
Linear Skin Defects with Multiple Congenital Anomalies (LSDMCA): An Unconventional Mitochondrial Disorder



Abstract
1. Introduction: An Historical Perspective
2. The Molecular Basis of LSDMCA
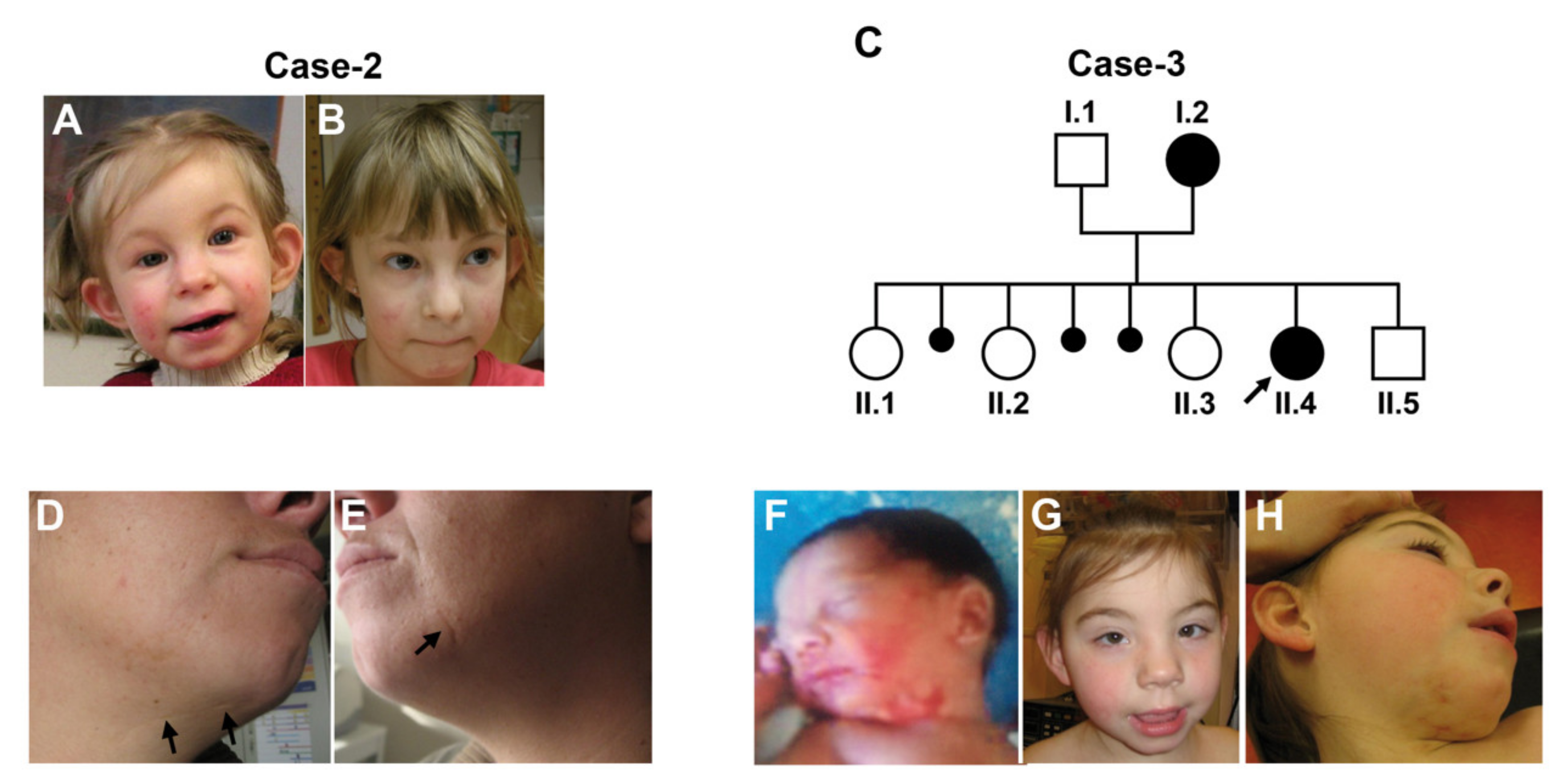
3. The Clinical Spectrum of LSDMCA
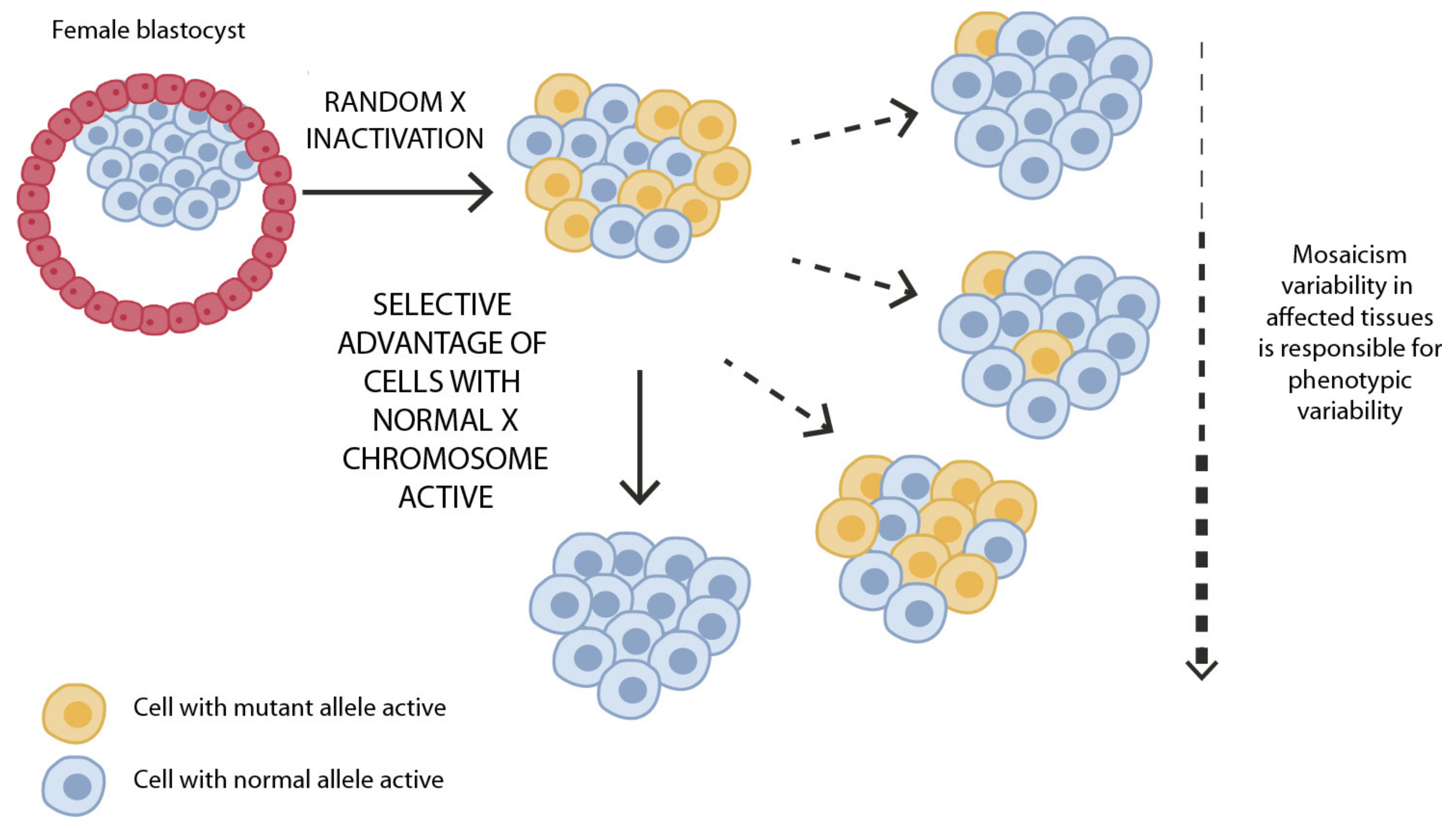
4. The Role of X-Chromosome and X-Chromosome Inactivation
5. LSDMCA as an Unconventional Mitochondrial Disorder
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Lindsay, E.A.; Grillo, A.; Ferrero, G.B.; Roth, E.J.; Magenis, E.; Grompe, M.; Hultén, M.; Gould, C.; Baldini, A.; Zoghbi, H.Y.; et al. Microphthalmia with linear skin defects (MLS) syndrome: Clinical, cytogenetic and molecular characterization. Am. J. Med. Genet. 1994, 49, 229–234. [Google Scholar] [CrossRef] [PubMed]
- Wimplinger, I.; Morleo, M.; Rosenberger, G.; Iaconis, D.; Orth, U.; Meinecke, P.; Lerer, I.; Ballabio, A.; Gal, A.; Franco, B.; et al. Mutations of the Mitochondrial Holocytochrome c–Type Synthase in X-Linked Dominant Microphthalmia with Linear Skin Defects Syndrome. Am. J. Hum. Genet. 2006, 79, 878–889. [Google Scholar] [CrossRef] [PubMed]
- Indrieri, A.; Van Rahden, V.A.; Tiranti, V.; Morleo, M.; Iaconis, D.; Tammaro, R.; D’Amato, I.; Conte, I.; Maystadt, I.; Demuth, S.; et al. Mutations in COX7B cause microphthalmia with linear skin lesions, an unconventional mitochondrial disease. Am. J. Hum. Genet. 2012, 91, 942–949. [Google Scholar] [CrossRef] [PubMed]
- Van Rahden, V.A.; Fernandez-Vizarra, E.; Alawi, M.; Brand, K.; Fellmann, F.; Horn, D.; Zeviani, M.; Kutsche, K. Mutations in NDUFB11, Encoding a Complex I Component of the Mitochondrial Respiratory Chain, Cause Microphthalmia with Linear Skin Defects Syndrome. Am. J. Hum. Genet. 2015, 96, 640–650. [Google Scholar] [CrossRef]
- Morleo, M.; Pramparo, T.; Perone, L.; Gregato, G.; Le Caignec, C.; Mueller, R.F.; Ogata, T.; Raas-Rothschild, A.; De Blois, M.C.; Wilson, L.C.; et al. Microphthalmia with linear skin defects (MLS) syndrome: Clinical, cytogenetic, and molecular characterization of 11 cases. Am. J. Med. Genet. 2005, 137, 190–198. [Google Scholar] [CrossRef]
- Wapenaar, M.C.; Bassi, M.T.; Schaefer, L.; Grillo, A.; Ferrero, G.B.; Chinault, A.C.; Ballabio, A.; Zoghbi, H.Y. The genes for X-linked ocular albinism (OA1) and microphthalmia with linear skin defects (MLS): Cloning and characterization of the critical regions. Hum. Mol. Genet. 1993, 2, 947–952. [Google Scholar] [CrossRef]
- Wapenaar, M.C.; Schiaffino, M.V.; Bassi, M.T.; Schaefer, L.; Chinault, A.C.; Zoghbi, H.Y.; Ballabio, A. A YAC-based binning strategy facilitating the rapid assembly of cosmid contigs: 1.6 Mb of overlapping cosmids in Xp22. Hum. Mol. Genet. 1994, 3, 1155–1161. [Google Scholar] [CrossRef]
- Quaderi, N.A.; Schweiger, S.; Gaudenz, K.; Franco, B.; Rugarli, E.I.; Berger, W.; Feldman, G.J.; Volta, M.; Andolfi, G.; Gilgenkrantz, S.; et al. Opitz G/BBB syndrome, a defect of midline development, is due to mutations in a new RING finger gene on Xp22. Nat. Genet. 1997, 17, 285–291. [Google Scholar] [CrossRef]
- Schaefer, L.; Ballabio, A.; Zoghbi, H.Y. Cloning and characterization of a putative human holocytochrome c-type synthetase gene (HCCS) isolated from the critical region for microphthalmia with linear skin defects (MLS). Genomics 1996, 34, 166–172. [Google Scholar] [CrossRef]
