Exploring the Regulatory Potential of Long Non-Coding RNA in Feed Efficiency of Indicine Cattle

 ,
, 
Abstract
1. Introduction
2. Materials and Methods
2.1. Data Acquisition
2.2. Identification of New Transcripts
2.3. Identification of lncRNA
2.4. General Classification of lncRNA
2.5. lncRNA and miRNA
2.6. lncRNA Expression
2.7. lncRNA and mRNA
2.8. Functional Analysis
3. Results
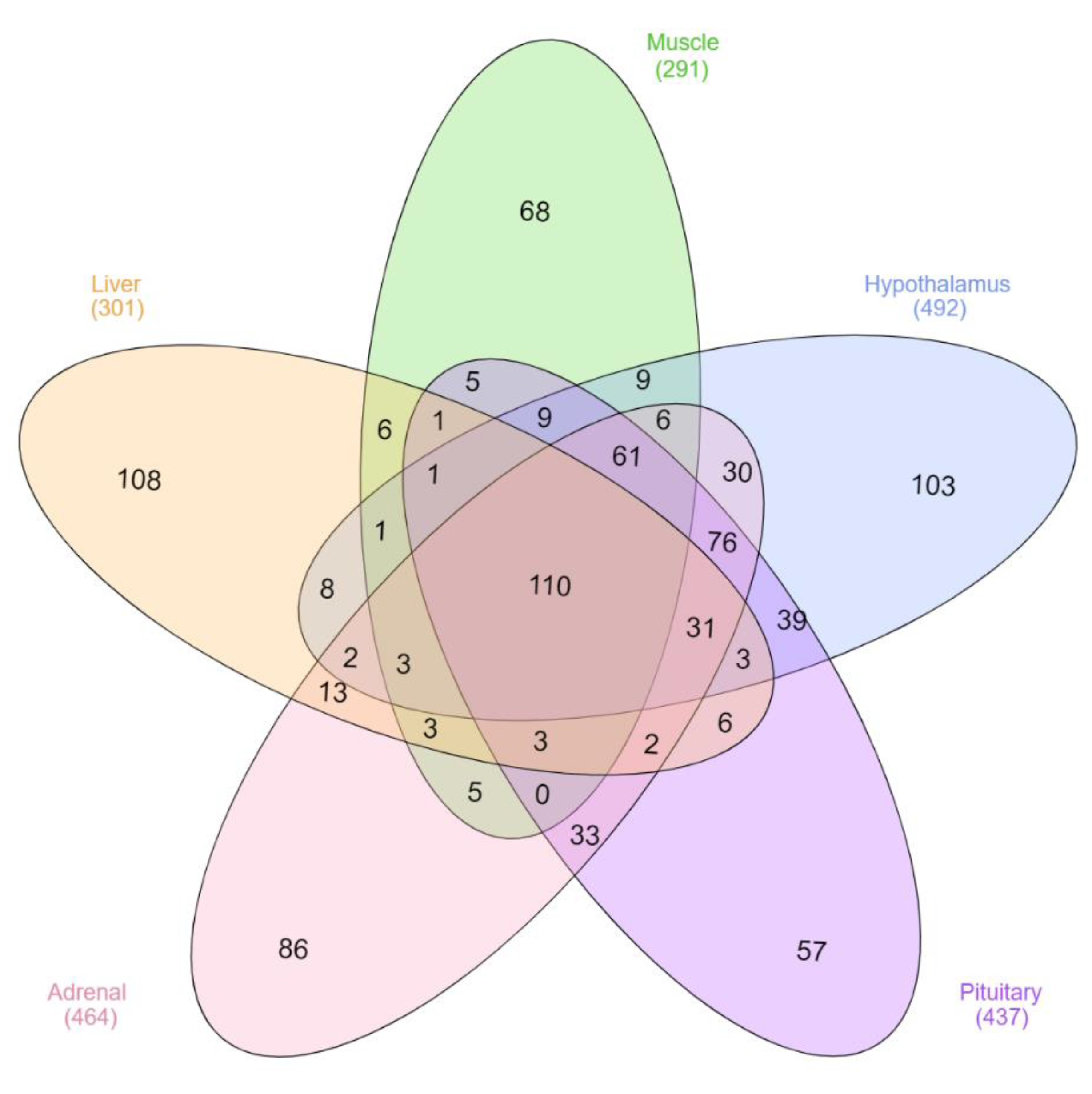
3.1. New lncRNA
3.2. Characteristics of New lncRNA
3.3. Differentially Expressed lncRNA
3.4. Key lncRNA
3.5. Possible Functions of Relevant lncRNA for Feed Efficiency
3.6. lncRNA Co-Expression Networks
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Salviano-Silva, A.; Lobo-Alves, S.; Almeida, R.; Malheiros, D.; Petzl-Erler, M. Besides pathology: Long non-coding RNA in cell and tissue homeostasis. Non-Coding RNA 2018, 4, 3. [Google Scholar] [CrossRef] [PubMed]
- Weikard, R.; Demasius, W.; Kuehn, C. Mining long noncoding RNA in livestock. Anim. Genet. 2017, 48, 3–18. [Google Scholar] [CrossRef] [PubMed]
- Deniz, E.; Erman, B. Long noncoding RNA (lincRNA), a new paradigm in gene expression control. Funct. Integr. Genom. 2017, 17, 135–143. [Google Scholar] [CrossRef] [PubMed]
- Quinn, J.J.; Chang, H.Y. Unique features of long non-coding RNA biogenesis and function. Nat. Rev. Genet. 2016, 17, 47–62. [Google Scholar] [CrossRef] [PubMed]
- Batista, P.J.; Chang, H.Y. Long noncoding RNAs: Cellular address codes in development and disease. Cell 2013, 152, 1298–1307. [Google Scholar] [CrossRef]
- Zheng, X.; Ning, C.; Zhao, P.; Feng, W.; Jin, Y.; Zhou, L.; Yu, Y.; Liu, J. Integrated analysis of long noncoding RNA and mRNA expression profiles reveals the potential role of long noncoding RNA in different bovine lactation stages. J. Dairy Sci. 2018, 101, 11061–11073. [Google Scholar] [CrossRef]
- Wang, H.; Wang, X.; Li, X.; Wang, Q.; Qing, S.; Zhang, Y.; Gao, M. A novel long non-coding RNA regulates the immune response in MAC—T cells and contributes to bovine mastitis. FEBS J. 2019, 286, 1780–1795. [Google Scholar] [CrossRef] [PubMed]
- Tong, C.; Chen, Q.; Zhao, L.; Ma, J.; Ibeagha-Awemu, E.M.; Zhao, X. Identification and characterization of long intergenic noncoding RNAs in bovine mammary glands. BMC Genom. 2017, 18, 468. [Google Scholar] [CrossRef]
- Sun, X.; Li, M.; Sun, Y.; Cai, H.; Lan, X.; Huang, Y.; Bai, Y.; Qi, X.; Chen, H. The developmental transcriptome sequencing of bovine skeletal muscle reveals a long noncoding RNA, lncMD, promotes muscle differentiation by sponging miR-125b. Biochim. Biophys. Acta. Mol. Cell Res. 2016, 1863, 2835–2845. [Google Scholar] [CrossRef]
- Li, Q.; Qiao, J.; Zhang, Z.; Shang, X.; Chu, Z.; Fu, Y.; Chu, M. Identification and analysis of differentially expressed long non-coding RNAs of Chinese Holstein cattle responses to heat stress. Anim. Biotechnol. 2020, 31, 9–16. [Google Scholar] [CrossRef]
