Molecular Evidence for Hybrid Origin and Phenotypic Variation of Rosa Section Chinenses

 ,
,
Abstract
1. Introduction
2. Materials and Methods
2.1. Experimental Materials
2.2. Measurement and Analysis of Phenotypic Traits
2.3. DNA Extraction, Amplification, and Sequencing
2.4. Analysis of Genetic Diversity
3. Results
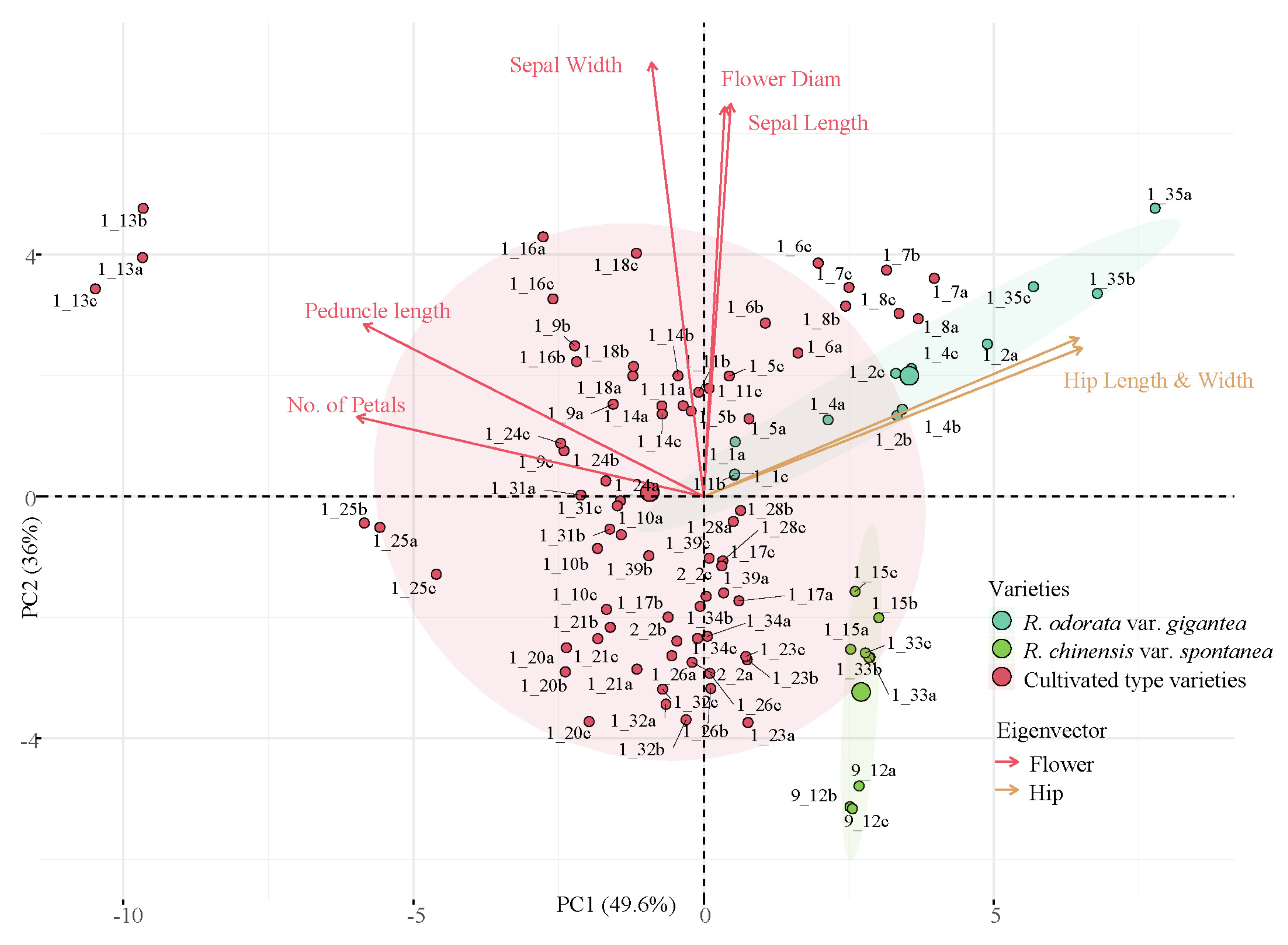
3.1. Variance Analysis of Phenotypic Traits
3.2. Genetic Diversity of SSR Markers
3.3. Screening of SCGs
3.4. Phylogenetic Analysis
4. Discussion
4.1. Origin of Cultivation of the China Rose
4.2. Variation of Phenotypic Characters of Cultivated Type Varieties
4.3. Role of Rose Germplasm Resources
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Wang, G.L. China’s Ancient Rose; Science Press: Beijing, China, 2015. [Google Scholar]
- Wissemann, V. Conventional taxonomy (wild roses). In Reference Module in Life Sciences; Reference Module in Life Sciences; Elsevier: Amsterdam, The Netherlands, 2017. [Google Scholar]
- Ku, C.Z.; Robertson, K.R. Flora of China; Science Press: Beijing, China, 2003. [Google Scholar]
- Roberts, A.V. Encyclopedia of Rose Science; Academic Press: San Diego, CA, USA, 2003. [Google Scholar]
- Meng, J.; Fougere-Danezan, M.; Zhang, L.B.; Li, D.Z.; Yi, T.S. Untangling the hybrid origin of the Chinese tea roses: Evidence from DNA sequences of single-copy nuclear and chloroplast genes. Plant Syst. Evol. 2011, 297, 157–170. [Google Scholar] [CrossRef]
- Miller, A.J.; Gross, B.L. From forest to field: Perennial fruit crop domestication. Am. J. Bot. 2011, 98, 1389–1414. [Google Scholar] [CrossRef]
- Wei, X.M.; Gao, X.F.; Zhang, L.B. A systematic study of Rosa sericea (Rosaceae) complex: Are R. omeiensis and R. sericea conspecific? J. Syst. Evol. 2008, 46, 919–928. [Google Scholar] [CrossRef]
- Ahmadi, S.J.; Mortazaeinezhad, F.; Zeinali, H.; Askari-Khorasgani, O.; Pessarakli, M. Evaluation of various Rosa damascena mill. genotypes grown under rainfed semi-arid condition. Commun. Soil Sci. Plant Anal. 2019, 50, 2534–2543. [Google Scholar] [CrossRef]
- Vaezi, J.; Arjmandi, A.A.; Sharghi, H.R. Origin of Rosa x binaloudensis (Rosaceae), a new natural hybrid species from Iran. PhytoTaxa 2019, 411, 23–38. [Google Scholar] [CrossRef]
- Meng, J.; Li, D.; Yi, T.; Yang, J.; Zhao, X. Development and characterization of microsatellite loci for Rosa odorata var. gigantea Rehder & E. H. Wilson (Rosaceae). Conserv. Genet. 2009, 10, 1973–1976. [Google Scholar] [CrossRef]
