Exome-Sequence Analyses of Four Multi-Incident Multiple Sclerosis Families

 ,
, 
Abstract
1. Introduction
2. Methods and Patients
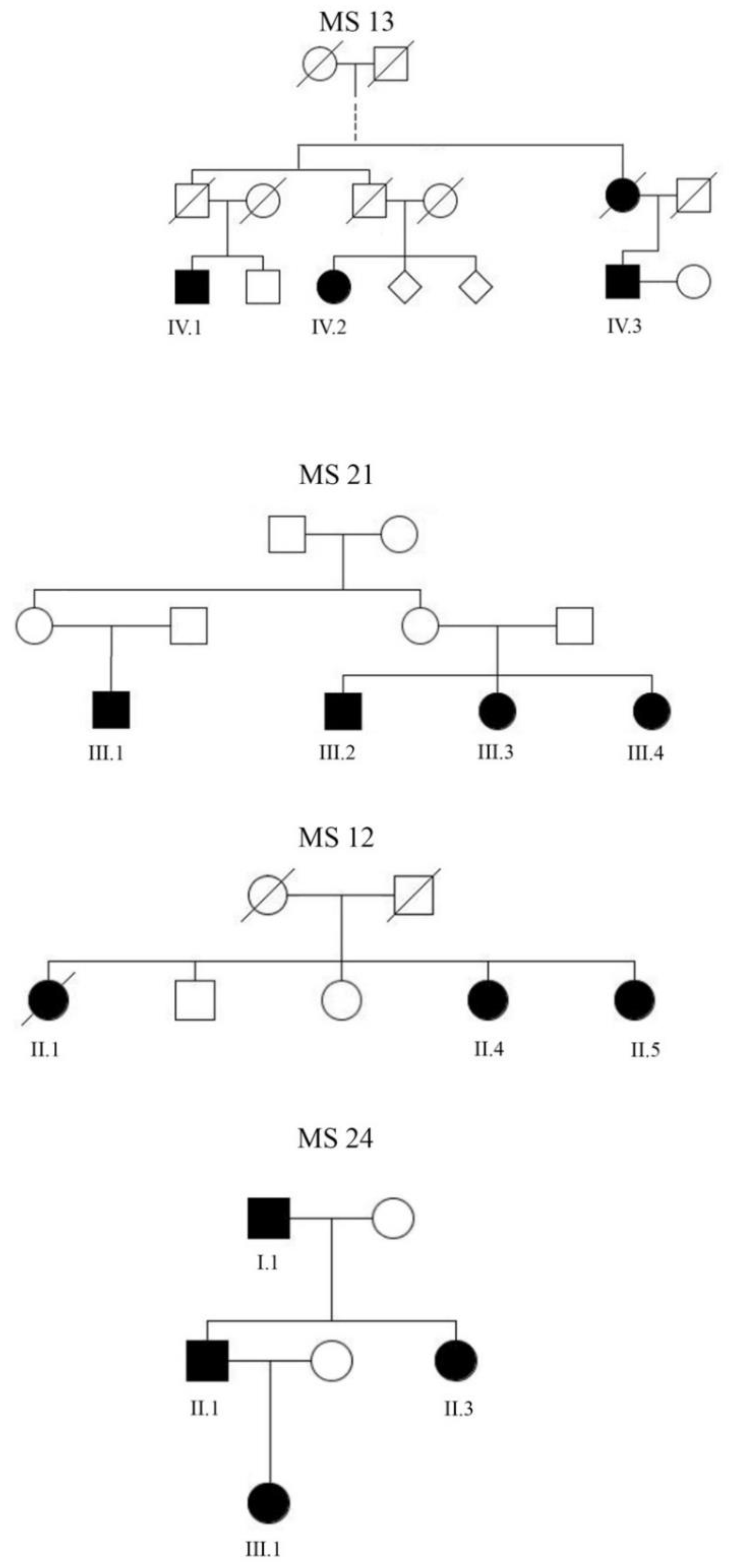
2.1. Participants
2.2. Sequencing
2.3. Literature Search
3. Results
4. Discussion
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- O’Gorman, C.; Lin, R.; Stankovich, J.; Broadley, S. Genetic Susceptibility to Multiple Sclerosis: Modelling the Risk with Family Data and Exploring the Effects of Latitude (P05. 129); AAN Enterprises: Haryana, India, 2013. [Google Scholar]
- Eichhorst, H. Über infantile und hereditäre multiple Sklerose. Virchows Arch. 1896, 146, 173–192. [Google Scholar] [CrossRef]
- Sawcer, S.; Franklin, R.J.; Ban, M. Multiple sclerosis genetics. Lancet. Neurol. 2014, 13, 700–709. [Google Scholar] [CrossRef]
- The International Multiple Sclerosis Genetics Consortium & The Wellcome Trust Case Control Consortium 2; Sawcer, S.; Hellenthal, G.; Pirinen, M.; Spencer, C.C.; Patsopoulos, N.A.; Moutsianas, L.; Dilthey, A.; Su, Z. Genetic risk and a primary role for cell-mediated immune mechanisms in multiple sclerosis. Nature 2011, 476, 214–219. [Google Scholar] [CrossRef] [PubMed]
- International Multiple Sclerosis Genetics Consortium. Multiple sclerosis genomic map implicates peripheral immune cells and microglia in susceptibility. Science 2019, 365. [Google Scholar] [CrossRef]
- Hollenbach, J.A.; Oksenberg, J.R. The immunogenetics of multiple sclerosis: A comprehensive review. J. Autoimmun. 2015, 64, 13–25. [Google Scholar] [CrossRef]
- Lill, C.M. Recent advances and future challenges in the genetics of multiple sclerosis. Front. Neurol. 2014, 5, 130. [Google Scholar] [CrossRef]
- Mitrovič, M.; Patsopoulos, N.A.; Beecham, A.H.; Dankowski, T.; Goris, A.; Dubois, B.; D’Hooghe, M.B.; Lemmens, R.; Van Damme, P.; Søndergaard, H.B.; et al. International Multiple Sclerosis Genetics Consortium. Electronic address, c.c.y.e.; International Multiple Sclerosis Genetics, C. Low-Frequency and Rare-Coding Variation Contributes to Multiple Sclerosis Risk. Cell 2018, 175, 1679–1687.e1677. [Google Scholar] [CrossRef]
- Kuokkanen, S.; Gschwend, M.; Rioux, J.D.; Daly, M.J.; Terwilliger, J.D.; Tienari, P.J.; Wikstrom, J.; Palo, J.; Stein, L.D.; Hudson, T.J.; et al. Genomewide scan of multiple sclerosis in Finnish multiplex families. Am. J. Hum. Genet. 1997, 61, 1379–1387. [Google Scholar] [CrossRef]
