A Systematic Review of Extreme Phenotype Strategies to Search for Rare Variants in Genetic Studies of Complex Disorders




Abstract
1. Introduction
2. Materials and Methods
2.1. Study Design
2.2. Search Strategies
2.3. Research Question and Selection Criteria
- Population: Patients with a complex disease or condition.
- Intervention: Selection of individuals according to extreme phenotype criteria (i.e., early onset, fast progression of disease, very high or very low scores in psychometric tests, or extreme levels of a biomarker).
- Comparison: Genetic association studies (genome-wide association studies (GWAS), WES, genotyping, Sanger sequencing, or targeted sequencing).
- Outcome: genetic findings reported (rare variants, candidate genes, or pathways associated with the condition of interest).
- Study design: case–control, case report, case–cohort, or trios.
2.4. Exclusion Criteria
- Studies in non-human models.
- Studies not published in English.
- Studies with a publication date 10 years.
2.5. Quality Assessment of Selected Studies
2.6. Data Extraction and Synthesis
2.7. Risk of Bias
3. Results
3.1. Selection and Characteristics of EP Studies
3.2. Synthesized Findings of EP Studies
4. Discussion
4.1. Summary of the Main Findings
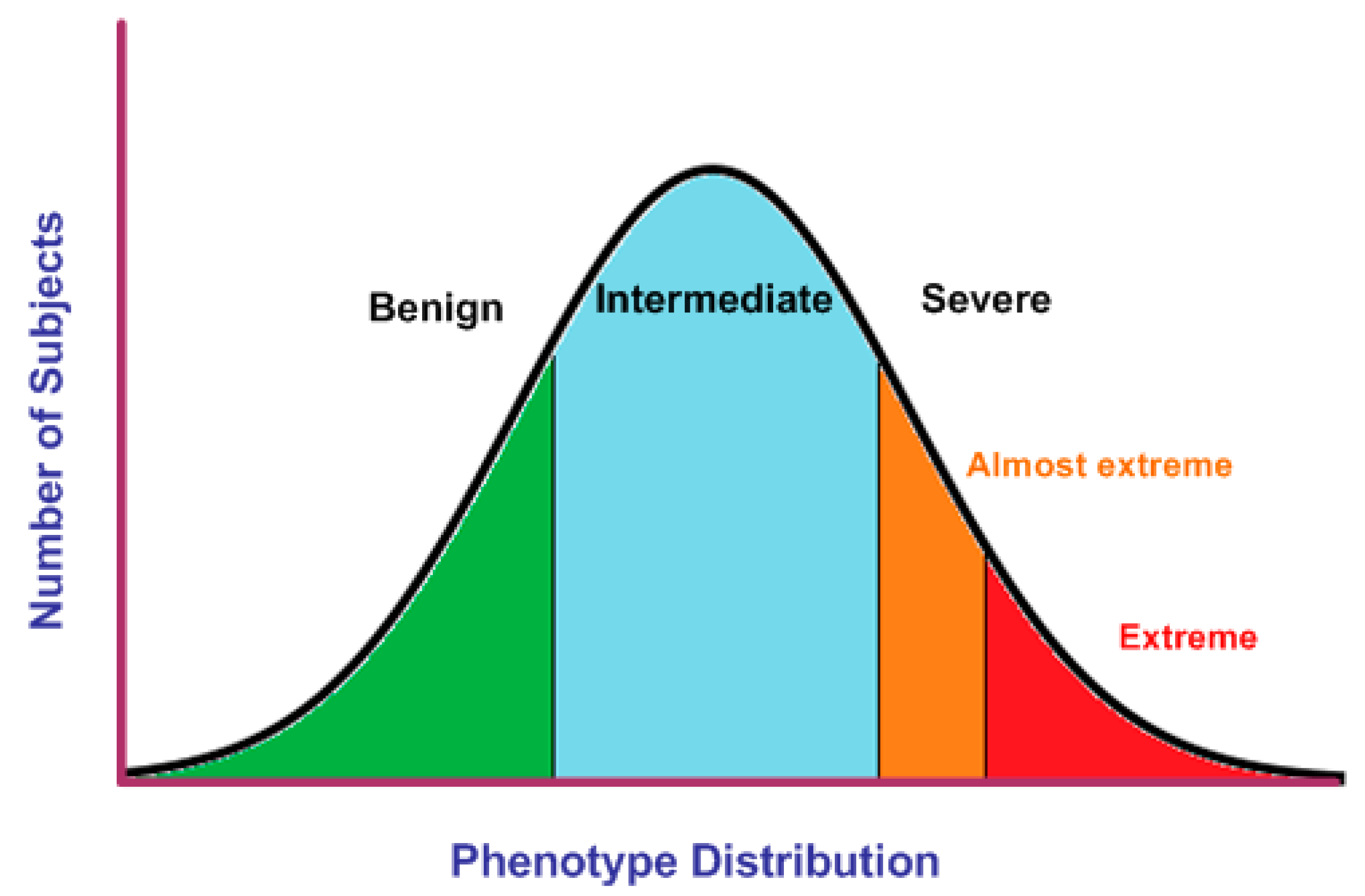
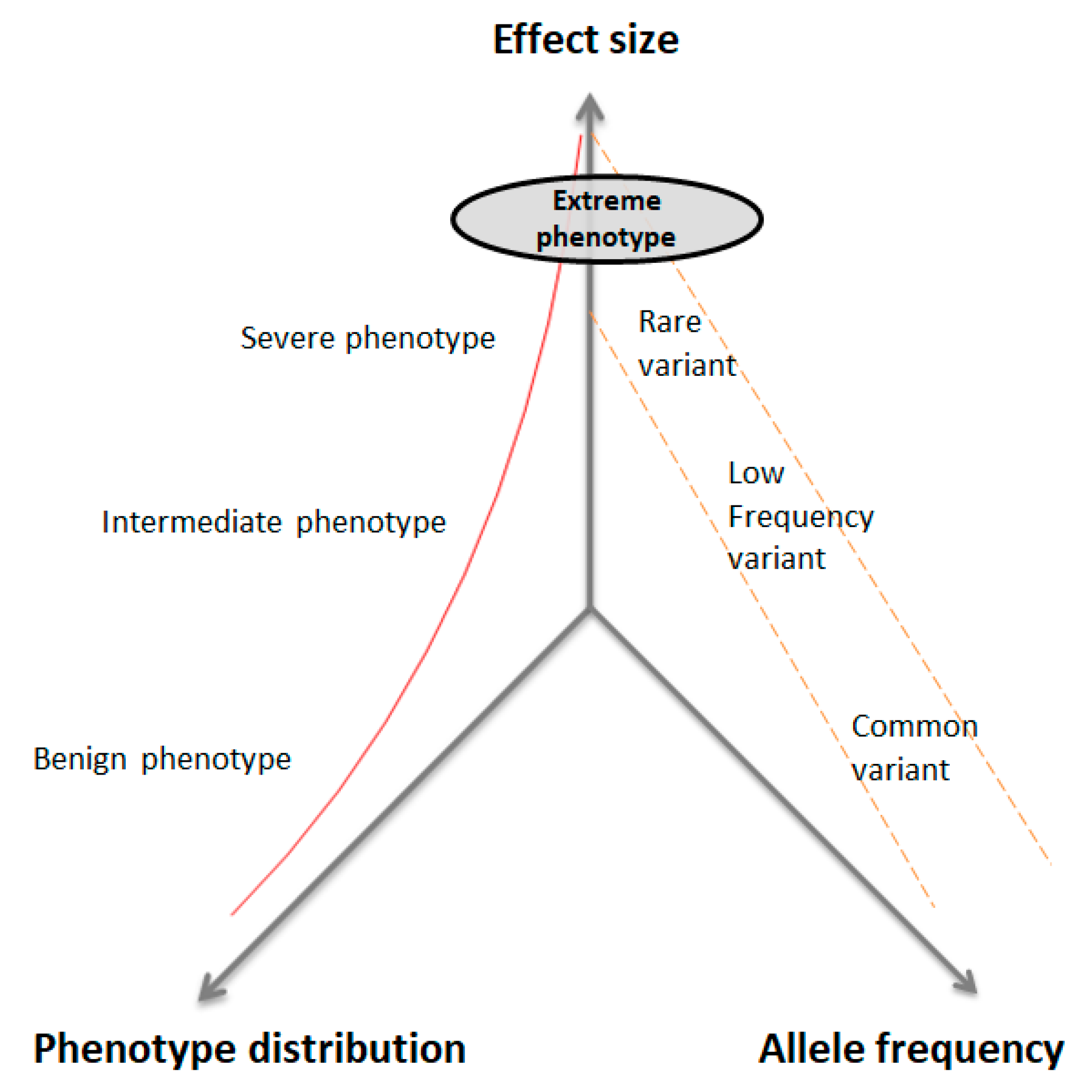
4.2. Selection of EP in Quantitative Traits
4.3. Familial Disorders and EP Strategy
4.4. An EP Strategy to Investigate the Genetic Contribution to Tinnitus
4.5. Limitations
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Pérez-Gracia, J.L.; Gúrpide, A.; Ruiz-Ilundain, M.G.; Alegría, C.A.; Colomer, R.; García-Foncillas, J.; Bermejo, I.M. Selection of extreme phenotypes: The role of clinical observation in translational research. Clin. Transl. Oncol. 2010, 12, 174–180. [Google Scholar] [CrossRef] [PubMed]
- Zimmermann, E.; Gamborg, M.; Sørensen, T.I.A.; Baker, J.L. Sex differences in the association between birth weight and adult type 2 diabetes. Diabetes 2015, 64, 4220–4225. [Google Scholar] [CrossRef] [PubMed]
- Gibson, G. Rare and common variants: Twenty arguments. Nat. Rev. Genet. 2012, 13, 135–145. [Google Scholar] [CrossRef] [PubMed]
- Lohmann, K.; Klein, C. Next generation sequencing and the future of genetic diagnosis. Neurotherapeutics 2014, 11, 699–707. [Google Scholar] [CrossRef]
- Srivastava, S.; Cohen, J.S.; Vernon, H.; Barañano, K.; McClellan, R.; Jamal, L.; Naidu, S.; Fatemi, A. Clinical whole exome sequencing in child neurology practice. Ann. Neurol. 2014, 76, 473–483. [Google Scholar] [CrossRef]
- Jansen, I.E.; Ye, H.; Heetveld, S.; Lechler, M.C.; Michels, H.; Seinstra, R.I.; Lubbe, S.J.; Drouet, V.; Lesage, S.; Majounie, E.; et al. Discovery and functional prioritization of Parkinson ’ s disease candidate genes from large-scale whole exome sequencing. Genome Biol. 2017, 18, 1–26. [Google Scholar] [CrossRef]