- Mahmoudi, B.; Fayazi, J.; Roshanfekr, H.; Sari, M.; Bakhtiarizadeh, M.R. Genome-wide identification and characterization of novel long non-coding RNA in Ruminal tissue affected with sub-acute Ruminal acidosis from Holstein cattle. Vet. Res. Commun. 2020, 44, 19–27. [Google Scholar] [CrossRef] [PubMed]
- Gao, Y.; Li, S.; Lai, Z.; Zhou, Z.; Wu, F.; Huang, Y.; Lan, X.; Lei, C.; Chen, H.; Dang, R. Analysis of Long Non-Coding RNA and mRNA expression profiling in immature and mature bovine (Bos taurus) testes. Front. Genet. 2019, 10, 646. [Google Scholar] [CrossRef] [PubMed]
- Tizioto, P.C.; Coutinho, L.L.; Decker, J.E.; Schnabel, R.D.; Rosa, K.O.; Oliveira, P.S.; Souza, M.M.; Mourão, G.B.; Tullio, R.R.; Chaves, A.S.; et al. Global liver gene expression differences in Nelore steers with divergent residual feed intake phenotypes. BMC Genom. 2015, 16, 242. [Google Scholar] [CrossRef] [PubMed]
- Paradis, F.; Yue, S.; Grant, J.R.; Stothard, P.; Basarab, J.A.; Fitzsimmons, C. Transcriptomic analysis by RNA sequencing reveals that hepatic interferon-induced genes may be associated with feed efficiency in beef heifers. J. Anim. Sci. 2015, 93, 3331–3341. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Gondro, C.; Quinn, K.; Herd, R.M.; Parnell, P.F.; Vanselow, B. Global gene expression profiling reveals genes expressed differentially in cattle with high and low residual feed intake. Anim. Genet. 2011, 42, 475–490. [Google Scholar] [CrossRef]
- Weber, K.L.; Welly, B.T.; Van Eenennaam, A.L.; Young, A.E.; Port-Neto, L.R.; Reverter, A.; Rincon, G. Identification of Gene networks for residual feed intake in Angus cattle using genomic prediction and RNA-seq. PLoS ONE 2016, 11, 1–19. [Google Scholar] [CrossRef]
- De Lima, A.O.; Koltes, J.E.; Diniz, W.J.S.; de Oliveira, P.S.N.; Cesar, A.S.M.; Tizioto, P.C.; Afonso, J.; de Souza, M.M.; Petrini, J.; Rocha, M.I.P.; et al. Potential biomarkers for feed efficiency-related traits in nelore cattle identified by co-expression network and integrative genomics analyses. Front. Genet. 2020, 11, 189. [Google Scholar] [CrossRef]
- Koufariotis, L.T.; Chen, Y.-P.P.P.; Chamberlain, A.; Vander Jagt, C.; Hayes, B.J. A catalogue of novel bovine long noncoding RNA across 18 tissues. PLoS ONE 2015, 10, e0141225. [Google Scholar] [CrossRef]
- Signal, B.; Gloss, B.S.; Dinger, M.E. Computational approaches for functional prediction and characterisation of long noncoding RNAs. Trends Genet. 2016, 32, 620–637. [Google Scholar] [CrossRef]
- Nolte, W.; Weikard, R.; Brunner, R.M.; Albrecht, E.; Hammon, H.M.; Reverter, A.; Kühn, C. Biological network approach for the identification of regulatory long non-coding RNAs associated with metabolic efficiency in cattle. Front. Genet. 2019, 10, 1130. [Google Scholar] [CrossRef]