- Panwar, S.; Singh, K.P.; Namita, N.A.M.I.T.A.; Sonah, H.; Deshmukh, R.K.; Sharma, T.R. Identification and characterization of microsatellites in ESTs of Rosa species: Insight in development of SSR markers. Indian J. Agric. Sci. 2015, 85, 137–141. [Google Scholar]
- Scariot, V.; Akkak, A.; Botta, R. Characterization and genetic relationships of wild species and old garden roses based on microsatellite analysis. J. Am. Soc. Hortic. Sci. 2006, 131, 66–73. [Google Scholar] [CrossRef]
- Liorzou, M.; Pernet, A.; Li, S.; Chastellier, A.; Thouroude, T.; Michel, G.; Malecot, V.; Gaillard, S.; Briee, C.; Foucher, F.; et al. Nineteenth century French rose (Rosa sp.) germplasm shows a shift over time from a European to an Asian genetic background. J. Exp. Bot. 2016, 67, 4711–4725. [Google Scholar] [CrossRef] [PubMed]
- Joly, S.; Starr, J.R.; Lewis, W.H.; Bruneau, A. Polyploid and hybrid evolution in roses east of the Rocky Mountains. Am. J. Bot. 2006, 93, 412–425. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Z.M.; Gao, X.F.; Fougere-Danezan, M. Phylogeny of Rosa sections Chinenses and Synstylae (Rosaceae) based on chloroplast and nuclear markers. Mol. Phylogenetics Evol. 2015, 87, 50–64. [Google Scholar] [CrossRef] [PubMed]
- Martin, W.F.; Cerff, R. Physiology, phylogeny, early evolution, and GAPDH. Protoplasma 2017, 254, 1823–1834. [Google Scholar] [CrossRef] [PubMed]
- Hibrand Saint-Oyant, L.; Ruttink, T.; Hamama, L.; Kirov, I.; Lakhwani, D.; Zhou, N.N.; Bourke, P.M.; Daccord, N.; Leus, L.; Schulz, D.; et al. A high-quality genome sequence of Rosa chinensis to elucidate ornamental traits. Nat. Plants 2018, 4, 473–484. [Google Scholar] [CrossRef] [PubMed]
- Raymond, O.; Gouzy, J.; Just, J.; Badouin, H.; Verdenaud, M.; Lemainque, A.; Vergne, P.; Moja, S.; Choisne, N.; Pont, C.; et al. The Rosa genome provides new insights into the domestication of modern roses. Nat. Genet. 2018, 50, 772–777. [Google Scholar] [CrossRef]
- Debray, K.; Marie-Magdelaine, J.; Ruttink, T.; Clotault, J.; Foucher, F.; Malecot, V. Identification and assessment of variable single-copy orthologous (SCO) nuclear loci for low-level phylogenomics: A case study in the genus Rosa (Rosaceae). BMC Evol. Biol. 2019, 19. [Google Scholar] [CrossRef] [PubMed]
- Sang, T. Utility of low-copy nuclear gene sequences in plant phylogenetics. Crit. Rev. Biochem. Mol. Biol. 2002, 37, 121–147. [Google Scholar] [CrossRef]
- Reboud, X.; Zeyl, C. Organelle inheritance in plants. Heredity 1994, 72, 132–140. [Google Scholar] [CrossRef]