- Johnson, M.B.; Patel, K.A.; De Franco, E.; Houghton, J.A.; McDonald, T.J.; Ellard, S.; Flanagan, S.E.; Hattersley, A.T. A type 1 diabetes genetic risk score can discriminate monogenic autoimmunity with diabetes from early-onset clustering of polygenic autoimmunity with diabetes. Diabetologia 2018, 61, 862–869. [Google Scholar] [CrossRef]
- Verma, S.S.; Ritchie, M.D. Another round of “clue” to uncover the mystery of complex traits. Genes 2018, 9, 61. [Google Scholar] [CrossRef]
- Craddock, N.; Kendler, K.; Neale, M.; Nurnberger, J.; Purcell, S.; Rietschel, M.; Perlis, R.; Santangelo, S.L.; Schulze, T.; Smoller, J.W.; et al. Dissecting the phenotype in genome-wide association studies of psychiatric illness. Br. J. Psychiatry 2009, 195, 97–99. [Google Scholar]
- Bamshad, M.J.; Ng, S.B.; Bigham, A.W.; Tabor, H.K.; Emond, M.J.; Nickerson, D.A.; Shendure, J. Exome sequencing as a tool for Mendelian disease gene discovery. Nat. Rev. Genet. 2011, 12, 745–755. [Google Scholar] [CrossRef]
- Bjørnland, T.; Bye, A.; Ryeng, E.; Wisløff, U.; Langaas, M. Improving power of genetic association studies by extreme phenotype sampling: A review and some new results. arXiv 2017, arXiv:1701.01286. [Google Scholar]
- Barnett, I.J.; Lee, S.; Lin, X. Detecting rare variant effects using extreme phenotype sampling in sequencing association studies. Genet. Epidemiol. 2013, 37, 142–151. [Google Scholar] [CrossRef]
- Emond, M.J.; Louie, T.; Emerson, J.; Zhao, W.; Mathias, R.A.; Knowles, M.R.; Wright, F.A.; Rieder, M.J.; Tabor, H.K.; Nickerson, D.A.; et al. Exome sequencing of extreme phenotypes identifies DCTN4 as a modifier of chronic Pseudomonas aeruginosa infection in cystic fibrosis. Nat. Genet. 2012, 44, 886–889. [Google Scholar] [CrossRef] [PubMed]
- Bjørnland, T.; Langaas, M.; Grill, V.; Mostad, I.L. Assessing gene-environment interaction effects of FTO, MC4R and lifestyle factors on obesity using an extreme phenotype sampling design: Results from the HUNT study. PLoS ONE 2017, 12, e0175071. [Google Scholar] [CrossRef] [PubMed]
- Goldberg-Stern, H.; Aharoni, S.; Afawi, Z.; Bennett, O.; Appenzeller, S.; Pendziwiat, M.; Kuhlenbäumer, G.; Basel-Vanagaite, L.; Shuper, A.; Korczyn, A.D.; et al. Broad phenotypic heterogeneity due to a novel SCN1A mutation in a family with genetic epilepsy with febrile seizures plus. J. Child Neurol. 2014, 29, 221–226. [Google Scholar] [CrossRef] [PubMed]
- Shtir, C.; Aldahmesh, M.A.; Al-Dahmash, S.; Abboud, E.; Alkuraya, H.; Abouammoh, M.A.; Nowailaty, S.R.; Al-Thubaiti, G.; Naim, E.A.; ALYounes, B.; et al. Exome-based case—Control association study using extreme phenotype design reveals novel candidates with protective effect in diabetic retinopathy. Hum. Genet. 2016, 135, 193–200. [Google Scholar] [CrossRef]