- Schwarz, Q.P.; Cox, T.C. Complementation of a yeast CYC3 deficiency identifies an X-linked mammalian activator of apocytochrome c. Genomics 2002, 79, 51–57. [Google Scholar] [CrossRef] [PubMed]
- Prakash, S.K.; Paylor, R.; Jenna, S.; Lamarche-Vane, N.; Armstrong, D.L.; Xu, B.; Mancini, M.A.; Zoghbi, H.Y. Functional analysis of ARHGAP6, a novel GTPase-activating protein for RhoA. Hum. Mol. Genet. 2000, 9, 477–488. [Google Scholar] [CrossRef] [PubMed]
- Prakash, S.K.; Cormier, T.A.; McCall, A.E.; Garcia, J.J.; Sierra, R.; Haupt, B.; Zoghbi, H.Y.; Van Den Veyver, I.B. Loss of holocytochrome c-type synthetase causes the male lethality of X-linked dominant microphthalmia with linear skin defects (MLS) syndrome. Hum. Mol. Genet. 2002, 11, 3237–3248. [Google Scholar] [CrossRef] [PubMed]
- Van Rahden, V.A.; Rau, I.; Fuchs, S.; Kosyna, F.K.; de Almeida, H.L.; Fryssira, H.; Isidor, B.; Jauch, A.; Joubert, M.; Lachmeijer, A.M.A.; et al. Clinical spectrum of females with HCCS mutation: From no clinical signs to a neonatal lethal form of the microphthalmia with linear skin defects (MLS) syndrome. Orphanet J. Rare Dis. 2014, 9, 53. [Google Scholar] [CrossRef] [PubMed]
- Wimplinger, I.; Shaw, G.M.; Kutsche, K. HCCS loss-of-function missense mutation in a female with bilateral microphthalmia and sclerocornea: A novel gene for severe ocular malformations? Mol. Vis. 2007, 13, 1475–1482. [Google Scholar] [PubMed]
- Ogata, T.; Wakui, K.; Muroya, K.; Ohashi, H.; Matsuo, N.; Brown, D.M.; Ishii, T.; Fukushima, Y. Microphthalmia with linear skin defects syndrome in a mosaic female infant with monosomy for the Xp22 region: Molecular analysis of the Xp22 breakpoint and the X-inactivation pattern. Hum. Genet. 1998, 103, 51–56. [Google Scholar] [CrossRef]
- Bernard, D.G.; Gabilly, S.T.; Dujardin, G.; Merchant, S.; Hamel, P.P. Overlapping specificities of the mitochondrial cytochrome c and c1 heme lyases. J. Biol. Chem. 2003, 278, 49732–49742. [Google Scholar] [CrossRef] [PubMed]
- Dumont, M.E.; Ernst, J.F.; Hampsey, D.M.; Sherman, F. Identification and sequence of the gene encoding cytochrome c heme lyase in the yeast Saccharomyces cerevisiae. EMBO J. 1987, 6, 235–241. [Google Scholar] [CrossRef]
- Zollner, A.; Rodel, G.; Haid, A. Molecular cloning and characterization of the Saccharomyces cerevisiae CYT2 gene encoding cytochrome-c1-heme lyase. Eur. J. BioChem. 1992, 207, 1093–1100. [Google Scholar] [CrossRef]
- Indrieri, A.; Conte, I.; Chesi, G.; Romano, A.; Quartararo, J.; Tatè, R.; Ghezzi, D.; Zeviani, M.; Goffrini, P.; Ferrero, I.; et al. The impairment of HCCS leads to MLS syndrome by activating a non-canonical cell death pathway in the brain and eyes. EMBO Mol. Med. 2013, 5, 280–293. [Google Scholar] [CrossRef]
- Drenckhahn, J.D.; Schwarz, Q.P.; Gray, S.; Laskowski, A.; Kiriazis, H.; Ming, Z.; Harvey, R.P.; Du, X.J.; Thorburn, D.R.; Cox, T.C. Compensatory growth of healthy cardiac cells in the presence of diseased cells restores tissue homeostasis during heart development. Dev. Cell 2008, 15, 521–533. [Google Scholar] [CrossRef]
- Li, P.; Nijhawan, D.; Budihardjo, I.; Srinivasula, S.M.; Ahmad, M.; Alnemri, E.S.; Wang, X. Cytochrome c and dATP-dependent formation of Apaf-1/caspase-9 complex initiates an apoptotic protease cascade. Cell 1997, 91, 479–489. [Google Scholar] [CrossRef]
