Deletion in the Bardet–Biedl Syndrome Gene TTC8 Results in a Syndromic Retinal Degeneration in Dogs

 , , , , , , , , , , and
, , , , , , , , , , and
Abstract
1. Introduction
2. Materials and Methods
2.1. Animals and Samples
2.2. Ophthalmic Examination, Confocal Scanning Ophthalmoscopy (cSLO), Optical Coherence Tomography (OCT) and Electroretinography (ERG)
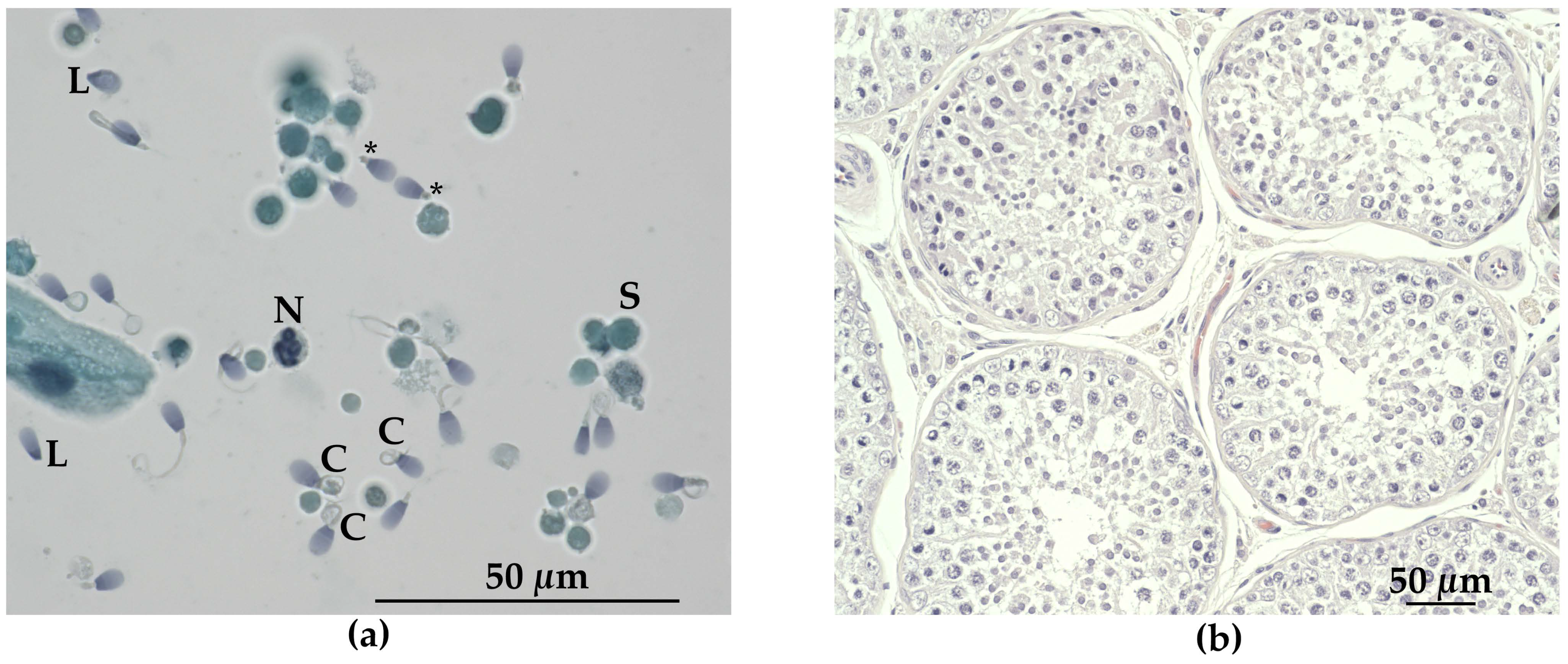
2.3. Clinical Andrological Examination and Semen Analysis
2.4. Echocardiography and Electrocardiographic (ECG) Examinations
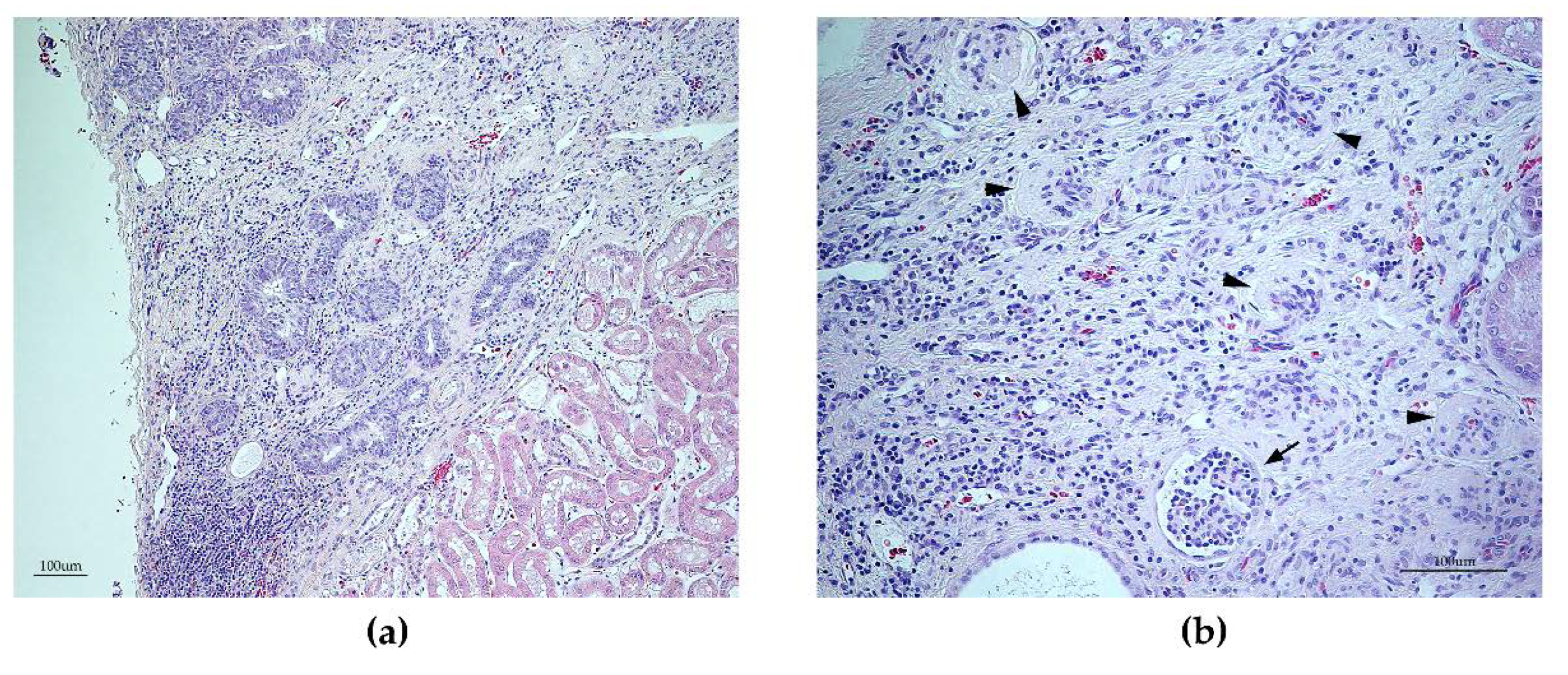
2.5. Histopathological Examinations
2.6. Total RNA Extraction
2.7. Long-Read cDNA Sequencing with Oxford Nanopore Sequencing Technology (ONT)
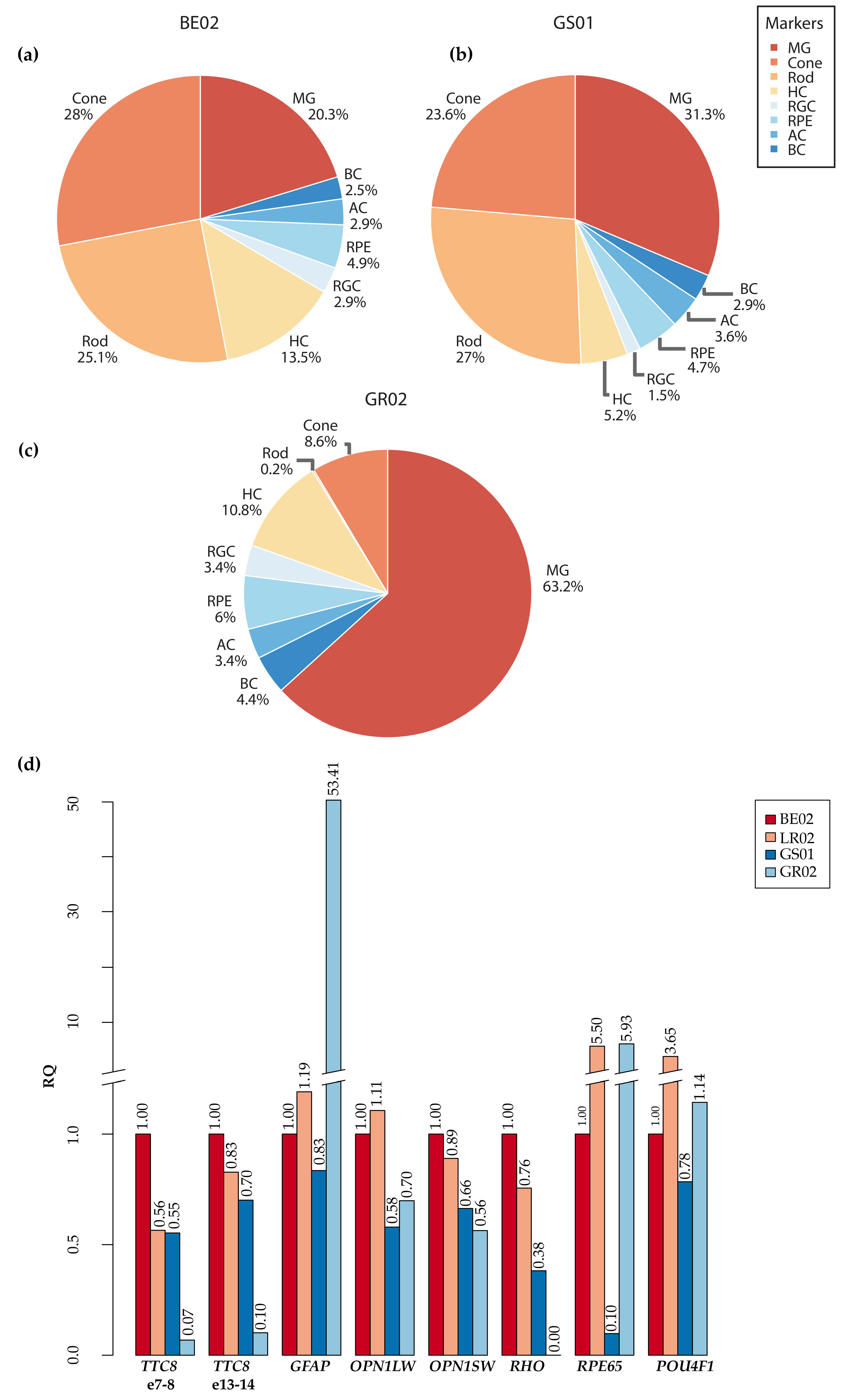
2.8. Quantitative RT-qPCR
2.9. Fluorescence Histochemistry
3. Results
3.1. Desciption of Primary and Secondary Bardet-Biedl Syndrome Characteristics
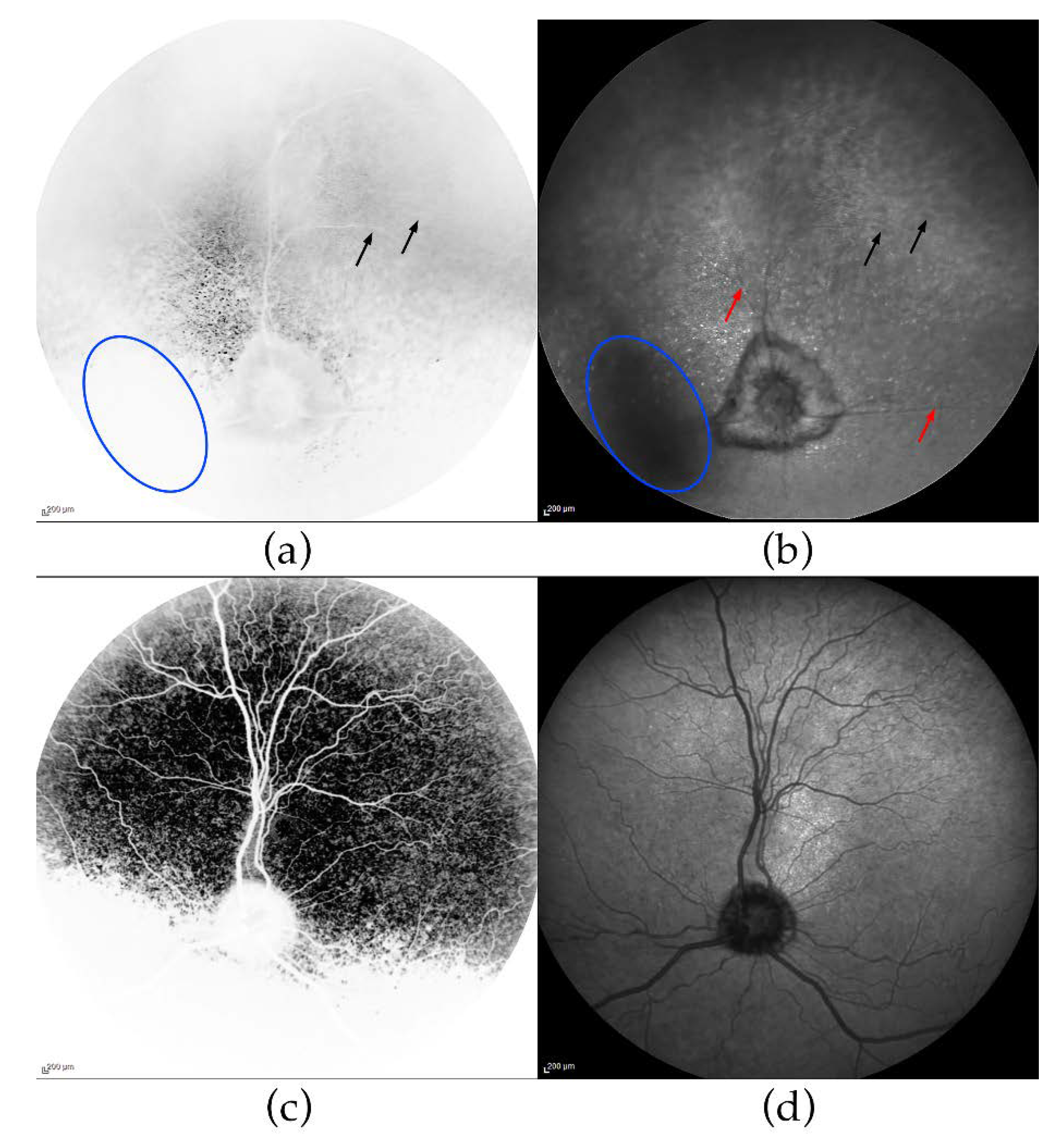
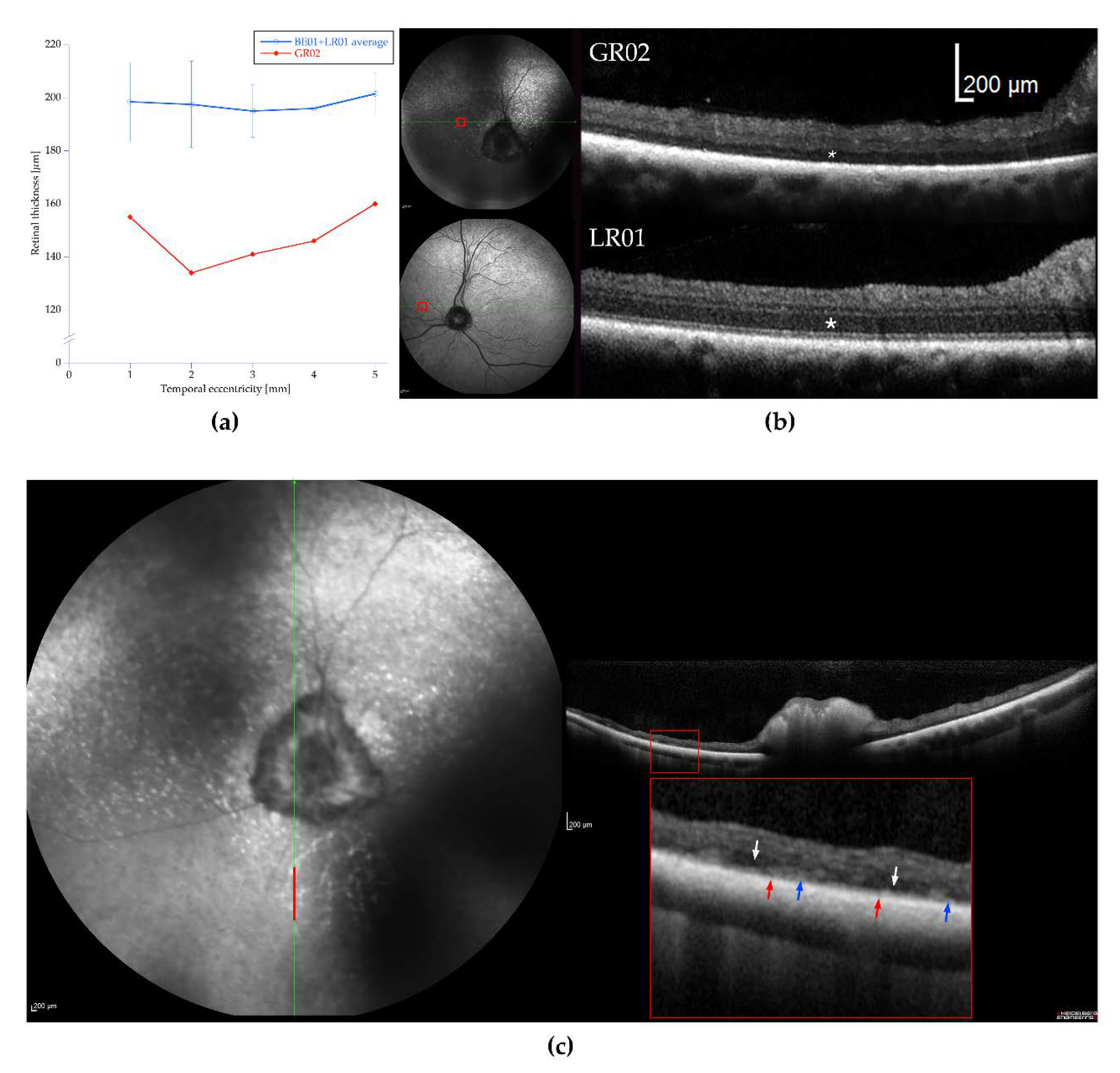
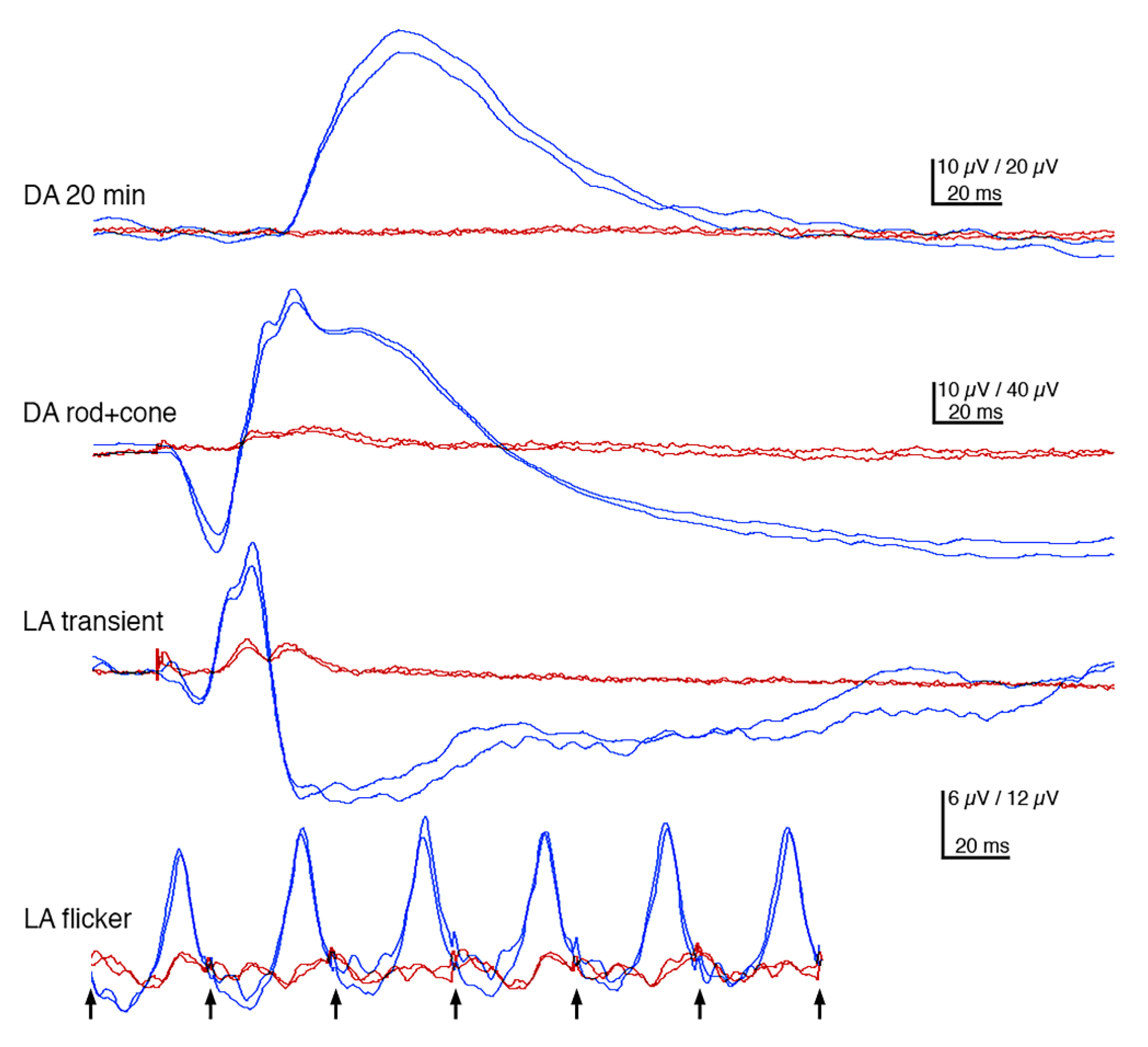
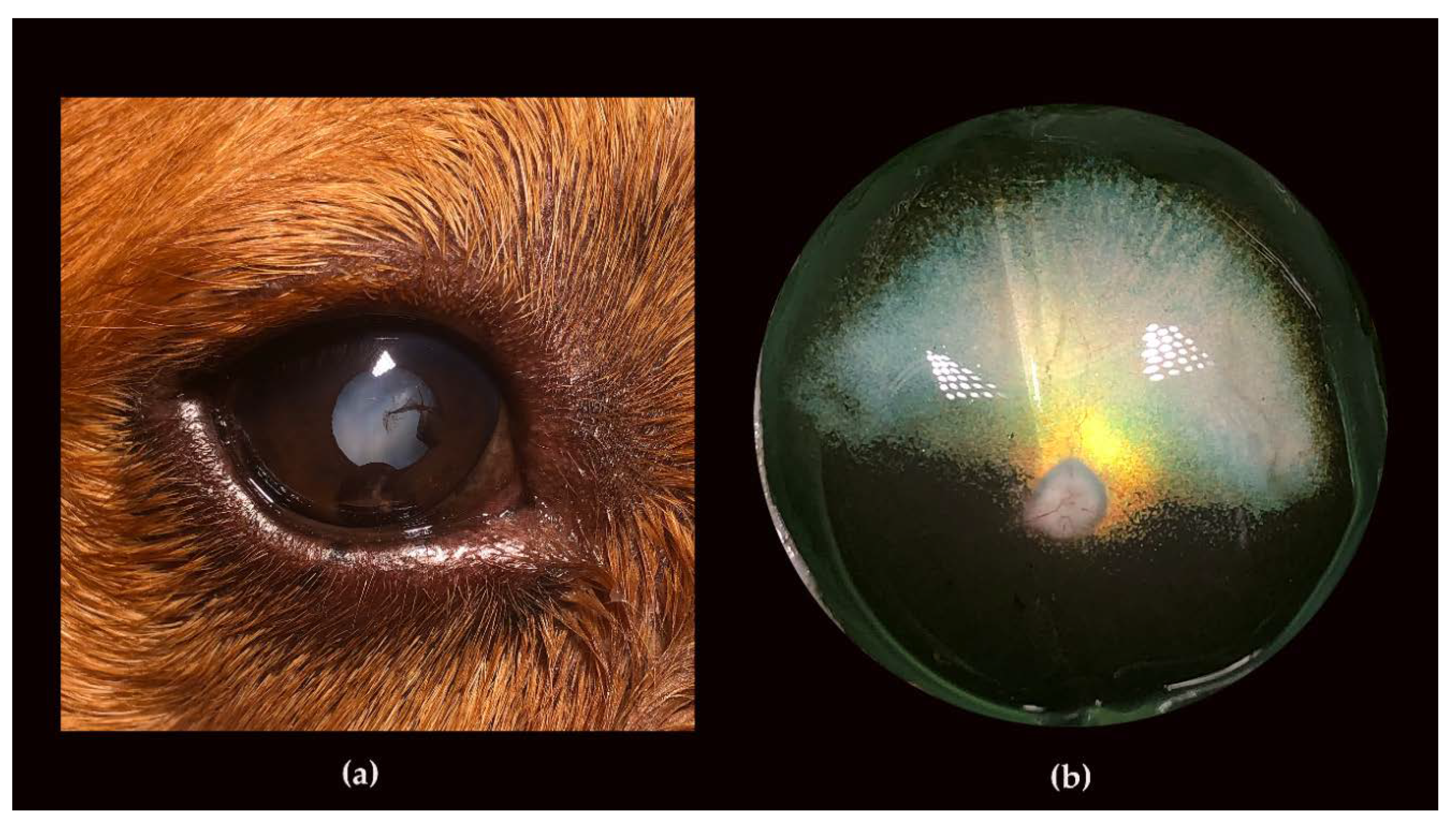
3.2. Ophthalmic Examinations of the Affected Sib-Pair
3.3. Andrological Examination
3.4. Cardiac Examination
3.5. Necropsy Findings
3.6. Histopathology Findings
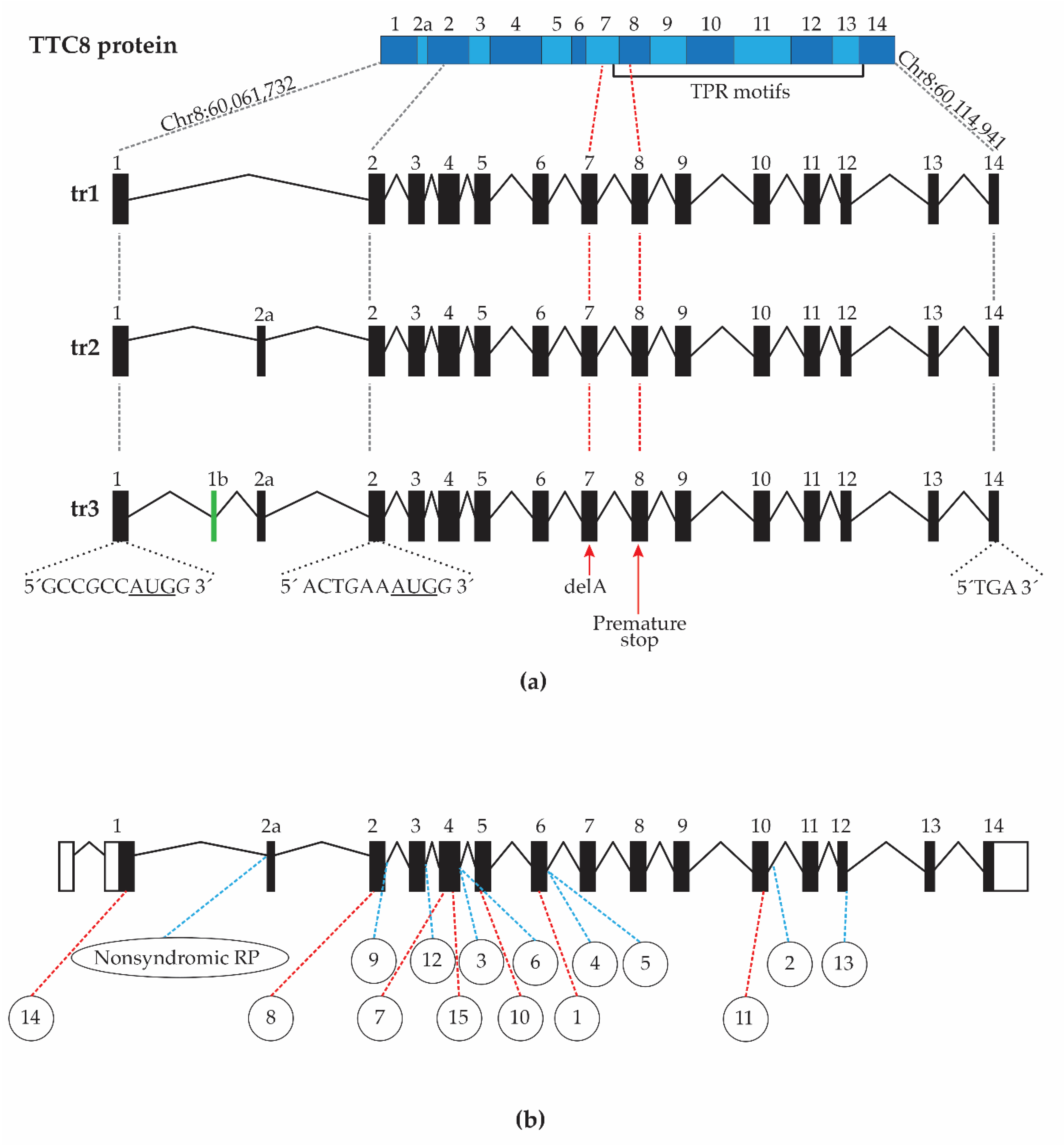
3.7. Characterization of Canine TTC8 Transcripts in the Retina
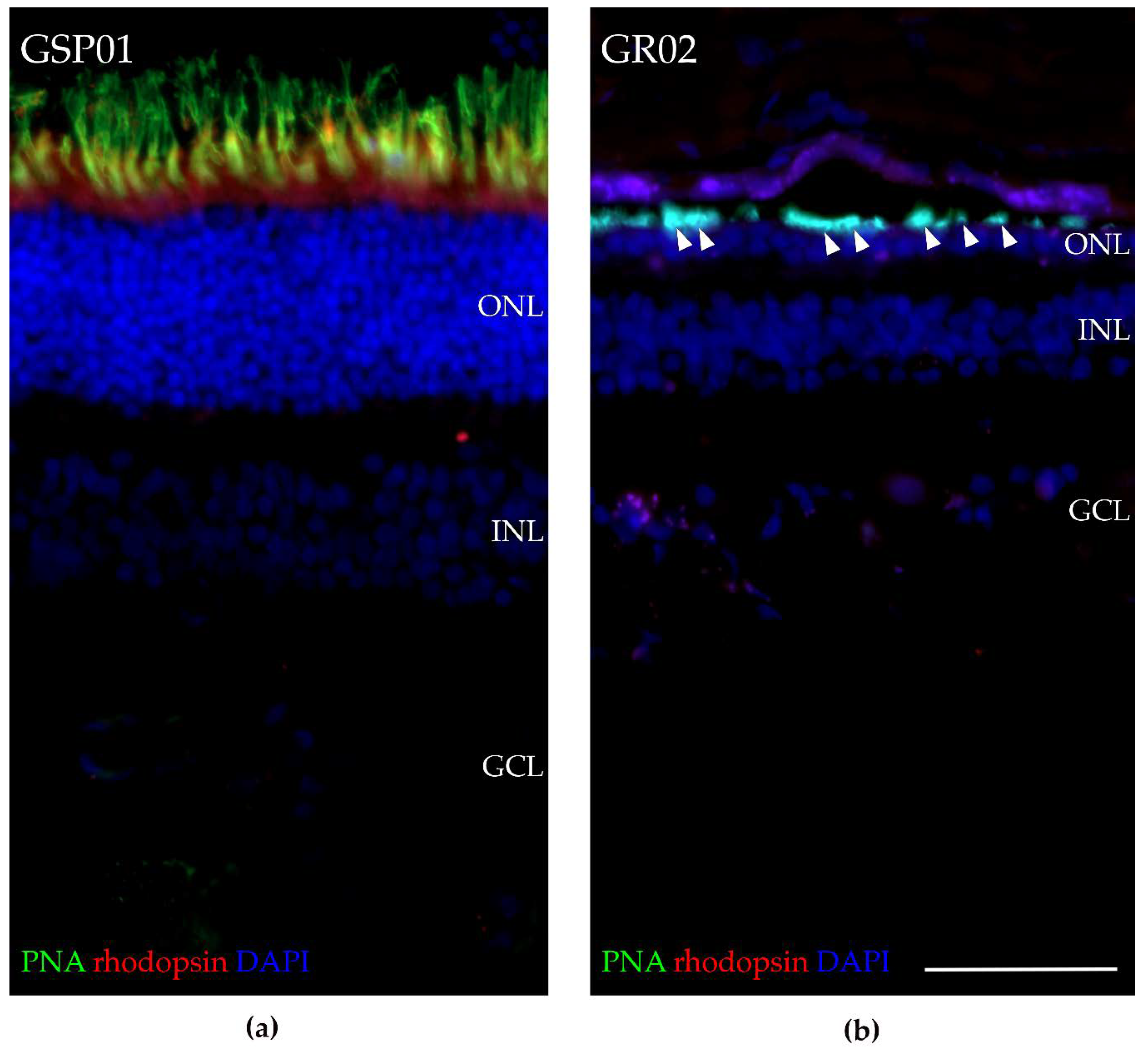
3.8. Fluorescence Histochemical Analysis of the TTC8delA Retina
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Downs, L.M.; Wallin-Håkansson, B.; Bergström, T.; Mellersh, C.S. A novel mutation in TTC8 is associated with progressive retinal atrophy in the golden retriever. Canine Genet. Epidemiol. 2014, 1, 4. [Google Scholar] [CrossRef] [PubMed]
- Ansley, S.J.; Badano, J.L.; Blacque, O.E.; Hill, J.; Hoskins, B.E.; Leitch, C.C.; Kim, J.C.; Ross, A.J.; Eichers, E.R.; Teslovich, T.M.; et al. Basal body dysfunction is a likely cause of pleiotropic Bardet-Biedl syndrome. Nature 2003, 425, 628–633. [Google Scholar] [CrossRef] [PubMed]
- Stone, E.M.; Andorf, J.L.; Whitmore, S.S.; DeLuca, A.P.; Giacalone, J.C.; Streb, L.M.; Braun, T.A.; Mullins, R.F.; Scheetz, T.E.; Sheffield, V.C.; et al. Clinically Focused Molecular Investigation of 1000 Consecutive Families with Inherited Retinal Disease. Ophthalmology 2017, 124, 1314–1331. [Google Scholar] [CrossRef] [PubMed]
- Bardet, G. Sur un Syndrome D’obesite Infantile avec Polydactylie et Retinite Pigmentaire (Contribution a L’etude des Formes Cliniques de L’obesite Hypophysaire); University of Paris: Paris, France, 1920. [Google Scholar]
- Biedl, A. Ein Geschwisterpaar mit adiposo-genitaler Dystrophie. Dtsch. Med. Wochenschr. 1922, 48, 1630. [Google Scholar]
- Forsythe, E.; Beales, P.L. Bardet-Biedl syndrome. Eur. J. Hum. Genet. 2013, 21, 8–13. [Google Scholar] [CrossRef]
- Beales, P.L.; Elcioglu, N.; Woolf, A.S.; Parker, D.; Flinter, F.A. New criteria for improved diagnosis of Bardet-Biedl syndrome: Results of a population survey. J. Med. Genet. 1999, 36, 437–446. [Google Scholar]
- Forsythe, E.; Beales, P.L. Bardet-Biedl Syndrome. In GeneReviews((R)); Adam, M.P., Ardinger, H.H., Pagon, R.A., Wallace, S.E., Bean, L.J.H., Stephens, K., Amemiya, A., Eds.; University of Washington: Seattle, WA, USA, 1993. [Google Scholar]
- Niederlova, V.; Modrak, M.; Tsyklauri, O.; Huranova, M.; Stepanek, O. Meta-analysis of genotype-phenotype associations in Bardet-Biedl syndrome uncovers differences among causative genes. Hum. Mutat 2019, 40, 2068–2087. [Google Scholar] [CrossRef]
- Wormser, O.; Gradstein, L.; Yogev, Y.; Perez, Y.; Kadir, R.; Goliand, I.; Sadka, Y.; El Riati, S.; Flusser, H.; Nachmias, D.; et al. SCAPER localizes to primary cilia and its mutation affects cilia length, causing Bardet-Biedl syndrome. Eur. J. Hum. Genet. 2019, 27, 928–940. [Google Scholar] [CrossRef]