- Husson, T.; Duboc, J.B.; Quenez, O.; Charbonnier, C.; Rotharmel, M.; Cuenca, M.; Jegouzo, X.; Richard, A.C.; Frebourg, T.; Deleuze, J.F.; et al. Identification of potential genetic risk factors for bipolar disorder by whole-exome sequencing. Transl. Psychiatry 2018, 8, 1–7. [Google Scholar] [CrossRef]
- Blue, E.; Louie, T.L.; Chong, J.X.; Hebbring, S.J.; Barnes, K.C.; Rafaels, N.M.; Knowles, M.R.; Gibson, R.L.; Bamshad, M.J.; Emond, M.J. Variation in cilia protein genes and progression of lung disease in cystic fibrosis. Ann. Am. Thorac. Soc. 2018, 15, 440–448. [Google Scholar] [CrossRef]
- Manolio, T.A.; Collins, F.S.; Cox, N.J.; Goldstein, D.B.; Hindorff, L.A.; Hunter, D.J.; McCarthy, M.I.; Ramos, E.M.; Cardon, L.R.; Chakravarti, A.; et al. Finding the missing heritability of complex diseases. Nature 2009, 461, 747–753. [Google Scholar] [CrossRef]
- Moher, D. Preferred reporting items for systematic review and meta-analysis protocols (PRISMA-P) 2015 statement David. Syst. Rev. 2015, 4, 1. [Google Scholar] [CrossRef]
- Higgins, J.P.; Altman, D.G.; Gøtzsche, P.C.; Jüni, P.; Moher, D.; Oxman, A.D.; Savović, J.; Schulz, K.F.; Weeks, L.; Sterne, J.A. The Cochrane Collaboration’s tool for assessing risk of bias in randomised trials. BMJ 2011, 343, d5928. [Google Scholar] [CrossRef] [PubMed]
- Bruse, S.; Moreau, M.; Bromberg, Y.; Jang, J.H.; Wang, N.; Ha, H.; Picchi, M.; Lin, Y.; Langley, R.J.; Qualls, C.; et al. Whole exome sequencing identifies novel candidate genes that modify chronic obstructive pulmonary disease susceptibility. Hum. Genom. 2016, 10, 1. [Google Scholar] [CrossRef] [PubMed]
- Pullabhatla, V.; Roberts, A.L.; Lewis, M.J.; Mauro, D.; Morris, D.L.; Odhams, C.A.; Tombleson, P.; Liljedahl, U.; Vyse, S.; Simpson, M.A.; et al. De novo mutations implicate novel genes in systemic lupus erythematosus. Hum. Mol. Genet. 2018, 27, 421–429. [Google Scholar] [CrossRef] [PubMed]
- Johar, A.; Sarmiento-Monroy, J.C.; Rojas-Villarraga, A.; Silva-Lara, M.F.; Patel, H.R.; Mantilla, R.D.; Velez, J.I.; Schulte, K.M.; Mastronardi, C.; Arcos-Burgos, M.; et al. Definition of mutations in polyautoimmunity. J. Autoimmun. 2016, 72, 65–72. [Google Scholar] [CrossRef]
- Kunkle, B.W.; Vardarajan, B.N.; Naj, A.C.; Whitehead, P.L.; Rolati, S.; Slifer, S.; Carney, R.M.; Cuccaro, M.L.; Vance, J.M.; Gilbert, J.R.; et al. Early-onset Alzheimer disease and candidate risk genes involved in endolysosomal transport. JAMA Neurol. 2017, 74, 1113–1122. [Google Scholar] [CrossRef]
- Liu, Y.; Kheradmand, F.; Davis, C.F.; Scheurer, M.E.; Wheeler, D.; Tsavachidis, S.; Armstrong, G.; Simpson, C.; Mandal, D.; Kupert, E.; et al. Focused analysis of exome sequencing data for rare germline mutations in familial and sporadic lung cancer. J. Thorac. Oncol. 2016, 11, 52–61. [Google Scholar] [CrossRef]
- Johar, A.S.; Mastronardi, C.; Rojas-Villarraga, A.; Patel, H.R.; Chuah, A.; Peng, K.; Higgins, A.; Milburn, P.; Palmer, S.; Silva-Lara, M.F.; et al. Novel and rare functional genomic variants in multiple autoimmune syndrome and Sjögren’s syndrome. J. Transl. Med. 2015, 13, 173. [Google Scholar] [CrossRef]