- Rodriguez, J.; Lazebnik, Y. Caspase-9 and APAF-1 form an active holoenzyme. Genes Dev. 1999, 13, 3179–3184. [Google Scholar] [CrossRef]
- Wang, C.; Youle, R.J. The Role of Mitochondria in Apoptosis. Annu. Rev. Genet. 2009, 43, 95–118. [Google Scholar] [CrossRef]
- Kiryu-Seo, S.; Gamo, K.; Tachibana, T.; Tanaka, K.; Kiyama, H. Unique anti-apoptotic activity of EAAC1 in injured motor neurons. EMBO J. 2006, 25, 3411–3421. [Google Scholar] [CrossRef]
- Morleo, M.; Franco, B. Dosage compensation of the mammalian X chromosome influences the phenotypic variability of X-linked dominant male-lethal disorders. J. Med. Genet. 2008, 45, 401–408. [Google Scholar] [CrossRef]
- Tsukihara, T.; Aoyama, H.; Yamashita, E.; Tomizaki, T.; Yamaguchi, H.; Shinzawa-Itoh, K.; Nakashima, R.; Yaono, R.; Yoshikawa, S. The whole structure of the 13-subunit oxidized cytochrome c oxidase at 2.8 A. Science 1996, 272, 1136–1144. [Google Scholar] [CrossRef]
- Fornuskova, D.; Stiburek, L.; Wenchich, L.; Vinsova, K.; Hansikova, H.; Zeman, J. Novel insights into the assembly and function of human nuclear-encoded cytochrome c oxidase subunits 4, 5a, 6a, 7a and 7b. Biochem. J. 2010, 428, 363–374. [Google Scholar] [CrossRef]
- Kadenbach, B.; Hüttemann, M. The subunit composition and function of mammalian cytochrome c oxidase. Mitochondrion 2015, 24, 64–76. [Google Scholar] [CrossRef] [PubMed]
- Indrieri, A.; Grimaldi, C.; Zucchelli, S.; Tammaro, R.; Gustincich, S.; Franco, B. Synthetic long non-coding RNAs [SINEUPs] rescue defective gene expression in vivo. Sci. Rep. 2016, 6, 27315. [Google Scholar] [CrossRef] [PubMed]
- Carroll, J.; Shannon, R.J.; Fearnley, I.M.; Walker, J.E.; Hirst, J. Definition of the nuclear encoded protein composition of bovine heart mitochondrial complex I: Identification of two new subunits. J. Biol. Chem. 2002. [Google Scholar] [CrossRef] [PubMed]
- Vinothkumar, K.R.; Zhu, J.; Hirst, J. Architecture of mammalian respiratory complex I. Nature 2014, 515, 80–84. [Google Scholar] [CrossRef]
- Fiedorczuk, K.; Sazanov, L.A. Mammalian Mitochondrial Complex I Structure and Disease-Causing Mutations. Trends Cell Biol. 2018, 28, 835–867. [Google Scholar] [CrossRef] [PubMed]
- Vergult, S.; Leroy, B.; Claerhout, I.; Menten, B. Familial cases of a submicroscopic Xp22.2 deletion: Genotype-phenotype correlation in microphthalmia with linear skin defects syndrome. Mol. Vis. 2013, 19, 311–318. [Google Scholar] [PubMed]
- Al-Gazali, L.I.; Mueller, R.F.; Caine, A.; Antoniou, A.; McCartney, A.; Fitchett, M.; Dennis, N.R. Two 46,XX,t(X;Y) females with linear skin defects and congenital microphthalmia: A new syndrome at Xp22.3. J. Med. Genet. 1990, 27, 59–63. [Google Scholar] [CrossRef] [PubMed]
- Friedman, P.A.; Rao, K.W.; Teplin, S.W.; Aylsworth, A.S. Provisional mapping deletion of the focal dermal hypoplasia (FDH) gene to Xp22.31. Am. J. Hum. Genet. Suppl. 1988, 43, A450. [Google Scholar]
- Temple, I.K.; Hurst, J.A.; Hing, S.; Butler, L.; Baraitser, M. De novo deletion of Xp22.2-pter in a female with linear skin lesions of the face and neck, microphthalmia, and anterior chamber eye anomalies. J. Med. Genet. 1990, 27, 56–58. [Google Scholar] [CrossRef]