- Forsythe, E.; Kenny, J.; Bacchelli, C.; Beales, P.L. Managing Bardet-Biedl Syndrome-Now and in the Future. Front. Pediatr. 2018, 6, 23. [Google Scholar] [CrossRef]
- Lindstrand, A.; Davis, E.E.; Carvalho, C.M.; Pehlivan, D.; Willer, J.R.; Tsai, I.C.; Ramanathan, S.; Zuppan, C.; Sabo, A.; Muzny, D.; et al. Recurrent CNVs and SNVs at the NPHP1 Locus Contribute Pathogenic Alleles to Bardet-Biedl Syndrome. Am. J. Hum. Genet. 2014, 94, 745–754. [Google Scholar] [CrossRef]
- Lindstrand, A.; Frangakis, S.; Carvalho, C.M.; Richardson, E.B.; McFadden, K.A.; Willer, J.R.; Pehlivan, D.; Liu, P.; Pediaditakis, I.L.; Sabo, A.; et al. Copy-Number Variation Contributes to the Mutational Load of Bardet-Biedl Syndrome. Am. J. Hum. Genet. 2016, 99, 318–336. [Google Scholar] [CrossRef] [PubMed]
- Stoetzel, C.; Laurier, V.; Davis, E.E.; Muller, J.; Rix, S.; Badano, J.L.; Leitch, C.C.; Salem, N.; Chouery, E.; Corbani, S.; et al. BBS10 encodes a vertebrate-specific chaperonin-like protein and is a major BBS locus. Nat. Genet. 2006, 38, 521–524. [Google Scholar] [CrossRef] [PubMed]
- Katsanis, N. The oligogenic properties of Bardet–Biedl syndrome. Hum. Mol. Genet. 2004, 13, R65–R71. [Google Scholar] [CrossRef] [PubMed]
- Mykytyn, K.; Nishimura, D.Y.; Searby, C.C.; Shastri, M.; Yen, H.J.; Beck, J.S.; Braun, T.; Streb, L.M.; Cornier, A.S.; Cox, G.F.; et al. Identification of the gene (BBS1) most commonly involved in Bardet-Biedl syndrome, a complex human obesity syndrome. Nat. Genet. 2002, 31, 435–438. [Google Scholar] [CrossRef] [PubMed]
- Stoetzel, C.; Laurier, V.; Faivre, L.; Megarbane, A.; Perrin-Schmitt, F.; Verloes, A.; Bonneau, D.; Mandel, J.L.; Cossee, M.; Dollfus, H. BBS8 is rarely mutated in a cohort of 128 Bardet-Biedl syndrome families. J. Hum. Genet. 2006, 51, 81–84. [Google Scholar] [CrossRef] [PubMed]
- Loktev, A.V.; Zhang, Q.; Beck, J.S.; Searby, C.C.; Scheetz, T.E.; Bazan, J.F.; Slusarski, D.C.; Sheffield, V.C.; Jackson, P.K.; Nachury, M.V. A BBSome subunit links ciliogenesis, microtubule stability, and acetylation. Dev. Cell 2008, 15, 854–865. [Google Scholar] [CrossRef]
- Nachury, M.V.; Loktev, A.V.; Zhang, Q.; Westlake, C.J.; Peranen, J.; Merdes, A.; Slusarski, D.C.; Scheller, R.H.; Bazan, J.F.; Sheffield, V.C.; et al. A core complex of BBS proteins cooperates with the GTPase Rab8 to promote ciliary membrane biogenesis. Cell 2007, 129, 1201–1213. [Google Scholar] [CrossRef]
- Jin, H.; White, S.R.; Shida, T.; Schulz, S.; Aguiar, M.; Gygi, S.P.; Bazan, J.F.; Nachury, M.V. The conserved Bardet-Biedl syndrome proteins assemble a coat that traffics membrane proteins to cilia. Cell 2010, 141, 1208–1219. [Google Scholar] [CrossRef]
- Pretorius, P.R.; Baye, L.M.; Nishimura, D.Y.; Searby, C.C.; Bugge, K.; Yang, B.; Mullins, R.F.; Stone, E.M.; Sheffield, V.C.; Slusarski, D.C. Identification and functional analysis of the vision-specific BBS3 (ARL6) long isoform. PLoS Genet. 2010, 6, e1000884. [Google Scholar] [CrossRef]
- Estrada-Cuzcano, A.; Koenekoop, R.K.; Senechal, A.; De Baere, E.B.W.; de Ravel, T.; Banfi, S.; Kohl, S.; Ayuso, C.; Sharon, D.; Hoyng, C.B.; et al. BBS1 Mutations in a Wide Spectrum of Phenotypes Ranging From Nonsyndromic Retinitis Pigmentosa to Bardet-Biedl Syndrome. Arch. Ophthalmol. 2012, 130, 1425–1432. [Google Scholar] [CrossRef]
- Shevach, E.; Ali, M.; Mizrahi-Meissonnier, L.; McKibbin, M.; El-Asrag, M.; Watson, C.M.; Inglehearn, C.F.; Ben-Yosef, T.; Blumenfeld, A.; Jalas, C.; et al. Association Between Missense Mutations in the BBS2 Gene and Nonsyndromic Retinitis Pigmentosa. JAMA Ophthalmol. 2015, 133, 312–318. [Google Scholar] [CrossRef]
- Riazuddin, S.A.; Iqbal, M.; Wang, Y.; Masuda, T.; Chen, Y.; Bowne, S.; Sullivan, L.S.; Waseem, N.H.; Bhattacharya, S.; Daiger, S.P.; et al. A splice-site mutation in a retina-specific exon of BBS8 causes nonsyndromic retinitis pigmentosa. Am. J. Hum. Genet. 2010, 86, 805–812. [Google Scholar] [CrossRef]