- Hiekkala, M.E.; Vuola, P.; Artto, V.; Häppölä, P.; Häppölä, E.; Vepsäläinen, S.; Cuenca-Leon, E.; Lal, D.; Gormley, P.; Hämäläinen, E.; et al. The contribution of CACNA1A, ATP1A2 and SCN1A mutations in hemiplegic migraine: A clinical and genetic study in Finnish migraine families. Cephalalgia 2018, 38, 1849–1863. [Google Scholar] [CrossRef]
- Qiao, D.; Ameli, A.; Prokopenko, D.; Chen, H.; Kho, A.T.; Parker, M.M.; Morrow, J.; Hobbs, B.D.; Liu, Y.; Beaty, T.H.; et al. Whole exome sequencing analysis in severe chronic obstructive pulmonary disease. Hum. Mol. Genet. 2018, 27, 3801–3812. [Google Scholar] [CrossRef]
- Nuytemans, K.; Ortel, T.L.; Gomez, L.; Hofmann, N.; Alves, N.; Dueker, N.; Beecham, A.; Whitehead, P.; Estabrooks, S.H.; Kitchens, C.S.; et al. Variants in chondroitin sulfate metabolism genes in thrombotic storm. Thromb. Res. 2018, 161, 43–51. [Google Scholar] [CrossRef]
- Aubart, M.; Gazal, S.; Arnaud, P.; Benarroch, L.; Gross, M.S.; Buratti, J.; Boland, A.; Meyer, V.; Zouali, H.; Hanna, N.; et al. Association of modifiers and other genetic factors explain Marfan syndrome clinical variability. Eur. J. Hum. Genet. 2018, 26, 1759–1772. [Google Scholar] [CrossRef] [PubMed]
- Gregson, C.L.; Newell, F.; Leo, P.J.; Clark, G.R.; Paternoster, L.; Marshall, M.; Forgetta, V.; Morris, J.A.; Ge, B.; Bao, X.; et al. Genome-wide association study of extreme high bone mass: Contribution of common genetic variation to extreme BMD phenotypes and potential novel BMD-associated genes. Bone 2018, 114, 62–71. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.S.; Yang, S.K.; Hong, M.; Jung, S.; Kim, B.M.; Moon, J.W.; Park, S.H.; Ye, B.D.; Oh, S.H.; Kim, K.M.; et al. An intergenic variant rs9268877 between HLA-DRA and HLA-DRB contributes to the clinical course and long-term outcome of ulcerative colitis. J. Crohn’s Colitis 2018, 12, 1113–1121. [Google Scholar] [CrossRef] [PubMed]
- Tomaiuolo, R.; Bellia, C.; Caruso, A.; Di Fiore, R.; Quaranta, S.; Noto, D.; Cefalù, A.B.; Di Micco, P.; Zarrilli, F.; Castaldo, G.; et al. Prothrombotic gene variants as risk factors of acute myocardial infarction in young women. J. Transl. Med. 2012, 10, 1–5. [Google Scholar] [CrossRef]
- Shen, Y.; Xu, J.; Yang, X.; Liu, Y.; Ma, Y.; Yang, D.; Dong, Q.; Yang, Y. Evidence for the involvement of the proximal copy of the MAGEA9 gene in Xq28-linked CNV67 specific to spermatogenic failure. Biol. Reprod. 2017, 96, 610–616. [Google Scholar] [CrossRef]
- Uzun, A.; Schuster, J.; McGonnigal, B.; Schorl, C.; Dewan, A.; Padbury, J. Targeted sequencing and meta-analysis of preterm birth. PLoS ONE 2016, 11, e0155021. [Google Scholar] [CrossRef]
- Allen, A.S.; Bellows, S.T.; Berkovic, S.F.; Bridgers, J.; Burgess, R.; Cavalleri, G.; Chung, S.K.; Cossette, P.; Delanty, N.; Dlugos, D.; et al. Ultra-rare genetic variation in common epilepsies: A case-control sequencing study. Lancet Neurol. 2017, 16, 135–143. [Google Scholar] [CrossRef]