- Nolte, W.; Weikard, R.; Brunner, R.M.; Albrecht, E.; Hammon, H.M.; Reverter, A.; Kühn, C. Identification and annotation of potential function of regulatory antisense long non-coding RNAs related to feed efficiency in bos taurus bulls. Int. J. Mol. Sci. 2020, 21, 3292. [Google Scholar] [CrossRef] [PubMed]
- Porto-Neto, L.R.; Sonstegard, T.S.; Liu, G.E.; Bickhart, D.M.; Da Silva, M.V.B.; Machado, M.A.; Utsunomiya, Y.T.; Garcia, J.F.; Gondro, C.; Van Tassell, C.P. Genomic divergence of zebu and taurine cattle identified through high-density SNP genotyping. BMC Genom. 2013, 14, 876. [Google Scholar] [CrossRef] [PubMed]
- Utsunomiya, Y.T.; Milanesi, M.; Fortes, M.R.S.; Porto-Neto, L.R.; Utsunomiya, A.T.H.; Silva, M.V.G.B.; Garcia, J.F.; Ajmone-Marsan, P. Genomic clues of the evolutionary history of Bos indicus cattle. Anim. Genet. 2019, 50, 557–568. [Google Scholar] [CrossRef] [PubMed]
- Naval-Sánchez, M.; Porto-Neto, L.R.; Cardoso, D.F.; Hayes, B.J.; Daetwyler, H.D.; Kijas, J.; Reverter, A. Selection signatures in tropical cattle are enriched for promoter and coding regions and reveal missense mutations in the damage response gene HELB. Genet. Sel. Evol. 2020, 52, 27. [Google Scholar] [CrossRef] [PubMed]
- Alexandre, P.A.; Naval-Sanchez, M.; Porto-Neto, L.R.; Ferraz, J.B.S.; Reverter, A.; Fukumasu, H. Systems biology reveals NR2F6 and TGFB1 as key regulators of feed efficiency in beef cattle. Front. Genet. 2019, 10, 230. [Google Scholar] [CrossRef]
- Koch, R.M.; Swiger, L.A.; Chambers, D.; Gregory, K.E. Efficiency of feed use in beef cattle. J. Anim. Sci. 1963, 22, 486–494. [Google Scholar] [CrossRef]
- Alexandre, P.A.; Kogelman, L.J.A.; Santana, M.H.A.; Passarelli, D.; Pulz, L.H.; Fantinato-Neto, P.; Silva, P.L.; Leme, P.R.; Strefezzi, R.F.; Coutinho, L.L.L.; et al. Liver transcriptomic networks reveal main biological processes associated with feed efficiency in beef cattle. BMC Genom. 2015, 16, 1073. [Google Scholar] [CrossRef]
- Dobin, A.; Davis, C.A.; Schlesinger, F.; Drenkow, J.; Zaleski, C.; Jha, S.; Batut, P.; Chaisson, M.; Gingeras, T.R. STAR: Ultrafast universal RNA-seq aligner. Bioinformatics 2013, 29, 1543–1551. [Google Scholar] [CrossRef]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R. The sequence alignment/map format and SAMtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef]
- Ghosh, S.; Chan, C.-K.K. Analysis of RNA-Seq data using TopHat and cufflinks. Methods Mol. Biol. 2016, 1374, 339–361. [Google Scholar] [CrossRef]
- Trapnell, C. Tracking Transfrags through Multiple Samples. Available online: http://cole-trapnell-lab.github.io/cufflinks/cuffcompare/#tracking-transfrags-through-multiple-samples-outprefixtracking (accessed on 29 April 2020).