- Ofri, R.; Ekesten, B. Baseline retinal OCT measurements in normal female beagles: The effects of eccentricity, meridian, and age on retinal layer thickness. Vet. Ophthalmol. 2020, 23, 52–60. [Google Scholar] [CrossRef] [PubMed]
- Karlstam, L.; Hertil, E.; Zeiss, C.; Ropstad, E.O.; Bjerkås, E.; Dubielzig, R.R.; Ekesten, B. A slowly progressive retinopathy in the Shetland Sheepdog. Vet. Ophthalmol. 2011, 14, 227–238. [Google Scholar] [CrossRef] [PubMed]
- Ekesten, B.; Komáromy, A.M.; Ofri, R.; Petersen-Jones, S.M.; Narfström, K. Guidelines for clinical electroretinography in the dog: 2012 update. Doc. Ophthalmol. 2013, 127, 79–87. [Google Scholar] [CrossRef] [PubMed]
- Bane, A. Acrosomal abnormality associated with sterility in a boar. In Proceedings of the 4th International Congress of Animal Reproductives, The Hague, The Netherlands, 5–9 June 1961; pp. 810–817. [Google Scholar]
- Thomas, W.P.; Gaber, C.E.; Jacobs, G.J.; Kaplan, P.M.; Lombard, C.W.; Moise, N.S.; Moses, B.L. Recommendations for standards in transthoracic two-dimensional echocardiography in the dog and cat. Echocardiography Committee of the Specialty of Cardiology, American College of Veterinary Internal Medicine. J. Vet. Intern. Med. 1993, 7, 247–252. [Google Scholar] [CrossRef]
- Cornell, C.C.; Kittleson, M.D.; Della Torre, P.; Haggstrom, J.; Lombard, C.W.; Pedersen, H.D.; Vollmar, A.; Wey, A. Allometric scaling of M-mode cardiac measurements in normal adult dogs. J. Vet. Intern. Med. 2004, 18, 311–321. [Google Scholar] [CrossRef]
- Hansson, K.; Haggstrom, J.; Kvart, C.; Lord, P. Left atrial to aortic root indices using two-dimensional and M-mode echocardiography in cavalier King Charles spaniels with and without left atrial enlargement. Vet. Radiol. Ultrasound Off. J. Am. Coll. Vet. Radiol. Int. Vet. Radiol. Assoc. 2002, 43, 568–575. [Google Scholar] [CrossRef]
- Li, H. Minimap2: Pairwise alignment for nucleotide sequences. Bioinformatics 2018, 34, 3094–3100. [Google Scholar] [CrossRef]
- Kovaka, S.; Zimin, A.V.; Pertea, G.M.; Razaghi, R.; Salzberg, S.L.; Pertea, M. Transcriptome assembly from long-read RNA-seq alignments with StringTie2. Genome Biol. 2019, 20, 278. [Google Scholar] [CrossRef]
- Robinson, J.T.; Thorvaldsdóttir, H.; Winckler, W.; Guttman, M.; Lander, E.S.; Getz, G.; Mesirov, J.P. Integrative genomics viewer. Nat. Biotechnol 2011, 29, 24–26. [Google Scholar] [CrossRef] [PubMed]
- Thorvaldsdóttir, H.; Robinson, J.T.; Mesirov, J.P. Integrative Genomics Viewer (IGV): High-performance genomics data visualization and exploration. Brief. Bioinform. 2013, 14, 178–192. [Google Scholar] [CrossRef]
- Patro, R.; Duggal, G.; Love, M.I.; Irizarry, R.A.; Kingsford, C. Salmon provides fast and bias-aware quantification of transcript expression. Nat. Methods 2017, 14, 417–419. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.; Lowe, A.; Dharmat, R.; Lee, S.; Owen, L.A.; Wang, J.; Shakoor, A.; Li, Y.; Morgan, D.J.; Hejazi, A.A.; et al. Generation, transcriptome profiling, and functional validation of cone-rich human retinal organoids. Proc. Natl. Acad. Sci. USA 2019, 116, 10824–10833. [Google Scholar] [CrossRef] [PubMed]
- Kohl, S.; Coppieters, F.; Meire, F.; Schaich, S.; Roosing, S.; Brennenstuhl, C.; Bolz, S.; van Genderen, M.M.; Riemslag, F.C.; European Retinal Disease, C.; et al. A nonsense mutation in PDE6H causes autosomal-recessive incomplete achromatopsia. Am. J. Hum. Genet. 2012, 91, 527–532. [Google Scholar] [CrossRef] [PubMed]
- Nordstrom, K.; Larsson, T.A.; Larhammar, D. Extensive duplications of phototransduction genes in early vertebrate evolution correlate with block (chromosome) duplications. Genomics 2004, 83, 852–872. [Google Scholar] [CrossRef]
- Renninger, S.L.; Gesemann, M.; Neuhauss, S.C. Cone arrestin confers cone vision of high temporal resolution in zebrafish larvae. Eur. J. Neurosci. 2011, 33, 658–667. [Google Scholar] [CrossRef]
- Terakita, A. The opsins. Genome Biol. 2005, 6, 213. [Google Scholar] [CrossRef]