- Forsberg, L.A.; Rasi, C.; Razzaghian, H.R.; Pakalapati, G.; Waite, L.; Thilbeault, K.S.; Ronowicz, A.; Wineinger, N.E.; Tiwari, H.K.; Boomsma, D.; et al. Age-related somatic structural changes in the nuclear genome of human blood cells. Am. J. Hum. Genet. 2012, 90, 217–228. [Google Scholar] [CrossRef]
- Castellani, C.; Assael, B.M. Cystic fibrosis: A clinical view. Cell. Mol. Life Sci. 2016, 74, 129–140. [Google Scholar] [CrossRef]
- Fuseini, H.; Newcomb, D.C. Mechanisms driving gender differences in asthma. Curr. Allergy Asthma Rep. 2017, 17, 19. [Google Scholar] [CrossRef]
- Kang, G.; Lin, D.; Hakonarson, H.; Chen, J. Two-stage extreme phenotype sequencing design for discovering and testing common and rare genetic variants: Efficiency and power. Hum. Hered. 2012, 73, 139–147. [Google Scholar] [CrossRef] [PubMed]
- Li, D.; Lewinger, J.P.; Gauderman, W.J.; Murcray, C.E.; Conti, D. Using extreme phenotype sampling to identify the rare causal variants of quantitative traits in association studies. Genet. Epidemiol. 2011, 35, 790–799. [Google Scholar] [CrossRef] [PubMed]
- Guey, L.T.; Kravic, J.; Melander, O.; Burtt, N.P.; Laramie, J.M.; Lyssenko, V.; Jonsson, A.; Lindholm, E.; Tuomi, T.; Isomaa, B.; et al. Power in the phenotypic extremes: A simulation study of power in discovery and replication of rare variants. Genet. Epidemiol. 2011, 35, 236–246. [Google Scholar] [CrossRef] [PubMed]
- De Sanctis, L.; Corrias, A.; Romagnolo, D.; Di Palma, T.; Biava, A.; Borgarello, G.; Gianino, P.; Silvestro, L.; Zannini, M.; Dianzani, I. Familial PAX8 small deletion (c.989_992delACCC) associated with extreme phenotype variability. J. Clin. Endocrinol. Metab. 2004, 89, 5669–5674. [Google Scholar] [CrossRef]
- Roman-naranjo, P.; Gallego-Martinez, A.; Soto-Varela, A.; Aran, I.; del Carmen Moleon, M.; Espinosa-Sanchez, J.M.; Amor-Dorado, J.C.; Batuecas-Caletrio, A.; Perez-Vazquez, P.; Lopez-Escamez, J.A. Rare variants in the OTOG gene are a frequent cause of familial Meniere ’ s disease. Ear Hear 2020. [Google Scholar] [CrossRef]
- Gilles, A.; Camp, G.; Van de Heyning, P.; Fransen, E. A pilot genome-wide association study identifies potential metabolic pathways involved in tinnitus. Front. Neurosci. 2017, 11, 71. [Google Scholar] [CrossRef][Green Version]
- Lopez-Escamez, J.A.; Bibas, T.; Cima, R.F.; Van de Heyning, P.; Knipper, M.; Mazurek, B.; Szczepek, A.J.; Cederroth, C.R. Genetics of tinnitus: An emerging area for molecular diagnosis and drug development. Front. Neurosci. 2016, 10, 377. [Google Scholar] [CrossRef]
- Vona, B.; Nanda, I.; Shehata-Dieler, W.; Haaf, T. Genetics of tinnitus: Still in its infancy. Front. Neurosci. 2017, 11, 236. [Google Scholar] [CrossRef]