- Mescheriakova, J.Y.; Verkerk, A.J.; Amin, N.; Uitterlinden, A.G.; van Duijn, C.M.; Hintzen, R.Q. Linkage analysis and whole exome sequencing identify a novel candidate gene in a Dutch multiple sclerosis family. Mult. Scler. 2019, 25, 909–917. [Google Scholar] [CrossRef]
- Wang, Z.; Sadovnick, A.D.; Traboulsee, A.L.; Ross, J.P.; Bernales, C.Q.; Encarnacion, M.; Yee, I.M.; de Lemos, M.; Greenwood, T.; Lee, J.D. Nuclear receptor NR1H3 in familial multiple sclerosis. Neuron 2016, 90, 948–954. [Google Scholar] [CrossRef]
- Reinthaler, E.M.; Graf, E.; Zrzavy, T.; Wieland, T.; Hotzy, C.; Kopecky, C.; Pferschy, S.; Schmied, C.; Leutmezer, F.; Keilani, M.; et al. TPP2 mutation associated with sterile brain inflammation mimicking MS. Neurol. Genet. 2018, 4, e285. [Google Scholar] [CrossRef] [PubMed]
- Ban, M.; Caillier, S.; Mero, I.L.; Myhr, K.M.; Celius, E.G.; Aarseth, J.; Torkildsen, O.; Harbo, H.F.; Oksenberg, J.; Hauser, S.L.; et al. No evidence of association between mutant alleles of the CYP27B1 gene and multiple sclerosis. Ann. Neurol. 2013, 73, 430–432. [Google Scholar] [CrossRef] [PubMed]
- Antel, J.; Ban, M.; Baranzini, S.; Barcellos, L.; Barizzone, N.; Beecham, A.; Berge, T.; Bernardinelli, L.; Booth, D.; Bos, S. NR1H3 p. Arg415Gln is not associated to multiple sclerosis risk. Neuron 2016, 92, 333–335. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Vilarino-Guell, C.; Zimprich, A.; Martinelli-Boneschi, F.; Herculano, B.; Wang, Z.; Matesanz, F.; Urcelay, E.; Vandenbroeck, K.; Leyva, L.; Gris, D.; et al. Exome sequencing in multiple sclerosis families identifies 12 candidate genes and nominates biological pathways for the genesis of disease. PLoS Genet. 2019, 15, e1008180. [Google Scholar] [CrossRef] [PubMed]
- Polman, C.H.; Reingold, S.C.; Banwell, B.; Clanet, M.; Cohen, J.A.; Filippi, M.; Fujihara, K.; Havrdova, E.; Hutchinson, M.; Kappos, L.; et al. Diagnostic criteria for multiple sclerosis: 2010 revisions to the McDonald criteria. Ann. Neurol. 2011, 69, 292–302. [Google Scholar] [CrossRef] [PubMed]
- Haeussler, M.; Gerner, M.; Bergman, C.M. Annotating genes and genomes with DNA sequences extracted from biomedical articles. Bioinformatics 2011, 27, 980–986. [Google Scholar] [CrossRef]