- Mowat, F.M.; Petersen-Jones, S.M.; Williamson, H.; Williams, D.L.; Luthert, P.J.; Ali, R.R.; Bainbridge, J.W. Topographical characterization of cone photoreceptors and the area centralis of the canine retina. Mol. Vis. 2008, 14, 2518–2527. [Google Scholar]
- Kaewkhaw, R.; Kaya, K.D.; Brooks, M.; Homma, K.; Zou, J.; Chaitankar, V.; Rao, M.; Swaroop, A. Transcriptome Dynamics of Developing Photoreceptors in Three-Dimensional Retina Cultures Recapitulates Temporal Sequence of Human Cone and Rod Differentiation Revealing Cell Surface Markers and Gene Networks. Stem Cells 2015, 33, 3504–3518. [Google Scholar] [CrossRef]
- Chen, H.; Weber, A.J. Expression of glial fibrillary acidic protein and glutamine synthetase by Muller cells after optic nerve damage and intravitreal application of brain-derived neurotrophic factor. Glia 2002, 38, 115–125. [Google Scholar] [CrossRef] [PubMed]
- Rauen, T.; Taylor, W.R.; Kuhlbrodt, K.; Wiessner, M. High-affinity glutamate transporters in the rat retina: A major role of the glial glutamate transporter GLAST-1 in transmitter clearance. Cell Tissue Res. 1998, 291, 19–31. [Google Scholar] [CrossRef] [PubMed]
- Roesch, K.; Jadhav, A.P.; Trimarchi, J.M.; Stadler, M.B.; Roska, B.; Sun, B.B.; Cepko, C.L. The transcriptome of retinal Muller glial cells. J. Comp. Neurol. 2008, 509, 225–238. [Google Scholar] [CrossRef] [PubMed]
- Macosko, E.Z.; Basu, A.; Satija, R.; Nemesh, J.; Shekhar, K.; Goldman, M.; Tirosh, I.; Bialas, A.R.; Kamitaki, N.; Martersteck, E.M.; et al. Highly Parallel Genome-wide Expression Profiling of Individual Cells Using Nanoliter Droplets. Cell 2015, 161, 1202–1214. [Google Scholar] [CrossRef]
- Siegert, S.; Cabuy, E.; Scherf, B.G.; Kohler, H.; Panda, S.; Le, Y.Z.; Fehling, H.J.; Gaidatzis, D.; Stadler, M.B.; Roska, B. Transcriptional code and disease map for adult retinal cell types. Nat. Neurosci. 2012, 15, 487–495. [Google Scholar] [CrossRef]
- Barnstable, C.J.; Drager, U.C. Thy-1 antigen: A ganglion cell specific marker in rodent retina. Neuroscience 1984, 11, 847–855. [Google Scholar] [CrossRef]
- Erkman, L.; McEvilly, R.J.; Luo, L.; Ryan, A.K.; Hooshmand, F.; O’Connell, S.M.; Keithley, E.M.; Rapaport, D.H.; Ryan, A.F.; Rosenfeld, M.G. Role of transcription factors Brn-3.1 and Brn-3.2 in auditory and visual system development. Nature 1996, 381, 603–606. [Google Scholar] [CrossRef]
- Fremeau, R.T., Jr.; Voglmaier, S.; Seal, R.P.; Edwards, R.H. VGLUTs define subsets of excitatory neurons and suggest novel roles for glutamate. Trends Neurosci. 2004, 27, 98–103. [Google Scholar] [CrossRef]
- Huang, W.; Fileta, J.; Guo, Y.; Grosskreutz, C.L. Downregulation of Thy1 in retinal ganglion cells in experimental glaucoma. Curr. Eye Res. 2006, 31, 265–271. [Google Scholar] [CrossRef]
- Rodriguez, A.R.; de Sevilla Muller, L.P.; Brecha, N.C. The RNA binding protein RBPMS is a selective marker of ganglion cells in the mammalian retina. J. Comp. Neurol. 2014, 522, 1411–1443. [Google Scholar] [CrossRef]
- Ruiz-Ederra, J.; Garcia, M.; Hicks, D.; Vecino, E. Comparative study of the three neurofilament subunits within pig and human retinal ganglion cells. Mol. Vis. 2004, 10, 83–92. [Google Scholar] [PubMed]
- Carr, A.J.; Vugler, A.A.; Yu, L.; Semo, M.; Coffey, P.; Moss, S.E.; Greenwood, J. The expression of retinal cell markers in human retinal pigment epithelial cells and their augmentation by the synthetic retinoid fenretinide. Mol. Vis. 2011, 17, 1701–1715. [Google Scholar] [PubMed]
- Cherry, T.J.; Trimarchi, J.M.; Stadler, M.B.; Cepko, C.L. Development and diversification of retinal amacrine interneurons at single cell resolution. Proc. Natl. Acad. Sci. USA 2009, 106, 9495–9500. [Google Scholar] [CrossRef] [PubMed]