- Allanson, J.; Richter, S. Linear skin defects and congenital microphthalmia: A new syndrome at Xp22.2. J. Med. Genet. 1991, 28, 143–144. [Google Scholar] [CrossRef] [PubMed]
- Gericke, G.S.; Myburgh, E.; Bester, R.; van Rensberg, E.J.; Neethling, E. Further delineation of the Xp22.2-pter sundrome of linear skin lesions, microphthalmia and anterior chamber anomalies. Am. J. Hum. Genet. Suppl. 1991, 49, A271. [Google Scholar]
- Lindor, N.M.; Michels, V.V.; Hoppe, D.A.; Driscoll, D.J.; Leavitt, J.A. Xp22.3 microdeletion syndrome with microphthalmia, sclerocornea, linear skin defects, and congenital heart defects. Am. J. Med. Genet. 1992, 44, 61–65. [Google Scholar] [CrossRef] [PubMed]
- Naritomi, K.; Izumikawa, Y.; Nagataki, S.; Fukushima, Y.; Wakui, K.; Niikawa, N.; Hirayama, K. Combined Goltz and Aicardi syndromes in a terminal Xp deletion: Are they a contiguous gene syndrome? Am. J. Med. Genet. 1992, 43, 839–843. [Google Scholar] [CrossRef]
- Mucke, J.; Hoepffner, W.; Thamm, B.; Theile, H. MIDAS syndrome (microphthalmia, dermal aplasia and sclerocornea): An autonomous entity with linear skin defects within the spectrum of focal hypoplasias. Eur. J. Dermatol. 1995, 5, 197–203. [Google Scholar]
- Eng, A.; Lebel, R.R.; Elejalde, B.R.; Anderson, C.; Bennett, L. Linear facial skin defects associated with microphthalmia and other malformations, with chromosome deletion Xp22.1. J. Am. Acad. Dermatol. 1994, 31, 680–682. [Google Scholar] [CrossRef]
- Paulger, B.R.; Kraus, E.W.; Pulitzer, D.R.; Moore, C.M. Xp microdeletion syndrome characterized by pathognomonic linear skin defects on the head and neck. Pediatr. Dermatol. 1997, 14, 26–30. [Google Scholar] [CrossRef]
- Camacho, J.A.; Goodman, B.K.; Hamosh, A.; Hurko, O.; Thomas, G.H. MIDAS syndrome in a 46, XX new born with ambiguous genitalia and a cryptic, de novo X;Y translocation. Am. J. Hum. Genet. Suppl. 1997, 61, A93. [Google Scholar]
- Kono, T.; Migita, T.; Koyama, S.; Seki, I. Another observation of microphthalmia in an XX male: Microphthalmia with linear skin defects syndrome without linear skin lesions. J. Hum. Genet. 1999, 44, 63–68. [Google Scholar] [CrossRef]
- Al-Gazali, L.I.; Mueller, R.F.; Caine, A.; Nennis, N.; Fitchett, M.; Insley, L.; Goodfellow, P.G.; Hultén, M. An XX male and two t(X;Y) females with linear skin defects and congenital microphthalmia: A new syndrome at Xp22.3. J. Med. Genet. 1988, 25, 638–639. [Google Scholar]
- Donnenfeld, A.E.; Coyne, M.D.; Beauregard, L.J. Microphthalmia and chorioretinal lesions in a girl with an Xp22.2-pter deletion and partial 3p trisomy: Clinical observations relevant to Aicardi syndrome gene localization. Am. J. Med. Genet. 1990, 37, 182–186. [Google Scholar] [CrossRef] [PubMed]
- Thies, U.; Rao, V.V.N.G.; Engel, W.; Schmidtke, J. Physical mapping of two Xp markers DXS16 and DXS143. Hum. Genet. 1991, 86, 418–420. [Google Scholar] [CrossRef] [PubMed]
- Koyama, S.; Kono, T.; Uchida, K.; Noguchi, E.; Suzuki, K.; Yamasuge, M.; Otsuka, M.; Ito, M.; Suzuki, M.; Migita, T.; et al. XX male with ocular symptoms [Japanese]. Presented at the 34th Annual Meetingof the Japanese Teratology Society at Kochi, Japan. Congenit. Anom. 1993, 34, B–04. [Google Scholar]