- Rice, P.; Longden, L.; Bleasby, A. EMBOSS: The European molecular biology open software suite. Trends Genet. 2000, 16, 276–277. [Google Scholar] [CrossRef]
- Niazi, F.; Valadkhan, S. Computational analysis of functional long noncoding RNAs reveals lack of peptide-coding capacity and parallels with 3′ UTRs. RNA 2012, 18, 825–843. [Google Scholar] [CrossRef] [PubMed]
- Camacho, C.; Coulouris, G.; Avagyan, V.; Ma, N.; Papadopoulos, J.; Bealer, K.; Madden, T.L. BLAST+: Architecture and applications. BMC Bioinformatics 2009, 10, 421. [Google Scholar] [CrossRef] [PubMed]
- Kang, Y.J.; Yang, D.C.; Kong, L.; Hou, M.; Meng, Y.Q.; Wei, L.; Gao, G. CPC2: A fast and accurate coding potential calculator based on sequence intrinsic features. Nucleic Acids Res. 2017, 45, W12–W16. [Google Scholar] [CrossRef] [PubMed]
- Trapnell, C.; Hendrickson, D.G.; Sauvageau, M.; Goff, L.; Rinn, J.L.; Pachter, L. Differential analysis of gene regulation at transcript resolution with RNA-seq. Nat. Biotechnol. 2013, 31, 46–53. [Google Scholar] [CrossRef]
- Fang, S.; Zhang, L.; Guo, J.; Niu, Y.; Wu, Y.; Li, H.; Zhao, L.; Li, X.; Teng, X.; Sun, X.; et al. NONCODEV5: A comprehensive annotation database for long non-coding RNAs. Nucleic Acids Res. 2018, 46, D308–D314. [Google Scholar] [CrossRef]
- Enright, A.J.; John, B.; Gaul, U.; Tuschl, T.; Sander, C.; Marks, D.S. MicroRNA targets in drosophila. Genome Biol. 2003, 5, R12. [Google Scholar] [CrossRef]
- Hofacker, I.L. Vienna RNA secondary structure server. Nucleic Acids Res. 2003, 31, 3429–3431. [Google Scholar] [CrossRef]
- Liao, Y.; Smyth, G.K.; Shi, W. FeatureCounts: An efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics 2014, 30, 923–930. [Google Scholar] [CrossRef]
- Robinson, M.D.; McCarthy, D.J.; Smyth, G.K. edgeR: A Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 2010, 26, 139–140. [Google Scholar] [CrossRef]
- Cánovas, A.; Reverter, A.; DeAtley, K.L.; Ashley, R.L.; Colgrave, M.L.; Fortes, M.R.S.; Islas-Trejo, A.; Lehnert, S.; Porto-Neto, L.; Rincón, G.; et al. Multi-Tissue omics analyses reveal molecular regulatory networks for puberty in composite beef cattle. PLoS ONE 2014, 9, e0102551. [Google Scholar] [CrossRef] [PubMed]
- Vaquerizas, J.M.; Kummerfeld, S.K.; Teichmann, S.A.; Luscombe, N.M. A census of human transcription factors: Function, expression and evolution. Nat. Rev. Genet. 2009, 10, 252–263. [Google Scholar] [CrossRef] [PubMed]
- Heberle, H.; Meirelles, V.G.; da Silva, F.R.; Telles, G.P.; Minghim, R. InteractiVenn: A web-based tool for the analysis of sets through Venn diagrams. BMC Bioinformatics 2015, 16, 169. [Google Scholar] [CrossRef]
- Reverter, A.; Hudson, N.J.; Nagaraj, S.H.; Pérez-Enciso, M.; Dalrymple, B.P. Regulatory impact factors: Unraveling the transcriptional regulation of complex traits from expression data. Bioinformatics 2010, 26, 896–904. [Google Scholar] [CrossRef] [PubMed]
- Hudson, N.J.; Dalrymple, B.P.; Reverter, A. Beyond differential expression: The quest for causal mutations and effector molecules. BMC Genom. 2012, 13, 356. [Google Scholar] [CrossRef]
- Hu, Z.-L.; Park, C.A.; Wu, X.-L.; Reecy, J.M. Animal QTLdb: An improved database tool for livestock animal QTL/association data dissemination in the post-genome era. Nucleic Acids Res. 2013, 41, D871–D879. [Google Scholar] [CrossRef]
- Reverter, A.; Chan, E.K.F. Combining partial correlation and an information theory approach to the reversed engineering of gene co-expression networks. Bioinformatics 2008, 24, 2491–2497. [Google Scholar] [CrossRef]
- Backes, C.; Khaleeq, Q.T.; Meese, E.; Keller, A. MiEAA: MicroRNA enrichment analysis and annotation. Nucleic Acids Res. 2016, 44, W110–W116. [Google Scholar] [CrossRef]
- Hofacker, I.L.; Fontana, W.; Stadler, P.F.; Bonhoeffer, L.S.; Tacker, M.; Schuster, P. Fast folding and comparison of RNA secondary structures. Mon. Chem. Chem. Mon. 1989, 125, 167–188. [Google Scholar] [CrossRef]