- Shekhar, K.; Lapan, S.W.; Whitney, I.E.; Tran, N.M.; Macosko, E.Z.; Kowalczyk, M.; Adiconis, X.; Levin, J.Z.; Nemesh, J.; Goldman, M.; et al. Comprehensive Classification of Retinal Bipolar Neurons by Single-Cell Transcriptomics. Cell 2016, 166, 1308–1323 e1330. [Google Scholar] [CrossRef]
- Untergasser, A.; Cutcutache, I.; Koressaar, T.; Ye, J.; Faircloth, B.C.; Remm, M.; Rozen, S.G. Primer3—New capabilities and interfaces. Nucleic Acids Res. 2012, 40, e115. [Google Scholar] [CrossRef]
- Koressaar, T.; Remm, M. Enhancements and modifications of primer design program Primer3. Bioinformatics 2007, 23, 1289–1291. [Google Scholar] [CrossRef]
- Blom, E. A new sterilizing and hereditary defect (the “Dag defect”) located in the bull sperm tail. Nature 1966, 209, 739–740. [Google Scholar] [CrossRef]
- Laflamme, D. Development and validation of a body condition score system for dogs. Canine Pract. 1997, 22, 10–15. [Google Scholar]
- Kozak, M. An analysis of vertebrate mRNA sequences: Intimations of translational control. J. Cell Biol. 1991, 115, 887–903. [Google Scholar] [CrossRef]
- Hernández, G.; Osnaya, V.G.; Pérez-Martínez, X. Conservation and Variability of the AUG Initiation Codon Context in Eukaryotes. Trends Biochem. Sci. 2019, 44, 1009–1021. [Google Scholar] [CrossRef]
- Chakraborty, D.; Conley, S.M.; Pittler, S.J.; Naash, M.I. Role of RDS and Rhodopsin in Cngb1-Related Retinal Degeneration. Investig. Ophthalmol. Vis. Sci. 2016, 57, 787–797. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Downes, G.B.; Gautam, N. The G protein subunit gene families. Genomics 1999, 62, 544–552. [Google Scholar] [CrossRef] [PubMed]
- Winkler, P.A.; Ekenstedt, K.J.; Occelli, L.M.; Frattaroli, A.V.; Bartoe, J.T.; Venta, P.J.; Petersen-Jones, S.M. A large animal model for CNGB1 autosomal recessive retinitis pigmentosa. PLoS ONE 2013, 8, e72229. [Google Scholar] [CrossRef] [PubMed]
- Jain, V.; Ravindran, E.; Dhingra, N.K. Differential expression of Brn3 transcription factors in intrinsically photosensitive retinal ganglion cells in mouse. J. Comp. Neurol. 2012, 520, 742–755. [Google Scholar] [CrossRef]
- Damiani, D.; Alexander, J.J.; O’Rourke, J.R.; McManus, M.; Jadhav, A.P.; Cepko, C.L.; Hauswirth, W.W.; Harfe, B.D.; Strettoi, E. Dicer inactivation leads to progressive functional and structural degeneration of the mouse retina. J. Neurosci. 2008, 28, 4878–4887. [Google Scholar] [CrossRef]
- Moore, S.J.; Green, J.S.; Fan, Y.; Bhogal, A.K.; Dicks, E.; Fernandez, B.A.; Stefanelli, M.; Murphy, C.; Cramer, B.C.; Dean, J.C.; et al. Clinical and genetic epidemiology of Bardet-Biedl syndrome in Newfoundland: A 22-year prospective, population-based, cohort study. Am. J. Med. Genet. A 2005, 132A, 352–360. [Google Scholar] [CrossRef]
- Ullah, A.; Umair, M.; Yousaf, M.; Khan, S.A.; Nazim-Ud-Din, M.; Shah, K.; Ahmad, F.; Azeem, Z.; Ali, G.; Alhaddad, B.; et al. Sequence variants in four genes underlying Bardet-Biedl syndrome in consanguineous families. Mol. Vis. 2017, 23, 482–494. [Google Scholar]
- Smaoui, N.; Chaabouni, M.; Sergeev, Y.V.; Kallel, H.; Li, S.; Mahfoudh, N.; Maazoul, F.; Kammoun, H.; Gandoura, N.; Bouaziz, A.; et al. Screening of the eight BBS genes in Tunisian families: No evidence of triallelism. Investig. Ophthalmol. Vis. Sci. 2006, 47, 3487–3495. [Google Scholar] [CrossRef]
- Sato, S.; Morimoto, T.; Hotta, K.; Fujikado, T.; Nishida, K. A novel compound heterozygous mutation in TTC8 identified in a Japanese patient. Hum. Genome Var. 2019, 6, 14. [Google Scholar] [CrossRef]
- Janssen, S.; Ramaswami, G.; Davis, E.E.; Hurd, T.; Airik, R.; Kasanuki, J.M.; Van Der Kraak, L.; Allen, S.J.; Beales, P.L.; Katsanis, N.; et al. Mutation analysis in Bardet-Biedl syndrome by DNA pooling and massively parallel resequencing in 105 individuals. Hum. Genet. 2011, 129, 79–90. [Google Scholar] [CrossRef]
- Redin, C.; Le Gras, S.; Mhamdi, O.; Geoffroy, V.; Stoetzel, C.; Vincent, M.C.; Chiurazzi, P.; Lacombe, D.; Ouertani, I.; Petit, F.; et al. Targeted high-throughput sequencing for diagnosis of genetically heterogeneous diseases: Efficient mutation detection in Bardet-Biedl and Alström syndromes. J. Med. Genet. 2012, 49, 502–512. [Google Scholar] [CrossRef] [PubMed]