- Moe, R. Growth and flowering in roses. Acta Hortic. 1988, 218, 121–129. [Google Scholar] [CrossRef]
- Nadeem, M.; Khan, M.A.; Riaz, A.; Ahmad, R. Evaluation of growth and flowering potential of Rosa Hybrida cultivars under faisalabad climatic conditions. Pak. J. Agric. Sci. 2011, 48, 283–288. [Google Scholar]
- Liang, S.; Wu, X.; Byrne, D. Genetic analysis of flower size and production in diploid rose. J. Am. Soc. Hortic. Sci. 2017, 142, 306–313. [Google Scholar] [CrossRef]
- Lê, S.; Josse, J.; Husson, F. FactoMineR: A Package for Multivariate Analysis. J. Stat. Softw. 2008, 25, 1–18. [Google Scholar] [CrossRef]
- Yu, C. Construction of a Genetic Linkage Map and QTLs Analysis for Phenotypic Traits of Tetraploid Roses. PhD Thesis, Beijing Forestry University, Beijing, China, 2015. [Google Scholar]
- Hibrand-Saint Oyant, L.; Crespel, L.; Rajapakse, S.; Zhang, L.; Foucher, F. Genetic linkage maps of rose constructed with new microsatellite markers and locating QTL controlling flowering traits. Tree Genet. Genomes 2008, 4, 11–23. [Google Scholar] [CrossRef]
- Taberlet, P.; Gielly, L.; Pautou, G.; Bouvet, J. Universal primers for amplification of three non-coding regions of chloroplast DNA. Plant Mol. Biol. 1991, 17, 1105–1109. [Google Scholar] [CrossRef]
- Golenberg, E.M.; Clegg, M.T.; Durbin, M.L.; Doebley, J.; Ma, D.P. Evolution of a noncoding region of the chloroplast genome. Mol. Phylogenetics Evol. 1993, 2, 52–64. [Google Scholar] [CrossRef]
- Duarte, J.M.; Wall, P.K.; Edger, P.P.; Landherr, L.L.; Ma, H.; Pires, J.C.; Leebens-Mack, J.; DePamphilis, C.W. Identification of shared single copy nuclear genes in Arabidopsis, Populus, Vitis and Oryza and their phylogenetic utility across various taxonomic levels. BMC Evol. Biol. 2010, 10. [Google Scholar] [CrossRef]
- Camacho, C.; Coulouris, G.; Avagyan, V.; Ma, N.; Papadopoulos, J.; Bealer, K.; Madden, T.L. BLAST+: Architecture and applications. BMC Bioinform. 2009, 10, 421. [Google Scholar] [CrossRef]
- AppliedBiosystems. GeneMapper Software 5, Client Installation. Available online: https://www.thermofisher.com/order/catalog/product/4366847?SID=srch-hj-4366847#/4366847?SID=srch-hj-4366847 (accessed on 5 December 2019).
- Peakall, R.; Smouse, P.E. GenAlEx 6.5: Genetic analysis in Excel. Population genetic software for teaching and research-an update. Bioinformatics 2012, 28, 2537–2539. [Google Scholar] [CrossRef]
- Piry, S.; Luikart, G.; Cornuet, J.M. Computer note. BOTTLENECK: A computer program for detecting recent reductions in the effective size using allele frequency data. J. Hered. 1999, 90, 502–503. [Google Scholar] [CrossRef]