- Zhang, B.; Kirov, S.; Snoddy, J. WebGestalt: An integrated system for exploring gene sets in various biological contexts. Nucleic Acids Res. 2005, 33, W741–W748. [Google Scholar] [CrossRef]
- Kosinska-Selbi, B.; Mielczarek, M.; Szyda, J. Review: Long non-coding RNA in livestock. Animal 2020, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Kern, C.; Wang, Y.; Chitwood, J.; Korf, I.; Delany, M.; Cheng, H.; Medrano, J.F.; Van Eenennaam, A.L.; Ernst, C.; Ross, P.; et al. Genome-wide identification of tissue-specific long non-coding RNA in three farm animal species. BMC Genom. 2018, 19, 684. [Google Scholar] [CrossRef] [PubMed]
- Lloyd, K.C.K.; Adams, D.J.; Baynam, G.; Beaudet, A.L.; Bosch, F.; Boycott, K.M.; Braun, R.E.; Caulfield, M.; Cohn, R.; Dickinson, M.E.; et al. The deep genome project. Genome Biol. 2020, 21, 18. [Google Scholar] [CrossRef] [PubMed]
- Mukiibi, R.; Vinsky, M.; Keogh, K.A.; Fitzsimmons, C.; Stothard, P.; Waters, S.M.; Li, C. Transcriptome analyses reveal reduced hepatic lipid synthesis and accumulation in more feed efficient beef cattle. Sci. Rep. 2018, 8, 7303. [Google Scholar] [CrossRef] [PubMed]
- Salleh, M.S.; Mazzoni, G.; Höglund, J.K.; Olijhoek, D.W.; Lund, P.; Løvendahl, P.; Kadarmideen, H.N. RNA-Seq transcriptomics and pathway analyses reveal potential regulatory genes and molecular mechanisms in high- and low-residual feed intake in Nordic dairy cattle. BMC Genom. 2017, 18, 258. [Google Scholar] [CrossRef]
- Mani, V.; Harris, A.J.; Keating, A.F.; Weber, T.E.; Dekkers, J.C.M.; Gabler, N.K. Intestinal integrity, endotoxin transport and detoxification in pigs divergently selected for residual feed intake. J. Anim. Sci. 2013, 91, 2141–2150. [Google Scholar] [CrossRef]
- Ramayo-Caldas, Y.; Ballester, M.; Sánchez, J.P.; González-Rodríguez, O.; Revilla, M.; Reyer, H.; Wimmers, K.; Torrallardona, D.; Quintanilla, R. Integrative approach using liver and duodenum RNA-Seq data identifies candidate genes and pathways associated with feed efficiency in pigs. Sci. Rep. 2018, 8, 558. [Google Scholar] [CrossRef]
- Karisa, B.; Moore, S.; Plastow, G. Analysis of biological networks and biological pathways associated with residual feed intake in beef cattle. Anim. Sci. J. 2014, 85, 374–387. [Google Scholar] [CrossRef]
- Santana, M.H.A.; Rossi, P.; Almeida, R.; Cucco, D.C. Feed efficiency and its correlations with carcass traits measured by ultrasound in Nellore bulls. Livest. Sci. 2012, 145, 252–257. [Google Scholar] [CrossRef]
- Gomes, R.C.; Sainz, R.D.; Silva, S.L.; César, M.C.; Bonin, M.N.; Leme, P.R. Feedlot performance, feed efficiency reranking, carcass traits, body composition, energy requirements, meat quality and calpain system activity in Nellore steers with low and high residual feed intake. Livest. Sci. 2012, 150, 265–273. [Google Scholar] [CrossRef]
- Basarab, J.A.; Price, M.A.; Aalhus, J.L.; Okine, E.K.; Snelling, W.M.; Lyle, K.L. Residual feed intake and body composition in young growing cattle. Can. J. Anim. Sci. 2003, 83, 189–204. [Google Scholar] [CrossRef]
- Mader, C.J.; Montanholi, Y.R.; Wang, Y.J.; Miller, S.P.; Mandell, I.B.; McBride, B.W.; Swanson, K.C. Relationships among measures of growth performance and efficiency with carcass traits, visceral organ mass, and pancreatic digestive enzymes in feedlot cattle. J. Anim. Sci. 2009, 87, 1548–1557. [Google Scholar] [CrossRef] [PubMed]
- Oikonomopoulou, K.; Ricklin, D.; Ward, P.A.; Lambris, J.D. Interactions between coagulation and complement—Their role in inflammation. Semin. Immunopathol. 2012, 34, 151–165. [Google Scholar] [CrossRef] [PubMed]
- Tizioto, P.C.; Coutinho, L.L.; Oliveira, P.S.N.N.; Cesar, A.S.M.M.; Diniz, W.J.S.S.; Lima, A.O.; Rocha, M.I.; Decker, J.E.; Schnabel, R.D.; Mourão, G.B.; et al. Gene expression differences in Longissimus muscle of Nelore steers genetically divergent for residual feed intake. Sci. Rep. 2016, 6, 39493. [Google Scholar] [CrossRef]