- Harville, H.M.; Held, S.; Diaz-Font, A.; Davis, E.E.; Diplas, B.H.; Lewis, R.A.; Borochowitz, Z.U.; Zhou, W.; Chaki, M.; MacDonald, J.; et al. Identification of 11 novel mutations in eight BBS genes by high-resolution homozygosity mapping. J. Med. Genet. 2010, 47, 262–267. [Google Scholar] [CrossRef] [PubMed]
- M’Hamdi, O.; Redin, C.; Stoetzel, C.; Ouertani, I.; Chaabouni, M.; Maazoul, F.; M’Rad, R.; Mandel, J.L.; Dollfus, H.; Muller, J.; et al. Clinical and genetic characterization of Bardet-Biedl syndrome in Tunisia: Defining a strategy for molecular diagnosis. Clin. Genet. 2014, 85, 172–177. [Google Scholar] [CrossRef] [PubMed]
- Goyal, S.; Jager, M.; Robinson, P.N.; Vanita, V. Confirmation of TTC8 as a disease gene for nonsyndromic autosomal recessive retinitis pigmentosa (RP51). Clin. Genet. 2016, 89, 454–460. [Google Scholar] [CrossRef] [PubMed]
- Rizzo, J.F., III; Berson, E.L.; Lessell, S. Retinal and neurologic findings in the Laurence-Moon-Bardet-Biedl phenotype. Ophthalmology 1986, 93, 1452–1456. [Google Scholar] [CrossRef]
- Dilan, T.L.; Singh, R.K.; Saravanan, T.; Moye, A.; Goldberg, A.F.X.; Stoilov, P.; Ramamurthy, V. Bardet-Biedl syndrome-8 (BBS8) protein is crucial for the development of outer segments in photoreceptor neurons. Hum. Mol. Genet. 2018, 27, 283–294. [Google Scholar] [CrossRef]
- Bek, T.; Rosenberg, T. Clinical pathology and retinal vascular structure in the Bardet-Biedl syndrome. Br. J. Ophthalmol. 1995, 79, 76–80. [Google Scholar] [CrossRef]
- Daniels, A.B.; Sandberg, M.A.; Chen, J.; Weigel-DiFranco, C.; Fielding Hejtmancic, J.; Berson, E.L. Genotype-phenotype correlations in Bardet-Biedl syndrome. Arch. Ophthalmol. 2012, 130, 901–907. [Google Scholar] [CrossRef]
- Adkins, E.A.; Hendrix, D.V. Outcomes of dogs presented for cataract evaluation: A retrospective study. J. Am. Anim. Hosp. Assoc. 2005, 41, 235–240. [Google Scholar] [CrossRef]
- Zigler, J.S., Jr.; Hess, H.H. Cataracts in the Royal College of Surgeons rat: Evidence for initiation by lipid peroxidation products. Exp. Eye Res. 1985, 41, 67–76. [Google Scholar] [CrossRef]
- Deehr, A.J.; Dubielzig, R.R. A histopathological study of iridociliary cysts and glaucoma in Golden Retrievers. Vet. Ophthalmol. 1998, 1, 153–158. [Google Scholar] [CrossRef] [PubMed]
- Nachury, M.V. The molecular machines that traffic signaling receptors into and out of cilia. Curr. Opin. Cell Biol. 2018, 51, 124–131. [Google Scholar] [CrossRef] [PubMed]
- Ye, F.; Nager, A.R.; Nachury, M.V. BBSome trains remove activated GPCRs from cilia by enabling passage through the transition zone. J. Cell Biol. 2018, 217, 1847–1868. [Google Scholar] [CrossRef] [PubMed]
- Satir, P.; Pedersen, L.B.; Christensen, S.T. The primary cilium at a glance. J. Cell Sci. 2010, 123, 499. [Google Scholar] [CrossRef]
- Nonaka, S.; Shiratori, H.; Saijoh, Y.; Hamada, H. Determination of left-right patterning of the mouse embryo by artificial nodal flow. Nature 2002, 418, 96–99. [Google Scholar] [CrossRef]
- Nonaka, S.; Tanaka, Y.; Okada, Y.; Takeda, S.; Harada, A.; Kanai, Y.; Kido, M.; Hirokawa, N. Randomization of left-right asymmetry due to loss of nodal cilia generating leftward flow of extraembryonic fluid in mice lacking KIF3B motor protein. Cell 1998, 95, 829–837. [Google Scholar] [CrossRef]
- Olson, A.J.; Krentz, A.D.; Finta, K.M.; Okorie, U.C.; Haws, R.M. Thoraco-Abdominal Abnormalities in Bardet-Biedl Syndrome: Situs Inversus and Heterotaxy. J. Pediatr. 2019, 204, 31–37. [Google Scholar] [CrossRef]
- Elbedour, K.; Zucker, N.; Zalzstein, E.; Barki, Y.; Carmi, R. Cardiac abnormalities in the Bardet-Biedl syndrome: Echocardiographic studies of 22 patients. Am. J. Med. Genet. 1994, 52, 164–169. [Google Scholar] [CrossRef]