- Stratton, R.F.; Walter, C.A.; Paulgar, B.R.; Price, M.E.; Moore, C.M. Second 46,XX males with MLS syndrome. Am. J. Med. Genet. 1998, 76, 37–41. [Google Scholar] [CrossRef]
- Kayserili, H.; Cox, T.C.; Cox, L.L.; Basaran, S.; Kilic, G.; Ballabio, A.; Yuksel-Apak, M. Molecular characterisation of a new case of microphthalmia with linear skin defects (MLS). J. Med. Genet. 2001, 38, 411–417. [Google Scholar] [CrossRef][Green Version]
- Kutsche, K.; Werner, W.; Bartsch, O.; von der Wense, A.; Meinecke, P.; Gal, A. Microphthalmia with linear skin defects syndrome (MLS): A male with a mosaic paracentric inversion of Xp. CytoGenet. Genome Res. 2002, 99, 297–302. [Google Scholar] [CrossRef]
- Anguiano, A.; Yang, X.; Felix, J.K.; Hoo, J.J. Twin brothers with MIDAS syndrome and XX karyotype. Am. J. Med. Genet. 2003, 119A, 47–49. [Google Scholar] [CrossRef]
- Enright, F.; Campbell, P.; Stallings, R.L.; Hall, K.; Green, A.J.; Sweeney, E.; Barnes, L.; Watson, R. Xp22.3 microdeletion in a 19-year-old girl with clinical features of MLS syndrome. Pediatr. Dermatol. 2003, 20, 153–157. [Google Scholar] [CrossRef]
- Cape, C.J.; Zaidman, G.W.; Beck, A.D.; Kaufman, A.H. Phenotypic variation in ophthalmic manifestations of MIDAS syndrome (microphthalmia, dermal aplasia, and sclerocornea). Arch. OphthalMol. 2004, 122, 1070–1074. [Google Scholar] [CrossRef][Green Version]
- Cain, C.C.; Saul, D.; Attanasio, L.; Oehler, E.; Hamosh, A.; Blakemore, K.; Stetten, G. Microphthalmia with linear skin defects (MLS) syndrome evaluated by prenatal karyotyping, FISH and array comparative genomic hybridization. Prenat. Diagn. 2007, 27, 373–379. [Google Scholar] [CrossRef]
- Sharma, V.M.; Ruiz de Luzuriaga, A.M.; Waggoner, D.; Greenwald, M.; Stein, S.L. Microphthalmia with linear skin defects: A case report and review. Pediatr. Dermatol. 2008, 25, 548–552. [Google Scholar] [CrossRef] [PubMed]
- Hobson, G.M.; Gibson, C.W.; Aragon, M.; Yuan, Z.A.; Davis-Williams, A.; Banser, L.; Kirkham, J.; Brook, A.H. A large X-chromosomal deletion is associated with microphthalmia with linear skin defects (MLS) and amelogenesis imperfecta (XAI). Am. J. Med. Genet. A 2009, 149A, 1698–1705. [Google Scholar] [CrossRef] [PubMed]
- Alberry, M.S.; Juvanic, G.; Crolla, J.; Soothill, P.; Newbury-Ecob, R. Pseudotail as a feature of microphthalmia with linear skin defects syndrome. Clin. Dysmorphol. 2011, 20, 111–113. [Google Scholar] [CrossRef] [PubMed]
- Zumwalt, J.; Moorhead, C.; Golkar, L. Fourteen-month-old girl with facial skin thinning. Pediatr. Dermatol. 2012, 29, 217–218. [Google Scholar] [CrossRef]
- Steichen-Gersdorf, E.; Griesmaier, E.; Pientka, F.K.; Kotzot, D.; Kutsche, K. A severe form of the X-linked microphthalmia with linear skin defects syndrome in a female newborn. Clin. Dysmorphol. 2010, 19, 82–84. [Google Scholar] [CrossRef] [PubMed]
- Wimplinger, I.; Rauch, A.; Orth, U.; Schwarzer, U.; Trautmann, U.; Kutsche, K. Mother and daughter with a terminal Xp deletion: Implication of chromosomal mosaicism and X-inactivation in the high clinical variability of the microphthalmia with linear skin defects (MLS) syndrome. Eur. J. Med. Genet. 2007, 50, 421–431. [Google Scholar] [CrossRef] [PubMed]