- Maas, I.L.; Brüggemann, P.; Requena, T.; Bulla, J.; Edvall, N.K.; vBHjelmborg, J.; Szczepek, A.J.; Canlon, B.; Mazurek, B.; Lopez-Escamez, J.A.; et al. Genetic susceptibility to bilateral tinnitus in a Swedish twin cohort. Genet. Med. 2017, 19, 1007–1012. [Google Scholar] [CrossRef]
- Schlee, W.; Hall, D.A.; Canlon, B.; Cima, R.F.F.; Kleine, E.D.; Hauck, F.; Huber, A.; Gallus, S.; Kleinjung, T.; Kypraios, T.; et al. Innovations in Doctoral Training and Research on Tinnitus: The European School on Interdisciplinary Tinnitus Research (ESIT). Perspective 2018, 9, 1–7. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| No. | Question | Answer |
|---|---|---|
| Q1 | Is there a thorough description of the study design? | Yes/No |
| Q2 | Has the study described the method of sequencing/genotyping? | Yes/No |
| Q3 | Has the study provided information about population ancestry? | Yes/No |
| Q4 | Is there any information on the sex of the selected individuals? | Yes/No |
| Q5 | Is there any information on the age of disease onset? | Yes/No |
| Q6 | Has the study used extreme phenotype criteria for sample recruitment? | Yes/No |
| Q7 | Has the study performed sex-specific analysis for genetic associations? | Yes/No |
| Q8 | Has the study reported significant genetic findings? | Yes/No |
| Reference | Disease | EP Criteria | Study Design | Sequencing Method | Ancestry | Number of Patients | Onset | Sex | Genetic findings | AF (Ancestry-Dependent) | |
|---|---|---|---|---|---|---|---|---|---|---|---|
| Gene/ Pathway | SNP/Indel | ||||||||||
| Pullabhatla et al. (2017) [23] | Systemic lupus erythematosus | Proband with early onset and clinical features with poor outcome | Family trios, Replication cohort | WES | EU | 30 trios, 10995 | <25 y | Not reported | PRKCD | 3: 53223122 G>A | De novo variants and novel genes |
| C1QTNF4 | 11: 47611769 G> C | ||||||||||
| DNMT3A | 2: 25457236 G> A | ||||||||||
| Johar et al. (2016) [24] | Polyautoimmunity | Polyautoimmunity and familial autoimmunity | Case–control, Cross-sectional | WES | Colombian | 47 | Not reported | M,F | PLAUR | rs4760 | 0.1 |
| DHX34 | rs151213663 | 0.004 | |||||||||
| SRA1 | rs5871740, rs202193903 | Not found | |||||||||
| ABCB8 | 7:150744528:G>T, 7:150744370: CGT/- | Not found | |||||||||
| MLL4 | rs186268702 | 0.0007 | |||||||||
| Kunkle et al. (2017) [25] | Alzheimer’s disease | Early-onset Alzheimer’s disease, familial or sporadic | Case–control, Replication cohort | WES | NHW and Caribbean Hispanic | 93, 8570 | <65 y | M,F | RUFY1 | 5:179036506:T>G | 0.001 |
| RIN3 | 14:93022240:G>T | 0.0005 | |||||||||
| TCIRG | 11:67810477:C>T | 0.0007 | |||||||||
| PSD2 | 5:139216541:G>A, 5:139216759:G>A |
0.0006, 0.00005 | |||||||||
| Emond et al. (2012) [13] | Cystic fibrosis (CF) | CF with early onset of persistent Pseudomonas aeruginosa infection | Case–control, Replication cohort | WES | EU America, African American, White Hispanic, NHW, Asian, Aleut | 43, 696 | ≤2.5 y | M,F | DCTN4 |