- Fox, P.R. Pathology of myxomatous mitral valve disease in the dog. J. Vet. Cardiol. 2012, 14, 103–126. [Google Scholar] [CrossRef]
- Toomer, K.A.; Yu, M.; Fulmer, D.; Guo, L.; Moore, K.S.; Moore, R.; Drayton, K.D.; Glover, J.; Peterson, N.; Ramos-Ortiz, S.; et al. Primary cilia defects causing mitral valve prolapse. Sci. Transl. Med. 2019, 11. [Google Scholar] [CrossRef]
- Chew, T.; Haase, B.; Bathgate, R.; Willet, C.E.; Kaukonen, M.K.; Mascord, L.J.; Lohi, H.T.; Wade, C.M. A Coding Variant in the Gene Bardet-Biedl Syndrome 4 (BBS4) Is Associated with a Novel Form of Canine Progressive Retinal Atrophy. G3 2017, 7, 2327–2335. [Google Scholar] [CrossRef] [PubMed]
- Davis, R.E.; Swiderski, R.E.; Rahmouni, K.; Nishimura, D.Y.; Mullins, R.F.; Agassandian, K.; Philp, A.R.; Searby, C.C.; Andrews, M.P.; Thompson, S.; et al. A knockin mouse model of the Bardet-Biedl syndrome 1 M390R mutation has cilia defects, ventriculomegaly, retinopathy, and obesity. Proc. Natl. Acad. Sci. USA 2007, 104, 19422–19427. [Google Scholar] [CrossRef] [PubMed]
- Nishimura, D.Y.; Fath, M.; Mullins, R.F.; Searby, C.; Andrews, M.; Davis, R.; Andorf, J.L.; Mykytyn, K.; Swiderski, R.E.; Yang, B.; et al. Bbs2-null mice have neurosensory deficits, a defect in social dominance, and retinopathy associated with mislocalization of rhodopsin. Proc. Natl. Acad. Sci. USA 2004, 101, 16588–16593. [Google Scholar] [CrossRef]
- Zhang, Q.; Nishimura, D.; Seo, S.; Vogel, T.; Morgan, D.A.; Searby, C.; Bugge, K.; Stone, E.M.; Rahmouni, K.; Sheffield, V.C. Bardet-Biedl syndrome 3 (Bbs3) knockout mouse model reveals common BBS-associated phenotypes and Bbs3 unique phenotypes. Proc. Natl. Acad. Sci. USA 2011, 108, 20678–20683. [Google Scholar] [CrossRef] [PubMed]
- Mykytyn, K.; Mullins, R.F.; Andrews, M.; Chiang, A.P.; Swiderski, R.E.; Yang, B.; Braun, T.; Casavant, T.; Stone, E.M.; Sheffield, V.C. Bardet-Biedl syndrome type 4 (BBS4)-null mice implicate Bbs4 in flagella formation but not global cilia assembly. Proc. Natl. Acad. Sci. USA 2004, 101, 8664–8669. [Google Scholar] [CrossRef]
- Fath, M.A.; Mullins, R.F.; Searby, C.; Nishimura, D.Y.; Wei, J.; Rahmouni, K.; Davis, R.E.; Tayeh, M.K.; Andrews, M.; Yang, B.; et al. Mkks-null mice have a phenotype resembling Bardet-Biedl syndrome. Hum. Mol. Genet. 2005, 14, 1109–1118. [Google Scholar] [CrossRef]
- Zhang, Q.; Nishimura, D.; Vogel, T.; Shao, J.; Swiderski, R.; Yin, T.; Searby, C.; Carter, C.S.; Kim, G.; Bugge, K.; et al. BBS7 is required for BBSome formation and its absence in mice results in Bardet-Biedl syndrome phenotypes and selective abnormalities in membrane protein trafficking. J. Cell Sci. 2013, 126, 2372–2380. [Google Scholar] [CrossRef]
- Tadenev, A.L.D.; Kulaga, H.M.; May-Simera, H.L.; Kelley, M.W.; Katsanis, N.; Reed, R.R. Loss of Bardet–Biedl syndrome protein-8 (BBS8) perturbs olfactory function, protein localization, and axon targeting. Proc. Natl. Acad. Sci. USA 2011, 108, 10320. [Google Scholar] [CrossRef]
- Kulaga, H.M.; Leitch, C.C.; Eichers, E.R.; Badano, J.L.; Lesemann, A.; Hoskins, B.E.; Lupski, J.R.; Beales, P.L.; Reed, R.R.; Katsanis, N. Loss of BBS proteins causes anosmia in humans and defects in olfactory cilia structure and function in the mouse. Nat. Genet. 2004, 36, 994–998. [Google Scholar] [CrossRef]
- Bonnett, B.N.; Egenvall, A. Age patterns of disease and death in insured Swedish dogs, cats and horses. J. Comp. Pathol. 2010, 142 (Suppl. 1), S33–S38. [Google Scholar] [CrossRef]
- O’Dea, D.; Parfrey, P.S.; Harnett, J.D.; Hefferton, D.; Cramer, B.C.; Green, J. The importance of renal impairment in the natural history of Bardet-Biedl syndrome. Am. J. Kidney Dis. Off. J. Natl. Kidney Found. 1996, 27, 776–783. [Google Scholar] [CrossRef]
- Murphy, D.; Singh, R.; Kolandaivelu, S.; Ramamurthy, V.; Stoilov, P. Alternative Splicing Shapes the Phenotype of a Mutation in BBS8 To Cause Nonsyndromic Retinitis Pigmentosa. Mol. Cell Biol. 2015, 35, 1860–1870. [Google Scholar] [CrossRef]