- Margari, L.; Colonna, A.; Craig, F.; Gentile, M.; Giannella, G.; Lamanna, A.L.; Legrottaglie, A.R. Microphthalmia with Linear Skin Defects (MLS) associated with Autism Spectrum Disorder (ASD) in a patient with Familial 12.9Mb Terminal Xp deletion. BMC Pediatr. 2014, 14, 1–5. [Google Scholar] [CrossRef]
- Kluger, N.; Bouissou, A.; Tauzin, L.; Puechberty, J.; Dereure, O. Congenital Linear Streaks on the Face and Neck and Microphthalmia in an Infant Girl. Acta Derm. Venereol. 2014, 94, 342–343. [Google Scholar] [CrossRef] [PubMed]
- Prepeluh, N.; Korpar, B.; Zagorac, A.; Zagradišnik, B.; Golub, A.; Kokal, J.; Vokač, N. A mosaic form of microphthalmia with linear skin defects. BMC Pediatr. 2018, 18, 254. [Google Scholar] [CrossRef] [PubMed]
- Kumar, P.; Rajab, A. Microphthalmia with linear skin defects (MLS) syndrome: Familial presentation. Clin. Exp. Dermatol. 2018, 43, 196–197. [Google Scholar] [CrossRef]
- Satcher, K.G.; Maegawa, G.H.B.; Schoch, J.J. Microphthalmia and linear skin defects syndrome: Precise diagnosis guides prognosis. Pediatr. Dermatol. 2020, 37, 217–218. [Google Scholar] [CrossRef]
- Vendramini-Pittoli, S.; Candido-Souza, R.M.; Quiezi, R.G.; Zechi-Ceide, R.M.; Kokitsu-Nakata, N.M.; Jehee, F.S.; Ribeiro-Bicudo, L.A.; FitzPatrick, D.R.; Guion-Almeida, M.L.; Richieri-Costa, A. Microphthalmia, Linear Skin Defects, Callosal Agenesis, and Cleft Palate in a Patient with Deletion at Xp22.3p22.2. J. Pediatr. Genet. 2020, 09, 258–262. [Google Scholar] [CrossRef]
- De Almeida, H.L., Jr.; Rossi, G.; de Abreu, L.B.; Bergamaschi, C.; da Silva, A.B.; Kutsche, K. Dermatoscopic aspects of the Microphthalmia with Linear Skin Defects (MLS) Syndrome. An. Bras. Dermatol. 2014, 89, 180–181. [Google Scholar] [CrossRef]
- Durack, A.; Mehta, S.G.; Allen, L.E.; Ozanic Bulic, S.; Burrows, N.P. Linear skin defects and microphthalmia. Clin. Exp. Dermatol. 2018, 43, 860–862. [Google Scholar] [CrossRef]
- Kapur, R.; Tu, E.Y.; Toyran, S.; Shah, P.; Vangveeravong, S.; Lloyd, W.C., 3rd; Edward, D.P. Corneal pathology in microphthalmia with linear skin defects syndrome. Cornea 2008, 27, 734–738. [Google Scholar] [CrossRef]
- Feichtinger, R.G.; Sperl, W.; Bauer, J.W.; Kofler, B. Mitochondrial dysfunction: A neglected component of skin diseases. Exp. Dermatol. 2014, 23, 607–614. [Google Scholar] [CrossRef]
- Rea, G.; Homfray, T.; Till, J.; Roses-Noguer, F.; Buchan, R.J.; Wilkinson, S.; Wilk, A.; Walsh, R.; John, S.; McKee, S.; et al. Histiocytoid cardiomyopathy and microphthalmia with linear skin defects syndrome: Phenotypes linked by truncating variants in NDUFB11. Mol. Case Stud. 2017, 3, a001271. [Google Scholar] [CrossRef]
- Shehata, B.M.; Cundiff, C.A.; Lee, K.; Sabharwal, A.; Lalwani, M.K.; Davis, A.K.; Agrawal, V.; Sivasubbu, S.; Iannucci, G.J.; Gibson, G. Exome sequencing of patients with histiocytoid cardiomyopathy reveals a de novo NDUFB11 mutation that plays a role in the pathogenesis of histiocytoid cardiomyopathy. Am. J. Med. Genet. Part A 2015, 167, 2114–2121. [Google Scholar] [CrossRef]
- Kohda, M.; Tokuzawa, Y.; Kishita, Y.; Nyuzuki, H.; Moriyama, Y.; Mizuno, Y.; Hirata, T.; Yatsuka, Y.; Yamashita-Sugahara, Y.; Nakachi, Y.; et al. A Comprehensive Genomic Analysis Reveals the Genetic Landscape of Mitochondrial Respiratory Chain Complex Deficiencies. PLoS Genet. 2016, 12, e1005679. [Google Scholar] [CrossRef] [PubMed]