- Falush, D.; Stephens, M.; Pritchard, J.K. Inference of population structure using multilocus genotype data: Dominant markers and null alleles. Mol. Ecol. Notes 2007, 7, 574–578. [Google Scholar] [CrossRef]
- Earl, D.A.; VonHoldt, B.M. STRUCTURE HARVESTER: A website and program for visualizing STRUCTURE output and implementing the Evanno method. Conserv. Genet. Resour. 2012, 4, 359–361. [Google Scholar] [CrossRef]
- Jakobsson, M.; Rosenberg, N.A. CLUMPP: A cluster matching and permutation program for dealing with label switching and multimodality in analysis of population structure. Bioinformatics 2007, 23, 1801–1806. [Google Scholar] [CrossRef] [PubMed]
- Katoh, K.; Standley, D.M. MAFFT multiple sequence alignment software version 7: Improvements in performance and usability. Mol. Biol. Evol. 2013, 30, 772–780. [Google Scholar] [CrossRef] [PubMed]
- Rozas, J.; Ferrer-Mata, A.; Carlos Sanchez-DelBarrio, J.; Guirao-Rico, S.; Librado, P.; Ramos-Onsins, S.E.; Sanchez-Gracia, A. DnaSP 6: DNA sequence polymorphism analysis of large data sets. Mol. Biol. Evol. 2017, 34, 3299–3302. [Google Scholar] [CrossRef] [PubMed]
- Zhang, D.; Gao, F.; Jakovlic, I.; Zou, H.; Zhang, J.; Li, W.X.; Wang, G.T. PhyloSuite: An integrated and scalable desktop platform for streamlined molecular sequence data management and evolutionary phylogenetics studies. Mol. Ecol. Resour. 2020, 20, 348–355. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.B.; Li, D.Z.; Li, H.T. Highly effective sequencing whole chloroplast genomes of angiosperms by nine novel universal primer pairs. Mol. Ecol. Resour. 2014, 14, 1024–1031. [Google Scholar] [CrossRef]
- Jian, H.Y.; Zhang, Y.H.; Yan, H.J.; Qiu, X.Q.; Wang, Q.G.; Li, S.B.; Zhang, S.D. The complete chloroplast genome of a key ancestor of Modern Roses, Rosa chinensis var. spontanea, and a comparison with congeneric species. Molecules 2018, 23, 389. [Google Scholar] [CrossRef]
- Jeon, J.H.; Kim, S.C. Comparative analysis of the complete chloroplast genome sequences of three closely related East-Asian wild roses (Rosa sect. Synstylae; Rosaceae). Genes 2019, 10, 23. [Google Scholar] [CrossRef]
- Lanfear, R.; Frandsen, P.B.; Wright, A.M.; Senfeld, T.; Calcott, B. PartitionFinder 2: New methods for selecting partitioned models of evolution for molecular and morphological phylogenetic analyses. Mol. Biol. Evol. 2017, 34, 772–773. [Google Scholar] [CrossRef]