- Klein, D.C. The 2004 Aschoff/Pittendrigh Lecture: Theory of the Origin of the Pineal Gland—A Tale of Conflict and Resolution. J. Biol. Rhythm. 2004, 19, 264–279. [Google Scholar] [CrossRef]
- Lykke-Andersen, S.; Jensen, T.H. Nonsense-mediated mRNA decay: An intricate machinery that shapes transcriptomes. Nat. Rev. Mol. Cell Biol. 2015, 16, 665–677. [Google Scholar] [CrossRef]
- Blatch, G.L.; Lassle, M. The tetratricopeptide repeat: A structural motif mediating protein-protein interactions. Bioessays 1999, 21, 932–939. [Google Scholar] [CrossRef]
- Abd-El-Barr, M.M.; Sykoudis, K.; Andrabi, S.; Eichers, E.R.; Pennesi, M.E.; Tan, P.L.; Wilson, J.H.; Katsanis, N.; Lupski, J.R.; Wu, S.M. Impaired photoreceptor protein transport and synaptic transmission in a mouse model of Bardet-Biedl syndrome. Vis. Res. 2007, 47, 3394–3407. [Google Scholar] [CrossRef]
- Lewis, G.P.; Fisher, S.K. Up-Regulation of Glial Fibrillary Acidic Protein in Response to Retinal Injury: Its Potential Role in Glial Remodeling and a Comparison to Vimentin Expression. In International Review of Cytology; Academic Press: Cambridge, MA, USA, 2003; Volume 230, pp. 263–290. [Google Scholar]
- Jomary, C.; Ahir, A.; Agarwal, N.; Neal, M.J.; Jones, S.E. Spatio-temporal pattern of ocular clusterin mRNA expression in the rd mouse. Brain Res. Mol. Brain Res. 1995, 29, 172–176. [Google Scholar] [CrossRef]
- Wong, P.; Borst, D.E.; Farber, D.; Danciger, J.S.; Tenniswood, M.; Chader, G.J.; van Veen, T. Increased TRPM-2/clusterin mRNA levels during the time of retinal degeneration in mouse models of retinitis pigmentosa. Biochem. Cell Biol. Biochim. Biol. Cell. 1994, 72, 439–446. [Google Scholar] [CrossRef] [PubMed]
- Sarthy, P.V.; Fu, M.; Huang, J. Developmental expression of the glial fibrillary acidic protein (GFAP) gene in the mouse retina. Cell. Mol. Neurobiol. 1991, 11, 623–637. [Google Scholar] [CrossRef]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Dog | Reads Produced | Bases Called * | Quality Passed Reads | Quality Passed Bases * | Mean Read Length (bp) | N50 (bp) | Mean Read Quality |
|---|---|---|---|---|---|---|---|
| GR02 | 4,895,849 | 6.78 | 3,577,000 (73.1%) | 5.86 (86.3%) | 1637 | 2277 | 10.1 |
| BE02 | 10,284,735 | 12.36 | 7,819,514 (76%) | 10.68 (86.4%) | 1365 | 1756 | 10.6 |
| GS01 | 11,717,598 | 9.01 | 7,629,750 (65.1%) | 7.06 (78.4%) | 926 | 1135 | 9.5 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mäkeläinen, S.; Hellsand, M.; van der Heiden, A.D.; Andersson, E.; Thorsson, E.; S. Holst, B.; Häggström, J.; Ljungvall, I.; Mellersh, C.; Hallböök, F.; et al. Deletion in the Bardet–Biedl Syndrome Gene TTC8 Results in a Syndromic Retinal Degeneration in Dogs. Genes 2020, 11, 1090. https://doi.org/10.3390/genes11091090
Mäkeläinen S, Hellsand M, van der Heiden AD, Andersson E, Thorsson E, S. Holst B, Häggström J, Ljungvall I, Mellersh C, Hallböök F, et al. Deletion in the Bardet–Biedl Syndrome Gene TTC8 Results in a Syndromic Retinal Degeneration in Dogs. Genes. 2020; 11(9):1090. https://doi.org/10.3390/genes11091090
Chicago/Turabian StyleMäkeläinen, Suvi, Minas Hellsand, Anna Darlene van der Heiden, Elina Andersson, Elina Thorsson, Bodil S. Holst, Jens Häggström, Ingrid Ljungvall, Cathryn Mellersh, Finn Hallböök, and et al. 2020. "Deletion in the Bardet–Biedl Syndrome Gene TTC8 Results in a Syndromic Retinal Degeneration in Dogs" Genes 11, no. 9: 1090. https://doi.org/10.3390/genes11091090
APA StyleMäkeläinen, S., Hellsand, M., van der Heiden, A. D., Andersson, E., Thorsson, E., S. Holst, B., Häggström, J., Ljungvall, I., Mellersh, C., Hallböök, F., Andersson, G., Ekesten, B., & Bergström, T. F. (2020). Deletion in the Bardet–Biedl Syndrome Gene TTC8 Results in a Syndromic Retinal Degeneration in Dogs. Genes, 11(9), 1090. https://doi.org/10.3390/genes11091090