- Jing, L.; Hou, Y.; Wu, H.; Miao, Y.; Li, X.; Cao, J.; Brameld, J.M.; Parr, T.; Zhao, S. Transcriptome analysis of mRNA and miRNA in skeletal muscle indicates an important network for differential Residual Feed Intake in pigs. Sci. Rep. 2015, 5, 11953. [Google Scholar] [CrossRef]
- Carmelo, V.A.O.; Kadarmideen, H.N. Genome regulation and gene interaction networks inferred from muscle transcriptome underlying feed efficiency in pigs. Front. Genet. 2020, 11, 650. [Google Scholar] [CrossRef]
- Fukumasu, H.; Santana, M.H.; Alexandre, P.A.; Ferraz, J.B.S. Systems Biology Application in Feed Efficiency in Beef Cattle; Springer: Cham, Switzerland, 2016; Volume 2, pp. 79–95. [Google Scholar]
- Gondret, F.; Vincent, A.; Houée-Bigot, M.; Siegel, A.; Lagarrigue, S.; Causeur, D.; Gilbert, H.; Louveau, I. A transcriptome multi-tissue analysis identifies biological pathways and genes associated with variations in feed efficiency of growing pigs. BMC Genom. 2017, 18, 244. [Google Scholar] [CrossRef]
- Bush, J.A.; Kimball, S.R.; O’Connor, P.M.J.; Suryawan, A.; Orellana, R.A.; Nguyen, H.V.; Jefferson, L.S.; Davis, T.A. Translational control of protein synthesis in muscle and liver of growth hormone-treated pigs. Endocrinology 2003, 144, 1273–1283. [Google Scholar] [CrossRef][Green Version]
- Castro Bulle, F.C.P.; Paulino, P.V.; Sanches, A.C.; Sainz, R.D. Growth, carcass quality, and protein and energy metabolism in beef cattle with different growth potentials and residual feed intakes. J. Anim. Sci. 2007, 85, 928–936. [Google Scholar] [CrossRef]
- Widmann, P.; Reverter, A.; Weikard, R.; Suhre, K.; Hammon, H.M.; Albrecht, E.; Kuehn, C. Systems biology analysis merging phenotype, metabolomic and genomic data identifies Non-SMC Condensin I Complex, Subunit G (NCAPG) and cellular maintenance processes as major contributors to genetic variability in Bovine feed efficiency. PLoS ONE 2015, 10, e0124574. [Google Scholar] [CrossRef]
- Yu, X.; Zhang, Y.; Li, T.; Ma, Z.; Jia, H.; Chen, Q.; Zhao, Y.; Zhai, L.; Zhong, R.; Li, C.; et al. Long non-coding RNA Linc-RAM enhances myogenic differentiation by interacting with MyoD. Nat. Commun. 2017, 8, 14016. [Google Scholar] [CrossRef] [PubMed]
- Zhu, M.; Liu, J.; Xiao, J.; Yang, L.; Cai, M.; Shen, H.; Chen, X.; Ma, Y.; Hu, S.; Wang, Z.; et al. Lnc-mg is a long non-coding RNA that promotes myogenesis. Nat. Commun. 2017, 8, 14718. [Google Scholar] [CrossRef]
- Liu, P.; Jin, L.; Zhao, L.; Long, K.; Song, Y.; Tang, Q.; Ma, J.; Wang, X.; Tang, G.; Jiang, Y.; et al. Identification of a novel antisense long non-coding RNA PLA2G16-AS that regulates the expression of PLA2G16 in pigs. Gene 2018, 671, 78–84. [Google Scholar] [CrossRef] [PubMed]
- Zhan, S.; Dong, Y.; Zhao, W.; Guo, J.; Zhong, T.; Wang, L.; Li, L.; Zhang, H. Genome-wide identification and characterization of long non-coding RNAs in developmental skeletal muscle of fetal goat. BMC Genom. 2016, 17, 666. [Google Scholar] [CrossRef] [PubMed]
- Sui, Y.; Han, Y.; Zhao, X.; Li, D.; Li, G. Long non-coding RNA Irm enhances myogenic differentiation by interacting with MEF2D. Cell Death Dis. 2019, 10, 181. [Google Scholar] [CrossRef]
- Cônsolo, N.R.B.; Da Silva, J.; Buarque, V.L.M.; Higuera-Padilla, A.; Barbosa, L.C.G.S.; Zawadzki, A.; Colnago, L.A.; Saran Netto, A.; Gerrard, D.E.; Silva, S.L. Selection for growth and precocity alters muscle metabolism in nellore cattle. Metabolites 2020, 10, 58. [Google Scholar] [CrossRef]
- Nkrumah, J.D.; Keisler, D.H.; Crews, D.H.; Basarab, J.A.; Wang, Z.; Li, C.; Price, M.A.; Okine, E.K.; Moore, S.S. Genetic and phenotypic relationships of serum leptin concentration with performance, efficiency of gain, and carcass merit of feedlot cattle. J. Anim. Sci. 2007, 85, 2147–2155. [Google Scholar] [CrossRef]
- Widmann, P.; Reverter, A.; Fortes, M.R.S.; Weikard, R.; Suhre, K.; Hammon, H.; Albrecht, E.; Kuehn, C. A systems biology approach using metabolomic data reveals genes and pathways interacting to modulate divergent growth in cattle. BMC Genom. 2013, 14, 798. [Google Scholar] [CrossRef]