- Ronquist, F.; Teslenko, M.; van der Mark, P.; Ayres, D.L.; Darling, A.; Hohna, S.; Larget, B.; Liu, L.; Suchard, M.A.; Huelsenbeck, J.P. MrBayes 3.2: Efficient bayesian phylogenetic inference and model choice across a large model space. Syst. Biol. 2012, 61, 539–542. [Google Scholar] [CrossRef]
- Leigh, J.W.; Bryant, D. POPART: Full-feature software for haplotype network construction. Methods Ecol. Evol. 2015, 6, 1110–1116. [Google Scholar] [CrossRef]
- Bandelt, H.J.; Forster, P.; Rohl, A. Median-joining networks for inferring intraspecific phylogenies. Mol. Biol. Evol. 1999, 16, 37–48. [Google Scholar] [CrossRef]
- Smulders, M.J.M.; Arens, P.; Bourke, P.M.; Debener, T.; Linde, M.; De Riek, J.; Leus, L.; Ruttink, T.; Baudino, S.; Saint-Oyant, L.H.; et al. In the name of the rose: A roadmap for rose research in the genome era. Hortic. Res. 2019, 6. [Google Scholar] [CrossRef] [PubMed]
- Meng, J.; He, S.L.; Li, D.Z.; Yi, T.S. Nuclear genetic variation of Rosa odorata var. gigantea (Rosaceae): Population structure and conservation implications. Tree Genet. Genomes 2016, 12. [Google Scholar] [CrossRef]
- Wissemann, V.; Ritz, C.M. The genus Rosa (Rosoideae, Rosaceae) revisited: Molecular analysis of nrITS-1 and atpB-rbcL intergenic spacer (IGS) versus conventional taxonomy. Bot. J. Linn. Soc. 2005, 147, 275–290. [Google Scholar] [CrossRef]
- Bruneau, A.; Starr, J.R.; Joly, S. Phylogenetic relationships in the genus Rosa: New evidence from chloroplast DNA sequences and an appraisal of current knowledge. Syst. Bot. 2007, 32, 366–378. [Google Scholar] [CrossRef]
- Fougere-Danezan, M.; Joly, S.; Bruneau, A.; Gao, X.F.; Zhang, L.B. Phylogeny and biogeography of wild roses with specific attention to polyploids. Ann. Bot. 2015, 115, 275–291. [Google Scholar] [CrossRef]
- Nadeem, M.; Younis, A.; Riaz, A.; Lim, K.B. Crossability among modern roses and heterosis of quantitative and qualitative traits in hybrids. Hortic. Environ. Biotechnol. 2015, 56, 487–497. [Google Scholar] [CrossRef]
- Rehder, A. Bibliography of Cultivated Trees and Shrubs Hardy in the Cooler Temperate Regions of the Northern Hemisphere; Arnold Arboretum of Harvard University: Boston, MA, USA, 1949. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Accession Number | Collection Locality | Taxonomic Assignation | Section | Haplotype Assignation |
|---|---|---|---|---|
| 1_1 | Dali, Yunnan, China | Rosa odorata var. gigantea | Chinenses | H1 |
| 1_2 | Dali, Yunnan, China | R. odorata var. gigantea | Chinenses | H1 |
| 1_4 | Kunming, Yunnan, China | R. odorata var. gigantea | Chinenses | H1 |