- Pihlstrøm, L.; Wiethoff, S.; Houlden, H. Genetics of neurodegenerative diseases: An overview. In Handbook of Clinical Neurology; Elsevier: Amsterdam, The Netherlands, 2018; Volume 145, pp. 309–323. [Google Scholar]
- Sawcer, S.; Ban, M.; Maranian, M.; Yeo, T.W.; Compston, A.; Kirby, A.; Daly, M.J.; De Jager, P.L.; Walsh, E.; Lander, E.S.; et al. A high-density screen for linkage in multiple sclerosis. Am. J. Hum. Genet. 2005, 77, 454–467. [Google Scholar] [CrossRef]
- Patsopoulos, N.A. Genetics of Multiple Sclerosis: An Overview and New Directions. Cold Spring Harb. Perspect. Med. 2018, 8, a028951. [Google Scholar] [CrossRef]
- Vilariño-Güell, C.; Encarnacion, M.; Bernales, C.Q.; Sadovnick, A.D. Analysis of Canadian multiple sclerosis patients does not support a role for FKBP6 in disease. Mult. Scler. J. 2019, 25, 1011–1013. [Google Scholar] [CrossRef]
- MacArthur, D.G.; Manolio, T.A.; Dimmock, D.P.; Rehm, H.L.; Shendure, J.; Abecasis, G.R.; Adams, D.R.; Altman, R.B.; Antonarakis, S.E.; Ashley, E.A.; et al. Guidelines for investigating causality of sequence variants in human disease. Nature 2014, 508, 469–476. [Google Scholar] [CrossRef] [PubMed]
- Dong, M.; Su, X.; Dzikovski, B.; Dando, E.E.; Zhu, X.; Du, J.; Freed, J.H.; Lin, H. Dph3 is an electron donor for Dph1-Dph2 in the first step of eukaryotic diphthamide biosynthesis. J. Am. Chem. Soc. 2014, 136, 1754–1757. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.; Bachran, C.; Gupta, P.; Miller-Randolph, S.; Wang, H.; Crown, D.; Zhang, Y.; Wein, A.N.; Singh, R.; Fattah, R.; et al. Diphthamide modification on eukaryotic elongation factor 2 is needed to assure fidelity of mRNA translation and mouse development. Proc. Natl. Acad. Sci. USA 2012, 109, 13817–13822. [Google Scholar] [CrossRef] [PubMed]
- Arguelles, S.; Camandola, S.; Cutler, R.G.; Ayala, A.; Mattson, M.P. Elongation factor 2 diphthamide is critical for translation of two IRES-dependent protein targets, XIAP and FGF2, under oxidative stress conditions. Free Radic. Biol. Med. 2014, 67, 131–138. [Google Scholar] [CrossRef] [PubMed]
- Wucherpfennig, K.W.; Strominger, J.L. Molecular mimicry in T cell-mediated autoimmunity: Viral peptides activate human T cell clones specific for myelin basic protein. Cell 1995, 80, 695–705. [Google Scholar] [CrossRef]