- Lichtenstein, D.A.; Crispin, A.W.; Sendamarai, A.K.; Campagna, D.R.; Schmitz-Abe, K.; Sousa, C.M.; Kafina, M.D.; Schmidt, P.J.; Niemeyer, C.M.; Porter, J.; et al. A recurring mutation in the respiratory complex 1 protein NDUFB11 is responsible for a novel form of X-linked sideroblastic anemia. Blood 2016, 128, 1913–1917. [Google Scholar] [CrossRef]
- Torraco, A.; Bianchi, M.; Verrigni, D.; Gelmetti, V.; Riley, L.; Niceta, M.; Martinelli, D.; Montanari, A.; Guo, Y.; Rizza, T.; et al. A novel mutation in NDUFB11 unveils a new clinical phenotype associated with lactic acidosis and sideroblastic anemia. Clin. Genet. 2017, 91, 441–447. [Google Scholar] [CrossRef] [PubMed]
- Franco, B.; Meroni, G.; Parenti, G.; Levilliers, J.; Bernard, L.; Gebbia, M.; Cox, L.; Maroteaux, P.; Sheffield, L.; Rappold, G.A.; et al. A cluster of sulfatase genes on Xp22.3: Mutations in chondrodysplasia punctata (CDPX) and implications for warfarin embryopathy. Cell 1995, 81, 15–25. [Google Scholar] [CrossRef]
- Parenti, G.; Rizzolo, M.G.; Ghezzi, M.; Di Maio, S.; Sperandeo, M.P.; Incerti, B.; Franco, B.; Ballabio, A.; Andria, G. Variable penetrance of hypogonadism in a sibship with Kallmann syndrome due to a deletion of the KAL gene. Am. J. Med. Genet. 1995, 57, 476–478. [Google Scholar] [CrossRef] [PubMed]
- Franco, B.; Guioli, S.; Pragliola, A.; Incerti, B.; Bardoni, B.; Tonlorenzi, R.; Carrozzo, R.; Maestrini, E.; Pieretti, M.; Taillon-Miller, P.; et al. A gene deleted in Kallmann’s syndrome shares homology with neural cell adhesion and axonal path-finding molecules. Nature 1991, 353, 529–536. [Google Scholar] [CrossRef]
- Ferrante, M.I.; Feather, S.A.; Bulfone, A.; Wright, V.; Ghiani, M.; Selicorni, A.; Gammaro, L.; Scolari, F.; Woolf, A.S.; Sylvie, O.; et al. Identification of the Gene for Oral-Facial-Digital Type I Syndrome. Am. J. Hum. Genet. 2001, 68, 569–576. [Google Scholar] [CrossRef]
- Khonsari, R.H.; Seppala, M.; Pradel, A.; Dutel, H.; Clément, G.; Lebedev, O.; Ghafoor, S.; Rothova, M.; Tucker, A.; Maisey, J.G.; et al. The buccohypophyseal canal is an ancestral vertebrate trait maintained by modulation in sonic hedgehog signaling. BMC Biol. 2013, 11, 27. [Google Scholar] [CrossRef]
- Reinson, K.; Kovacs-Nagy, R.; Õiglane-Shlik, E.; Pajusalu, S.; Nõukas, M.; Wintjes, L.T.; van den Brandt, F.C.A.; Brink, M.; Acker, T.; Ahting, U.; et al. Diverse phenotype in patients with complex I deficiency due to mutations in NDUFB11. Eur. J. Med. Genet. 2019, 62, 103572. [Google Scholar] [CrossRef]
- Lyon, M.F. Gene action in the X-chromosome of the mouse (Mus musculus L.). Nature 1961, 190, 372–373. [Google Scholar] [CrossRef]
- Migeon, B.R. X-linked diseases: Susceptible females. Genet. Med. 2020, 22, 1156–1174. [Google Scholar] [CrossRef]
- Franco, B.; Ballabio, A. X-inactivation and human disease: X-linked dominant male-lethal disorders. Curr. Opin. Genet. Dev. 2006, 16, 254–259. [Google Scholar] [CrossRef]
- Munnich, A.; Rustin, P. Clinical spectrum and diagnosis of mitochondrial disorders. Am. J. Med. Genet. 2001, 106, 4–17. [Google Scholar] [CrossRef]
- DiMauro, S.; Tanji, K.; Schon, E.A. Mitochondrial Encephalomyopathies☆. In Reference Module in Neuroscience and Biobehavioral Psychology; Elsevier: Amsterdam, The Netherlands, 2017; ISBN 9780128093245. [Google Scholar]