- Reyer, H.; Oster, M.; Magowan, E.; Muráni, E.; Sauerwein, H.; Dannenberger, D.; Kuhla, B.; Ponsuksili, S.; Wimmers, K. Feed-efficient pigs exhibit molecular patterns allowing a timely circulation of hormones and nutrients. Physiol. Genom. 2018, 50, 729–734. [Google Scholar] [CrossRef]
- Ramayo-Caldas, Y.; Mármol-Sánchez, E.; Ballester, M.; Sánchez, J.P.; González-Prendes, R.; Amills, M.; Quintanilla, R. Integrating genome-wide co-association and gene expression to identify putative regulators and predictors of feed efficiency in pigs. Genet. Sel. Evol. 2019, 51, 48. [Google Scholar] [CrossRef]
- Hou, Y.; Hu, M.; Zhou, H.; Li, C.; Li, X.; Liu, X.; Zhao, Y.; Zhao, S. Neuronal signal transduction-involved genes in pig hypothalamus affect feed efficiency as revealed by transcriptome analysis. Biomed Res. Int. 2018, 2018, 1–10. [Google Scholar] [CrossRef]
- Cafe, L.M.; Robinson, D.L.; Ferguson, D.M.; Geesink, G.H.; Greenwood, P.L. Temperament and hypothalamic-pituitary-adrenal axis function are related and combine to affect growth, efficiency, carcass, and meat quality traits in Brahman steers. Domest. Anim. Endocrinol. 2011, 40, 230–240. [Google Scholar] [CrossRef] [PubMed]
- Kelly, A.K.; McGee, M.; Crews, D.H.; Fahey, A.G.; Wylie, A.R.; Kenny, D.A. Effect of divergence in residual feed intake on feeding behavior, blood metabolic variables, and body composition traits in growing beef heifers. J. Anim. Sci. 2010, 88, 109–123. [Google Scholar] [CrossRef] [PubMed]
- Richardson, E.C.; Herd, R.M.; Archer, J.A.; Arthur, P.F. Metabolic differences in Angus steers divergently selected for residual feed intake. Aust. J. Exp. Agric. 2004, 44, 441–452. [Google Scholar] [CrossRef]
- Foote, A.P.; Tait, R.G.; Keisler, D.H.; Hales, K.E.; Freetly, H.C. Leptin concentrations in finishing beef steers and heifers and their association with dry matter intake, average daily gain, feed efficiency, and body composition. Domest. Anim. Endocrinol. 2016, 55, 136–141. [Google Scholar] [CrossRef] [PubMed]
- Hoque, M.A.; Katoh, K.; Suzuki, K. Genetic associations of residual feed intake with serum insulin-like growth factor-I and leptin concentrations, meat quality, and carcass cross sectional fat area ratios in Duroc pigs. J. Anim. Sci. 2009, 87, 3069–3075. [Google Scholar] [CrossRef]
- Perkins, S.D.; Key, C.N.; Marvin, M.N.; Garrett, C.F.; Foradori, C.D.; Bratcher, C.L.; Kriese-Anderson, L.A.; Brandebourg, T.D. Effect of residual feed intake on hypothalamic gene expression and meat quality in Angus-sired cattle grown during the hot season1,2. J. Anim. Sci. 2014, 92, 1451–1461. [Google Scholar] [CrossRef]
- Perkins, S.D.; Key, C.N.; Garrett, C.F.; Foradori, C.D.; Bratcher, C.L.; Kriese-Anderson, L.A.; Brandebourg, T.D. Residual feed intake studies in Angus-sired cattle reveal a potential role for hypothalamic gene expression in regulating feed efficiency1,2. J. Anim. Sci. 2014, 92, 549–560. [Google Scholar] [CrossRef]
- Barrett, J.; Canning, B.; Dombrowsky, E.; Douglas, S.; Fong, T.; Heyward, C.; Leeman, S.; Remeshwar, P. Tachykinin receptors (version 2019.4) in the IUPHAR/BPS Guide to pharmacology database. IUPHAR/BPS Guid. Pharm. 2019, 2019. [Google Scholar] [CrossRef]
- Volkow, N.D.; Wang, G.J.; Baler, R.D. Reward, dopamine and the control of food intake: Implications for obesity. Trends Cogn. Sci. 2011, 15, 37–46. [Google Scholar] [CrossRef]
- Love, T.M. Oxytocin, motivation and the role of dopamine. Pharm. Biochem. Behav. 2014, 119, 49–60. [Google Scholar] [CrossRef] [PubMed]
- Wise, R.A. Dual Roles of Dopamine in food and drug seeking. Biol. Psychiatry 2013, 73, 819–826. [Google Scholar] [CrossRef] [PubMed]
- Guo, L.; Zheng, Z.; Ai, J.; Huang, B.; Li, X.A. Hepatic scavenger receptor bi protects against polymicrobial-induced sepsis through promoting LPS clearance in mice. J. Biol. Chem. 2014, 289, 14666–14673. [Google Scholar] [CrossRef] [PubMed]
- Fernández-Sánchez, A.; Madrigal-Santillán, E.; Bautista, M.; Esquivel-Soto, J.; Morales-González, A.; Esquivel-Chirino, C.; Durante-Montiel, I.; Sánchez-Rivera, G.; Valadez-Vega, C.; Morales-González, J.A. Inflammation, oxidative stress, and obesity. Int. J. Mol. Sci. 2011, 12, 3117–3132. [Google Scholar] [CrossRef]
