Analyzing Differentially Expressed Genes and Pathways Associated with Pistil Abortion in Japanese Apricot via RNA-Seq

 , , ,
, , ,
Abstract
1. Introduction
2. Materials and Methods
2.1. Experimental Material
2.2. RNA Extraction and cDNA Library Preparation
2.3. Quality Control and Reads Mapping
2.4. RNA-Seq Data Analysis
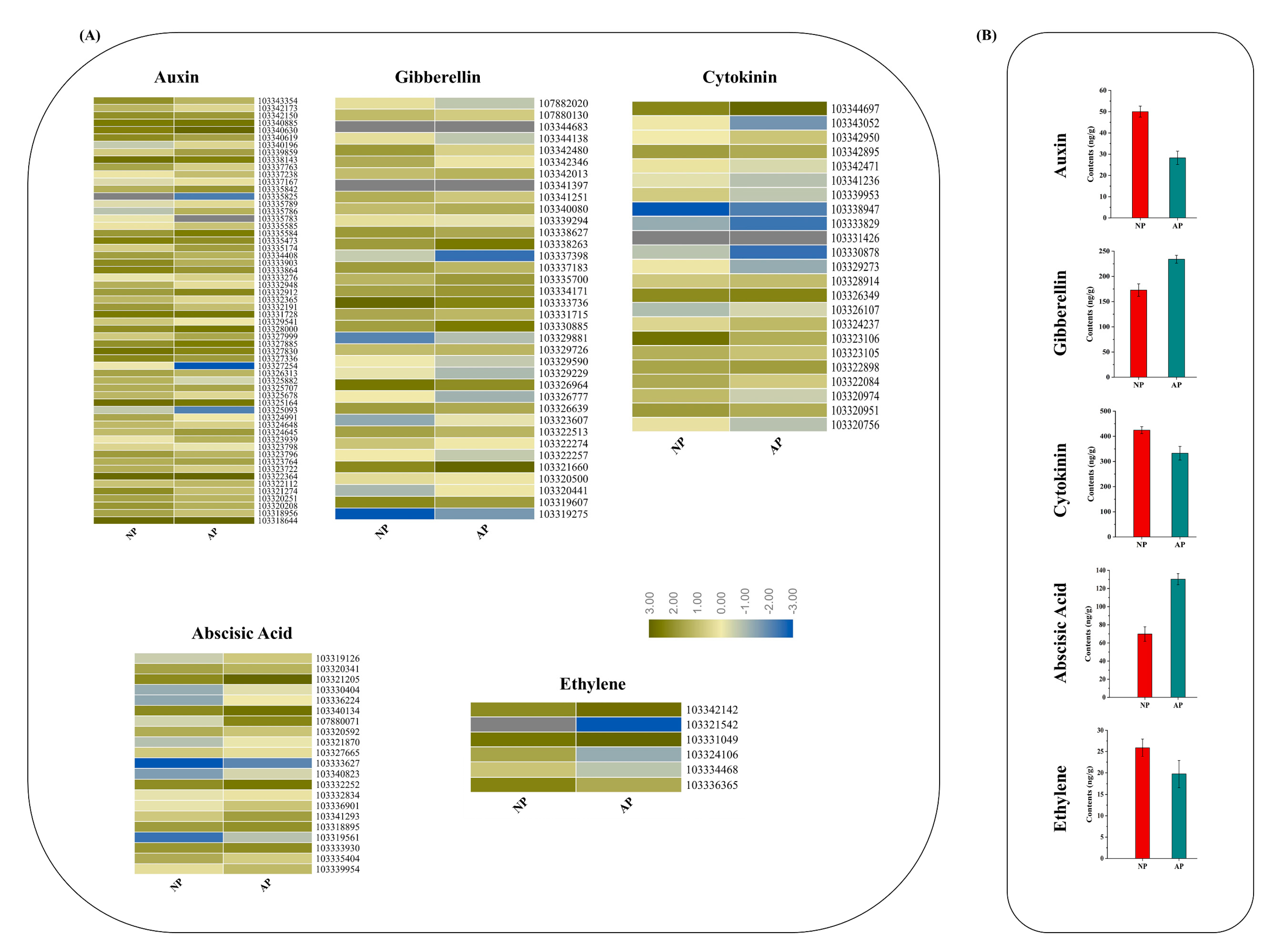
2.5. Endogenous Hormone Measurement
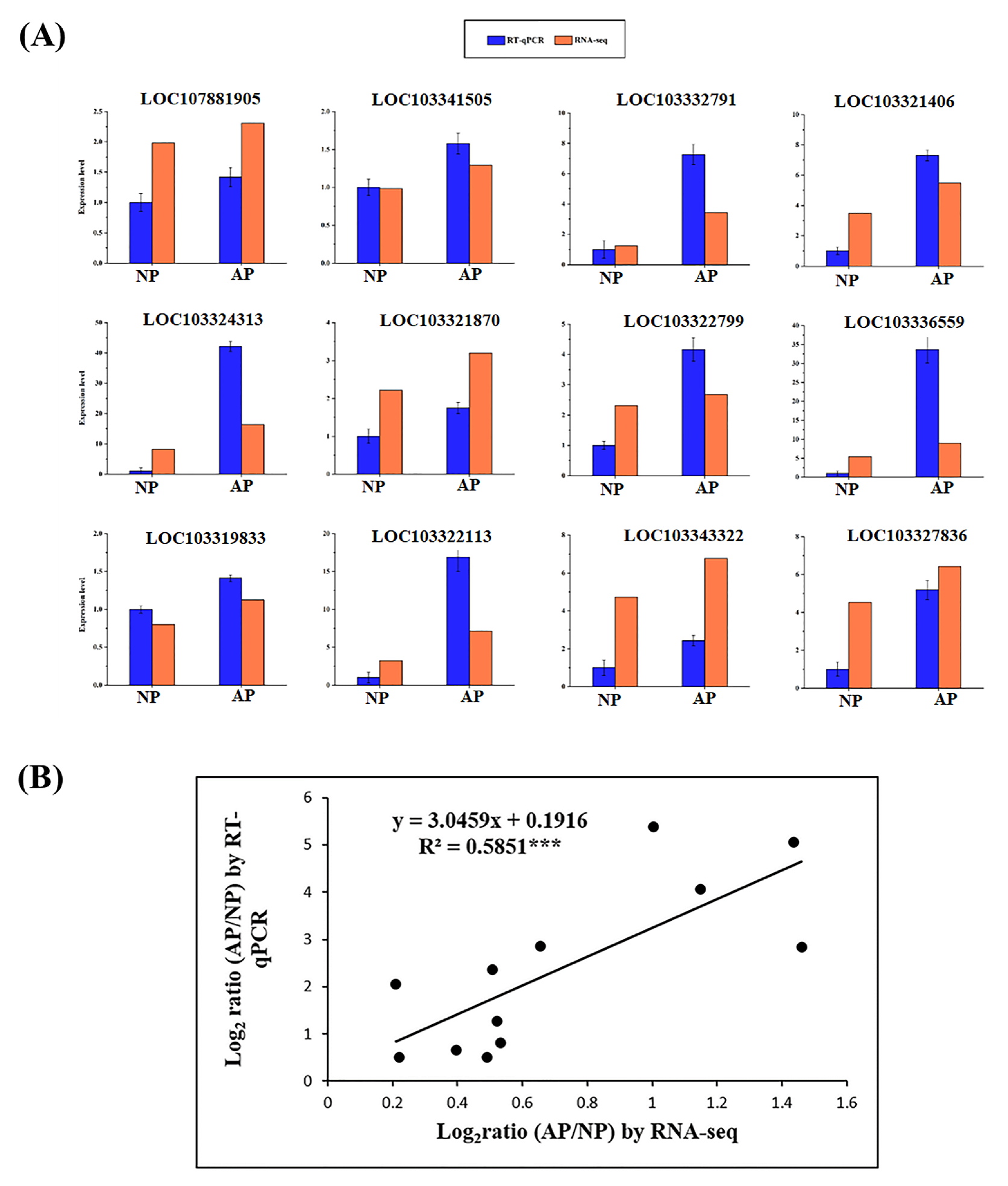
