.; Liu, Y.; Cai, S.; Gu, H.; Lu, S.; Zhang, H.; et al. HiPSC-derived retinal ganglion cells grow dendritic arbors and functional axons on a tissue-engineered scaffold. Acta Biomater. 2017, 54, 117–127. [Google Scholar] [CrossRef]
- Yang, T.C.; Chuang, J.H.; Buddhakosai, W.; Wu, W.J.; Lee, C.J.; Chen, W.S.; Yang, Y.P.; Li, M.C.; Peng, C.H.; Chen, S.J. Elongation of axon extension for human iPSC-derived retinal ganglion cells by a nano-imprinted scaffold. Int. J. Mol. Sci. 2017, 18, 2013. [Google Scholar] [CrossRef] [PubMed]
- Chen, T.C.; She, P.Y.; Chen, D.F.; Lu, J.H.; Yang, C.H.; Huang, D.S.; Chen, P.Y.; Lu, C.Y.; Cho, K.S.; Chen, H.F.; et al. Polybenzyl glutamate biocompatible scaffold promotes the efficiency of retinal differentiation toward retinal ganglion cell lineage from human-induced pluripotent stem cells. Int. J. Mol. Sci. 2019, 20, 178. [Google Scholar] [CrossRef] [PubMed]
- Hsu, C.; Lin, Y.; Yang, T.; Yarmishyn, A.A. P3HT:Bebq2-based photovoltaic device enhances differentiation of hiPSC-derived retinal ganglion cells. Int. J. Mol. Sci. 2019, 20, 2661. [Google Scholar] [CrossRef]
- Haderspeck, J.C.; Chuchuy, J.; Kustermann, S.; Liebau, S.; Loskill, P. Organ-on-a-chip technologies that can transform ophthalmic drug discovery and disease modeling. Expert Opin. Drug Discov. 2019, 14, 47–57. [Google Scholar] [CrossRef]
- Achberger, K.; Probst, C.; Haderspeck, J.C.; Bolz, S.; Rogal, J.; Chuchuy, J.; Nikolova, M.; Cora, V.; Antkowiak, L.; Haq, W.; et al. Merging organoid and organ-on-a-chip technology to generate complex multi-layer tissue models in a human retina-on-a-chip platform. eLife 2019, 8, e46188. [Google Scholar] [CrossRef]
- Mandai, M.; Watanabe, A.; Kurimoto, Y.; Hirami, Y.; Morinaga, C.; Daimon, T.; Fujihara, M.; Akimaru, H.; Sakai, N.; Shibata, Y.; et al. Autologous induced stem-cell-derived retinal cells for macular degeneration. N. Engl. J. Med. 2017, 376, 1038–1046. [Google Scholar] [CrossRef] [PubMed]
- Garber, K. RIKEN suspends first clinical trial involving induced pluripotent stem cells. Nat. Biotechnol. 2015, 33, 890–891. [Google Scholar] [CrossRef]
- Umekage, M.; Sato, Y.; Takasu, N. Overview: An iPS cell stock at CiRA. Inflamm. Regen. 2019, 39, 17. [Google Scholar] [CrossRef]
- Sugita, S.; Mandai, M.; Hirami, Y.; Takagi, S.; Maeda, T.; Fujihara, M.; Matsuzaki, M.; Yamamoto, M.; Iseki, K.; Hayashi, N.; et al. HLA-Matched Allogeneic iPS Cells-Derived RPE Transplantation for Macular Degeneration. J. Clin. Med. 2020, 9, 2217. [Google Scholar] [CrossRef] [PubMed]


| Authors | Differentiation Approach | Differentiation Factors | Culture Matrix | Markers for RGC Identification | Isolation Method | Time (d) | Efficiency | Ref. |
|---|---|---|---|---|---|---|---|---|
| Riazifar et al., 2014 | EBs, neural rosettes, neurospheres, RGCs | FBS, DAPT | Gelatin Laminin | Islet-1, TUJ-1, γ-synuclein, BRN3A, THY1 | N/A | 40 | 20–30% | [83] |
| Tanaka et al., 2015 | EBs, neurospheres, RGCs | IWR-1e, FBS, Matrigel, SAG, CHIR99021, N2, retinoid acid, BDNF | Poly-D-lysine/laminin | Islet-1, Tuj-1, γ-synuclein, Brn3b, Math5 | N/A | 35 | - | [84] |
| Ohlemacher et al., 2016 | EBs, neural rosettes, neurospheres, RGCs | N2, FBS, B27, BDNF | Poly-L-ornithine/laminin | ISLET1, BRN3, HUC/D, MELANOPSIN, TAU, MAP2, RBPMS | N/A | 40 | 36% | [86] |
| Teotia et al., 2017 | RPCs (Lamba et al., 2010), RGCs | RPCs: Noggin, Dkk-1, IGF-I RGCs: Shh, FGF8, DAPT, follistatin, cyclopamine, BDNF, NT4, CNTF, forskolin, cAMP | Matrigel | Islet-1, Tuj-1, Brn3, Thy1, Atoh7 | N/A | 36 | 20–30% | [87] |
| Lee et al., 2018 | EBs, neural rosettes, RGCs | Dorsomorphin, SB431542, XAV939, IGF-I, N2, B27 without vitamin A, bFGF, DAPT | Matrigel Poly-D-lysine/laminin | ISLET1, TUJ1, BRN3B, THY1, MATH5 | N/A | 40 | 45% | [88] |
| Chavali et al., 2020 | RPCs, RGCs | RPCs: B27 without vitamin A, N2, XAV939, SB431542, LDN193189, nicotinamide, IGF-I, bFGF, CHIR99021, RGCs: Shh/SAG, FGF8, follistatin, cyclopamine, DAPT, forskolin, cAMP, BDNF, NT4, CNTF | Matrigel (growth factor reduced) | TUJ1, BRN3A, BRN3B, γ-synuclein, THY1, MAP2, RBPMS | MACS (CD90.2 micro beads) | 40 | 95% | [89] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
García-López, M.; Arenas, J.; Gallardo, M.E. Hereditary Optic Neuropathies: Induced Pluripotent Stem Cell-Based 2D/3D Approaches. Genes 2021, 12, 112. https://doi.org/10.3390/genes12010112
García-López M, Arenas J, Gallardo ME. Hereditary Optic Neuropathies: Induced Pluripotent Stem Cell-Based 2D/3D Approaches. Genes. 2021; 12(1):112. https://doi.org/10.3390/genes12010112
Chicago/Turabian StyleGarcía-López, Marta, Joaquín Arenas, and M. Esther Gallardo. 2021. "Hereditary Optic Neuropathies: Induced Pluripotent Stem Cell-Based 2D/3D Approaches" Genes 12, no. 1: 112. https://doi.org/10.3390/genes12010112
APA StyleGarcía-López, M., Arenas, J., & Gallardo, M. E. (2021). Hereditary Optic Neuropathies: Induced Pluripotent Stem Cell-Based 2D/3D Approaches. Genes, 12(1), 112. https://doi.org/10.3390/genes12010112