rs11954652, rs35772018 | 0.048, 0.017 |
| Shtir et al. (2016) [16] | Diabetes | Diabetes for at least 10 years without diabetic retinopathy | Case–control, Cross-sectional | WES | Saudi | 43 | Not reported | M,F | FASTK | 7:150774771:C>T, 7:150777859:A>T |
0, 0 |
| LOC728699 |
rs149540491, rs117616768, 12:20704520:C>A |
0.05, 0.01, 0.02 | |||||||||
| Liu et al. (2016) [26] | Lung cancer | Familial or sporadic lung cancer cases, ever smokers or severe chronic obstructive pulmonary disease (COPD) | Case–control, Cross-sectional | WES | NHW | 48 sporadic 54 familial | 56 y familial 61 y sporadic | M,F | DBH | rs76856960 | 0.0034 |
| CCDC147 | rs41291850 | 0.0026 | |||||||||
| Husson et al. (2018) [17] | Bipolar I disorder | Family history of mood disorder and early onset | Case–control, Cross-sectional | WES | EU | 92 | mean: 24 y | M,F | >13 genes | >100 SNPs | 0.000015-0.009 |
| Johar et al. (2015) [27] | Multiple autoimmune syndrome | Multiple autoimmune syndrome with Sjögren’s syndrome | Case–control, Cross-sectional | WES | Colombian | 12 | 28–67 y | F | LRP1/STAT6 | 12:57522754:A>C | Novel mutation |
| Hiekkala et al. (2018) [28] | Hemiplegic migraine | ≥2 migraine attacks, completely reversible motor weakness | Case report, Cross-sectional | WES | Finnish | 293 | median: 12 y | M,F | ATP1A2 |
rs765909830, 1:160100376:G>A | 0, 0 |
| CACNA1A | rs121908212 | 0 | |||||||||
| Qiao et al. (2018) [29] | COPD | COPD cases with GOLD grade 3 or 4 | Case–control, Cross-sectional | WES | EU, NHW, African American | ≈1769 | >45 y, ≤65 y | M/F | jak-stat signaling pathway | - | Not reported |
| TBC1D10A, RFPL1 | Not reported | ||||||||||
| Bruse et al. (2016) [22] | COPD | COPD cases with GOLD grade 3 or 4 | Case–control, Cross-sectional | WES | NHW | 62 | Not reported | M/F | TACC2 | chr10:123842508, 10:123844900, 10:123903149, 10:123970638, 10:123987443, 10:123996970, 10:124009124 | 0.000008901, 0.000008796, 0.001851, 0.000008999, Not found 0.03476, 0.07 |
| Nuytemans et al. (2018) [30] | Thrombotic storm (TS) | Severe onset of ≥2 arterial, unusual clot location, refractory, reoccurrence | Case report, Cross-sectional | WES, Targeted sequencing | White and Indian | 26 (13 trios) | Not reported | M,F | STAB2 |
rs779748342, rs758868186, rs201799617, rs17034336, rs149382223 | Not found, Not found, 0.0002, 0.0441, 0.0008 |
| CHPF | 2:220405189:C>T | Not found | |||||||||
| CHST3 | rs145384892 | Not found | |||||||||
| SLC26A2 |
rs104893919, rs78676079 | Not found, 0.0076 | |||||||||
| CHST12 | rs17132399 | Not found | |||||||||
| CHPF2 |
rs776052782, rs117332591, rs377232422 | Not found, 0.0028, Not found | |||||||||
| CHST15 | rs34639461 | 0.011 | |||||||||
| PAPSS2 | rs45467596 | 0.0219 | |||||||||
| Aubart et al. (2018) [31] | Marfan syndrome | Severe aortic features (dissection or preventive thoracic aortic aneurysm rupture surgery at a young age) or sib pairs | Case–control, Cross-sectional | WES | EU | 51 EP and 8 sib-pairs | ≈10–30 y | M,F | COL4A1 |