| 1_35 | Kunming, Yunnan, China | R. odorata var. gigantea | Chinenses | H1 |
| 1_7 | Dali, Yunnan, China | R. odorata var. odorata | Chinenses | H1 |
| 1_28 | Dali, Yunnan, China | R. odorata var. odorata | Chinenses | H1 |
| 1_11 | LIjiang, Yunnan, China | R. odorata var. odorata | Chinenses | H1 |
| 1_34 | LIjiang, Yunnan, China | R. odorata var. odorata | Chinenses | H9 |
| 1_33 | - | Rosa chinensis var. spontanea | Chinenses | H5 |
| 1_15 | Wenchuan, Sichuan, China | R. chinensis var. spontanea | Chinenses | H5 |
| 9_12 | - | R. chinensis var. spontanea | Chinenses | H5 |
| 1_39 | - | R. chinensis var. chinensis | Chinenses | H2 |
| 2_2 | - | R. chinensis ’Old Blush’ | Chinenses | H7 |
| 1_5 | LIjiang, Yunnan, China | Rosa sect. Chinenses complex | Chinenses | H1 |
| 1_6 | LIjiang, Yunnan, China | R. sect. Chinenses complex | Chinenses | H1 |
| 1_8 | Kunming, Yunnan, China | R. sect. Chinenses complex | Chinenses | H6 |
| 1_9 | Kunming, Yunnan, China | R. sect. Chinenses complex | Chinenses | H10 |
| 1_10 | Kunming, Yunnan, China | R. sect. Chinenses complex | Chinenses | H2 |
| 1_13 | Puer, Yunnan, China | R. sect. Chinenses complex | Chinenses | H3 |
| 1_14 | Puer, Yunnan, China | R. sect. Chinenses complex | Chinenses | H4 |
| 1_16 | LIjiang, Yunnan, China | R. sect. Chinenses complex | Chinenses | H6 |
| 1_17 | LIjiang, Yunnan, China | R. sect. Chinenses complex | Chinenses | H2 |
| 1_18 | Tengchong, Yunnan, China | R. sect. Chinenses complex | Chinenses | H7 |
| 1_20 | Kunming, Yunnan, China | R. sect. Chinenses complex | Chinenses | H2 |
| 1_21 | - | R. sect. Chinenses complex | Chinenses | H2 |
| 1_23 | - | R. sect. Chinenses complex | Chinenses | H7 |
| 1_24 | Dali, Yunnan, China | R. sect. Chinenses complex | Chinenses | H1 |
| 1_25 | Dali, Yunnan, China | R. sect. Chinenses complex | Chinenses | H2 |
| 1_26 | - | R. sect. Chinenses complex | Chinenses | H7 |
| 1_31 | Dali, Yunnan, China | R. sect. Chinenses complex | Chinenses | H2 |
| 1_32 | - | R. sect. Chinenses complex | Chinenses | H2 |
| outgroup | ||||
| 24 | - | Rosa multiflora | Synstylae | H10 |
| 9 | - | Rosa rugosa | Cinnamomeae | H14 |
| 36 | - | Rosa banksiae f. lutea | Banksianae | H13 |
| 57 | - | Rosa sericea | Pimpinellifoliae | H12 |
| 10 | - | Rosa xanthina | Pimpinellifoliae | H11 |
| Traits | Median (mm) | p Value | |||||
|---|---|---|---|---|---|---|---|
| R. odorata var. gigantea | R. odorata var. odorata | R. chinensis var. spontanea | R. chinensis var. chinensis | Sect. Chinenses Complex | Median Test | K-S Test | |