- Chinnery, P.F. Mitochondrial Disorders Overview; University of Washington: Seattle, WA, USA, 1993. [Google Scholar]
- Birch-Machin, M.A. Mitochondria and skin disease. Clin. Exp. Dermatol. 2000, 25, 141–146. [Google Scholar] [CrossRef] [PubMed]
- Misgeld, T.; Schwarz, T.L. Mitostasis in Neurons: Maintaining Mitochondria in an Extended Cellular Architecture. Neuron 2017, 96, 651–666. [Google Scholar] [CrossRef] [PubMed]
- Yamaguchi, Y.; Miura, M. Programmed Cell Death in Neurodevelopment. Dev. Cell 2015, 32, 478–490. [Google Scholar] [CrossRef] [PubMed]
- Hagberg, H.; Mallard, C.; Rousset, C.I.; Thornton, C. Mitochondria: Hub of injury responses in the developing brain. Lancet Neurol. 2014, 13, 217–232. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
| Gene | Gene OMIM# | Nucleotide Change | Type of Mutation | Predicted Protein | Disease Symbol | Disease OMIM# | Ref |
|---|---|---|---|---|---|---|---|
| HCCS | 300056 | c.589C>T | Nonsense | p.R197* a | MLS/ | 309801 | [2,13,14] |
| c.649C>T | Missense | p.R217C | MIDAS | ||||
| c.475G>A | Missense | p.E159K | MCOPS7 | ||||
| c.[=/524_525delAG] | Frameshift | p.[=/E175Vfs*30] | LSDMCA1 | ||||
| COX7B | 300885 | c.196delC | Frameshift | p.L66Cfs*48 | LSDMCA2 | 300887 | [3] |
| c.41-2A>G | Frameshift | p.V14Gfs*19 | |||||
| c.55C>T | Nonsense | p.E19* | |||||
| NDUFB11 | 300403 | c.262C>T | Nonsense | p.Arg88* | LSDMCA3 | 300952 | [4] |
| c.402delG | Frameshift | p.Arg134Serfs*3 |
| LSDMCA/ Mutation | Skin Lesions | EYE | CNS Malformations | Intellectual Disabilities | Short Stature | Cardiac Anomalies | Ref | |||
|---|---|---|---|---|---|---|---|---|---|---|
| Micro/Anophthalmia | Corneal Abnormalities | Other | ||||||||
| LSDMCA1/ Xp22 R | # cases | 54/70 | 54/70 | 45/70 | 31/70 | 37/65 | 13/46 | 23/49 | 23/65 | [1,2,5,13,15,33,34,35,36,37,38,39,40,41,42,43,44,45,46,47,48,49,50,51,52,53,54,55,56,57,58,59,60,61,62,63,64,65,66,67,68,69,70,71] |
| % | 77 | 77 | 64 | 44 | 57 | 28 | 47 | 35 | ||
| LSDMCA1/ HCCS P | # cases | 2/5 | 5/5 | 5/5 | 3/5 | 3/5 | 3/5 | 1/5 | 2/5 | [2,13] |
| % | 40 | 100 | 100 | 60 | 60 | 60 | 20 | 40 | ||
| LSDMCA2/ COX7B P | # cases | 4/4 | 0/4 | 0/4 | 1/4 | 3/4 | 2/4 | 2/4 | 2/4 | [3] |
| % | 100 | 0 | 0 | 25 | 75 | 50 | 50 | 50 | ||
| LSDMCA3/ NDUFB11 P | # cases | 2/3 | 0/4 | 0/4 | 2/3 | 1/3 | 1/3 | 1/2 | 2/3 | [4] |
| % | 67 | 0 | 0 | 67 | 33 | 33 | 50 | 67 | ||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Indrieri, A.; Franco, B. Linear Skin Defects with Multiple Congenital Anomalies (LSDMCA): An Unconventional Mitochondrial Disorder. Genes 2021, 12, 263. https://doi.org/10.3390/genes12020263
Indrieri A, Franco B. Linear Skin Defects with Multiple Congenital Anomalies (LSDMCA): An Unconventional Mitochondrial Disorder. Genes. 2021; 12(2):263. https://doi.org/10.3390/genes12020263
Chicago/Turabian StyleIndrieri, Alessia, and Brunella Franco. 2021. "Linear Skin Defects with Multiple Congenital Anomalies (LSDMCA): An Unconventional Mitochondrial Disorder" Genes 12, no. 2: 263. https://doi.org/10.3390/genes12020263
APA StyleIndrieri, A., & Franco, B. (2021). Linear Skin Defects with Multiple Congenital Anomalies (LSDMCA): An Unconventional Mitochondrial Disorder. Genes, 12(2), 263. https://doi.org/10.3390/genes12020263