- Zhao, Z.; Bi, W.; Zhou, W.; VandeHaar, P.; Fritsche, L.G.; Lee, S. UK Biobank Whole-Exome Sequence Binary Phenome Analysis with Robust Region-Based Rare-Variant Test. Am. J. Hum. Genet. 2020, 106, 3–12. [Google Scholar] [CrossRef]
- Lemcke, S.; Muller, S.; Moller, S.; Schillert, A.; Ziegler, A.; Cepok-Kauffeld, S.; Comabella, M.; Montalban, X.; Rulicke, T.; Nandakumar, K.S.; et al. Nerve conduction velocity is regulated by the inositol polyphosphate-4-phosphatase II gene. Am. J. Pathol. 2014, 184, 2420–2429. [Google Scholar] [CrossRef]
- Goedde, R.; Sawcer, S.; Boehringer, S.; Miterski, B.; Sindern, E.; Haupts, M.; Schimrigk, S.; Compston, A.; Epplen, J.T. A genome screen for linkage disequilibrium in HLA-DRB1*15-positive Germans with multiple sclerosis based on 4666 microsatellite markers. Hum. Genet. 2002, 111, 270–277. [Google Scholar] [CrossRef]
- Bergsteinsdottir, K.; Yang, H.T.; Pettersson, U.; Holmdahl, R. Evidence for common autoimmune disease genes controlling onset, severity, and chronicity based on experimental models for multiple sclerosis and rheumatoid arthritis. J. Immunol. 2000, 164, 1564–1568. [Google Scholar] [CrossRef]
- Blankenhorn, E.P.; Butterfield, R.J.; Rigby, R.; Cort, L.; Giambrone, D.; McDermott, P.; McEntee, K.; Solowski, N.; Meeker, N.D.; Zachary, J.F.; et al. Genetic analysis of the influence of pertussis toxin on experimental allergic encephalomyelitis susceptibility: An environmental agent can override genetic checkpoints. J. Immunol. 2000, 164, 3420–3425. [Google Scholar] [CrossRef]
- Fujita, T.; Kitaura, F.; Fujii, H. A critical role of the Thy28-MYH9 axis in B cell-specific expression of the Pax5 gene in chicken B cells. PLoS ONE 2015, 10, e0116579. [Google Scholar] [CrossRef]
- Toyota, H.; Sudo, K.; Kojima, K.; Yanase, N.; Nagao, T.; Takahashi, R.H.; Iobe, H.; Kuwabara, T.; Kakiuchi, T.; Mizuguchi, J. Thy28 protects against anti-CD3-mediated thymic cell death in vivo. Apoptosis 2015, 20, 444–454. [Google Scholar] [CrossRef] [PubMed]
- Kleinewietfeld, M.; Manzel, A.; Titze, J.; Kvakan, H.; Yosef, N.; Linker, R.A.; Muller, D.N.; Hafler, D.A. Sodium chloride drives autoimmune disease by the induction of pathogenic T H 17 cells. Nature 2013, 496, 518–522. [Google Scholar] [CrossRef] [PubMed]
- Takeuchi, H. Does salt exacerbate multiple sclerosis? Clin. Exp. Neuroimmunol. 2013, 4, 5–6. [Google Scholar] [CrossRef]