| Hip length | 21.70 | 17.44 | 20.22 | 14.74 | 13.20 | <0.0001 | <0.0001 |
| Hip width | 21.93 | 16.16 | 19.06 | 14.11 | 12.94 | <0.0001 | <0.0001 |
| Peduncle length | 11.32 | 14.25 | 5.03 | 18.67 | 24.65 | <0.0001 | <0.0001 |
| Sepal length | 27.90 | 17.52 | 16.56 | 18.56 | 22.42 | 0.0021 | 0.0292 |
| Sepal width | 6.25 | 6.10 | 4.02 | 4.78 | 6.71 | 0.0038 | <0.0001 |
| Flower diameter | 80.05 | 78.32 | 56.97 | 62.14 | 67.14 | <0.0001 | <0.0001 |
| Number of petals | 5 | 29 | 5 | 22 | 28 | <0.0001 | <0.0001 |
| Pistil length | 3.24 | 3.43 | 2.35 | 3.80 | 5.19 | <0.0001 | <0.0001 |
| Stamen length | 7.88 | 7.06 | 5.84 | 5.40 | 6.43 | 0.1080 | 0.0091 |
| Locus Name | RefSeq | Length | Polymorphism Sites | Nucleotide Polymorphism | Favorite Model |
|---|---|---|---|---|---|
| matK | - | 533 bp | 10 (1.9%) | 0.00378 | K81uf + G |
| atpB-rbcL | - | 594 bp | 9 (1.5%) | 0.00171 | K81uf + G |
| trnL-trnF | - | 961 bp | 26 (2.7%) | 0.00412 | K81uf + G |
| ZIP4 | LOC112169932 | 720 bp | 53 (7.4%) | 0.01008 | HKY + I + G |
| AP5 | LOC112197902 | 1371 bp | 44 (3.2%) | 0.00127 | HKY + I + G |
| SQD1 | LOC112170325 | 767 bp | 35 (4.6%) | 0.00557 | HKY + I + G |
| ALG8 | LOC112182822 | 622 bp | 47 (7.6%) | 0.01134 | HKY + I + G |
| Test Combination | p Value of Wilcoxon Sign-Rank Test | |
|---|---|---|
| IAM | TPM | |
| All accessions | 0.06372 | 0.33026 |
| Cultivated type | 0.03534 | 0.08325 |
| Wild type | 0.84692 | 0.10700 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yang, C.; Ma, Y.; Cheng, B.; Zhou, L.; Yu, C.; Luo, L.; Pan, H.; Zhang, Q. Molecular Evidence for Hybrid Origin and Phenotypic Variation of Rosa Section Chinenses. Genes 2020, 11, 996. https://doi.org/10.3390/genes11090996
Yang C, Ma Y, Cheng B, Zhou L, Yu C, Luo L, Pan H, Zhang Q. Molecular Evidence for Hybrid Origin and Phenotypic Variation of Rosa Section Chinenses. Genes. 2020; 11(9):996. https://doi.org/10.3390/genes11090996
Chicago/Turabian StyleYang, Chenyang, Yujie Ma, Bixuan Cheng, Lijun Zhou, Chao Yu, Le Luo, Huitang Pan, and Qixiang Zhang. 2020. "Molecular Evidence for Hybrid Origin and Phenotypic Variation of Rosa Section Chinenses" Genes 11, no. 9: 996. https://doi.org/10.3390/genes11090996
APA StyleYang, C., Ma, Y., Cheng, B., Zhou, L., Yu, C., Luo, L., Pan, H., & Zhang, Q. (2020). Molecular Evidence for Hybrid Origin and Phenotypic Variation of Rosa Section Chinenses. Genes, 11(9), 996. https://doi.org/10.3390/genes11090996