- Kälin, S.; Heppner, F.L.; Bechmann, I.; Prinz, M.; Tschöp, M.H.; Yi, C.-X. Hypothalamic innate immune reaction in obesity. Nat. Rev. Endocrinol. 2015, 11, 339–351. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Tissue | DE IncRNA |
|---|---|
| Adrenal gland | TCONS_00223090, TCONS_00141903, TCONS_00214308, TCONS_00040537, TCONS_00119463, TCONS_00093659, TCONS_00180358, TCONS_00072894, TCONS_00034840, TCONS_00164459, TCONS_00027608, TCONS_00015370, TCONS_00127543 |
| Hypothalamus | TCONS_00222966, TCONS_00128697, TCONS_00016951, TCONS_00065862, TCONS_00106598, TCONS_00157676, TCONS_00083779, TCONS_00139694, TCONS_00141903 |
| Liver | TCONS_00106745, TCONS_00130767, TCONS_00061987, TCONS_00025987, TCONS_00128934, TCONS_00157869, TCONS_00222578, TCONS_00222972, TCONS_00188391, TCONS_00222966 |
| Muscle | TCONS_00140963, TCONS_00223154, TCONS_00128551, TCONS_00032445, TCONS_00095545, TCONS_00000271, TCONS_00141506, TCONS_00051404, TCONS_00120014, TCONS_00033623, TCONS_00203516, TCONS_00051406, TCONS_00167041, TCONS_00190543 |
| Pituitary gland | TCONS_00116172, TCONS_00032383, TCONS_00105367, TCONS_00077897, TCONS_00157315, TCONS_00202013, TCONS_00062811, TCONS_00009194, TCONS_00131281, TCONS_00150705, TCONS_00170772, TCONS_00116008, TCONS_00168127, TCONS_00188529, TCONS_00059814, TCONS_00223090, TCONS_00141903 |
| Tissue | Key IncRNA |
|---|---|
| Adrenal gland | TCONS_00106745, TCONS_00040537, TCONS_00006522, TCONS_00013774, TCONS_00022218, TCONS_00048225, TCONS_00064059, TCONS_00065193, TCONS_00065195, TCONS_00083522, TCONS_00088984, TCONS_00126728, TCONS_00154980, TCONS_00159584, TCONS_00171940, TCONS_00178323, TCONS_00182439, TCONS_00186763, TCONS_00193324, TCONS_00201789, TCONS_00219008 |
| Hypothalamus | TCONS_00214308, TCONS_00018896, TCONS_00028218, TCONS_00028219, TCONS_00033000, TCONS_00061315, TCONS_00068546, TCONS_00153695, TCONS_00157240, TCONS_00157945, TCONS_00164540, TCONS_00169707, TCONS_00176859, TCONS_00187047, TCONS_00198904 |
| Liver | TCONS_00056607, TCONS_00079733, TCONS_00090296, TCONS_00096860, TCONS_00111349, TCONS_00159585, TCONS_00185398, TCONS_00190687 |
| Muscle | TCONS_00140963, TCONS_00011978, TCONS_00028495, TCONS_00064224, TCONS_00103343, TCONS_00116181, TCONS_00119451, TCONS_00122105, TCONS_00135035, TCONS_00171719 |
| Pituitary gland | TCONS_00006521, TCONS_00012621, TCONS_00018857, TCONS_00024003, TCONS_00029744, TCONS_00045668, TCONS_00053912, TCONS_00056694, TCONS_00116405, TCONS_00140488, TCONS_00142880, TCONS_00149966, TCONS_00166200, TCONS_00184540, TCONS_00184673, TCONS_00202748, TCONS_00222510 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Alexandre, P.A.; Reverter, A.; Berezin, R.B.; Porto-Neto, L.R.; Ribeiro, G.; Santana, M.H.A.; Ferraz, J.B.S.; Fukumasu, H. Exploring the Regulatory Potential of Long Non-Coding RNA in Feed Efficiency of Indicine Cattle. Genes 2020, 11, 997. https://doi.org/10.3390/genes11090997
Alexandre PA, Reverter A, Berezin RB, Porto-Neto LR, Ribeiro G, Santana MHA, Ferraz JBS, Fukumasu H. Exploring the Regulatory Potential of Long Non-Coding RNA in Feed Efficiency of Indicine Cattle. Genes. 2020; 11(9):997. https://doi.org/10.3390/genes11090997
Chicago/Turabian StyleAlexandre, Pâmela A., Antonio Reverter, Roberta B. Berezin, Laercio R. Porto-Neto, Gabriela Ribeiro, Miguel H. A. Santana, José Bento S. Ferraz, and Heidge Fukumasu. 2020. "Exploring the Regulatory Potential of Long Non-Coding RNA in Feed Efficiency of Indicine Cattle" Genes 11, no. 9: 997. https://doi.org/10.3390/genes11090997
APA StyleAlexandre, P. A., Reverter, A., Berezin, R. B., Porto-Neto, L. R., Ribeiro, G., Santana, M. H. A., Ferraz, J. B. S., & Fukumasu, H. (2020). Exploring the Regulatory Potential of Long Non-Coding RNA in Feed Efficiency of Indicine Cattle. Genes, 11(9), 997. https://doi.org/10.3390/genes11090997