- Wang, W.; Ren, S.; Wang, Z.; Zhang, C.; Huang, J. Increased expression of TTC21A in lung adenocarcinoma infers favorable prognosis and high immune infiltrating level. Int. Immunopharmacol. 2020, 78, 106077. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
| Family | Patient | Age (years) | Disease Course | Age at Onset | Year of Last EDSS | EDSS | Av.Cov. | 20X |
|---|---|---|---|---|---|---|---|---|
| MS 12 | II. 1 | 70 * | SPMS | 43 | 2015 | 9 | 146 | 98.03 |
| II. 4 | 67 | RRMS | 45 | 2020 | 7 | 134 | 97.71 | |
| II. 5 | 63 | RRMS | 50 | 2015 | 6 | 147 | 96.32 | |
| MS 13 | IV. 1 | 67 | RRMS | 31 | 2019 | 1.5 | 132 | 97.56 |
| IV. 2 | 71 | SPMS | 18 | 2015 | 8 | 218 | 99.18 | |
| IV. 3 | 63 | RRMS | 42 | 2015 | 1.5 | 145 | 97.76 | |
| MS 21 | III. 1 | 20 | RRMS | 14 | 2017 | 2 | 143 | 99.04 |
| III. 2 | 39 | RRMS | 31 | 2017 | 0 | 147 | 99.07 | |
| III. 3 | 36 | RRMS | 25 | 2017 | 1 | 151 | 99.00 | |
| III. 4 | 32 | RRMS | 24 | 2020 | 1 | 158 | 98.99 | |
| MS 24 | I. 1 | 71 | RRMS | 48 | 2020 | 1.5 | 159 | 99.19 |
| II. 1 | 49 | RRMS | 35 | 2017 | 1 | 139 | 99.01 | |
| II. 3 | 45 | RRMS | 31 | 2014 | 0 | 134 | 98.73 | |
| III. 1 | 21 | RRMS | 18 | 2017 | 1 | 152 | 98.97 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zrzavy, T.; Leutmezer, F.; Kristoferitsch, W.; Kornek, B.; Schneider, C.; Rommer, P.; Berger, T.; Zimprich, A. Exome-Sequence Analyses of Four Multi-Incident Multiple Sclerosis Families. Genes 2020, 11, 988. https://doi.org/10.3390/genes11090988
Zrzavy T, Leutmezer F, Kristoferitsch W, Kornek B, Schneider C, Rommer P, Berger T, Zimprich A. Exome-Sequence Analyses of Four Multi-Incident Multiple Sclerosis Families. Genes. 2020; 11(9):988. https://doi.org/10.3390/genes11090988
Chicago/Turabian StyleZrzavy, Tobias, Fritz Leutmezer, Wolfgang Kristoferitsch, Barbara Kornek, Christine Schneider, Paulus Rommer, Thomas Berger, and Alexander Zimprich. 2020. "Exome-Sequence Analyses of Four Multi-Incident Multiple Sclerosis Families" Genes 11, no. 9: 988. https://doi.org/10.3390/genes11090988
APA StyleZrzavy, T., Leutmezer, F., Kristoferitsch, W., Kornek, B., Schneider, C., Rommer, P., Berger, T., & Zimprich, A. (2020). Exome-Sequence Analyses of Four Multi-Incident Multiple Sclerosis Families. Genes, 11(9), 988. https://doi.org/10.3390/genes11090988