c.4615C>T, c.1630G>C, c.4453T>C, |
0.02, 0.04, 0.003 |
| FBN1 | c.1585C>T | 0 | |||||||||
| SMAD3 | c.6424T>C | 0 | |||||||||
| Gregson et al. (2018) [32] | Bone mass density | Extremely high or moderately high bone mass density | Case–control, Replication cohort | GWAS | EU | 1258, 32965 | Not reported | M,F | WNT4/ZBTB40 | rs113784679 | 0.04 |
| Lee et al. (2018) [33] | Ulcerative colitis | Ulcerative colitis patients with good or poor prognosis | Case–control, Replication cohort | Genotyping | Korean | 881, 274 | 35.6 ± 13.9 y | M,F | HLA-DRA and HLA-DRB | rs9268877 | 0.000 |
| Tomaiuolo et al. (2012) [34] | Acute myocardial infarction (AMI) | AMI patients with first episode before or after 45 years of age | Case–control, Replication cohort | Genotyping | EU | 1653, 909 | Not reported | M,F | MTHFR C677T, FII G20210A, Factor V Leiden | -455G>A | - |
| Goldberg-Stern et al. (2013) [15] | Epilepsy with febrile seizures plus | Generalized epilepsy with febrile seizures plus, a proband with Dravet syndrome | Case-control, Cross-sectional | Sanger sequencing | Ashkenazi Jewish | 14 familial cases | infancy to 7 y | M,F | SCN1A | c.4114A>G: p.K1372E; exon 21 | - |
| Shen et al. (2017) [35] | Spermatogenic failure | Spermatogenic failure with azoospermia, mild oligozoospermia or severe oligozoospermia | Case–control, Cross sectional | Sanger sequencing | Chinese Han | 884 | Not reported | M | MAGEA9 | Deletion (chrX:149580739-149580850) | - |
| Uzun et al. (2016) [36] | Preterm birth | Patients delivering <34 weeks | Case report, Cross-sectional | Targeted Sequencing of 329 genes | African-American; Asian; Hispanic; White; Native American | 32 | Not reported | F | WASF3 | rs17084492 | 0.01357(NFE), 0.07(African) |
| AZU1 | rs28626600 |
0.1(NFE), 0.01662(African) | |||||||||
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Amanat, S.; Requena, T.; Lopez-Escamez, J.A. A Systematic Review of Extreme Phenotype Strategies to Search for Rare Variants in Genetic Studies of Complex Disorders. Genes 2020, 11, 987. https://doi.org/10.3390/genes11090987
Amanat S, Requena T, Lopez-Escamez JA. A Systematic Review of Extreme Phenotype Strategies to Search for Rare Variants in Genetic Studies of Complex Disorders. Genes. 2020; 11(9):987. https://doi.org/10.3390/genes11090987
Chicago/Turabian StyleAmanat, Sana, Teresa Requena, and Jose Antonio Lopez-Escamez. 2020. "A Systematic Review of Extreme Phenotype Strategies to Search for Rare Variants in Genetic Studies of Complex Disorders" Genes 11, no. 9: 987. https://doi.org/10.3390/genes11090987
APA StyleAmanat, S., Requena, T., & Lopez-Escamez, J. A. (2020). A Systematic Review of Extreme Phenotype Strategies to Search for Rare Variants in Genetic Studies of Complex Disorders. Genes, 11(9), 987. https://doi.org/10.3390/genes11090987