2.6. Quantitative Reverse Transcription PCR for Data Validation
3. Results
3.1. An Overview of RNA-Seq Libraries
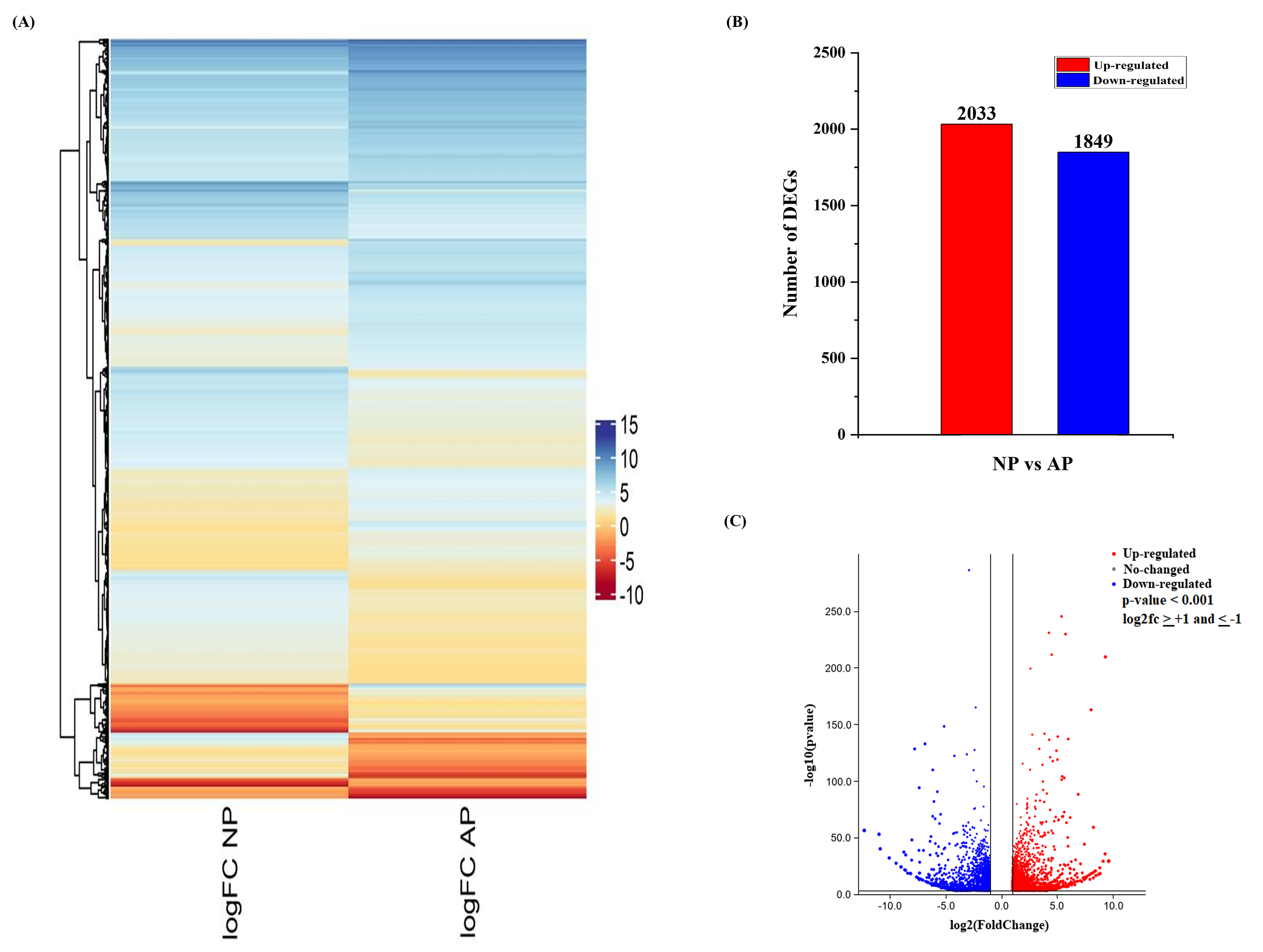
3.2. DEG Analysis of Normal and Abortive Pistils
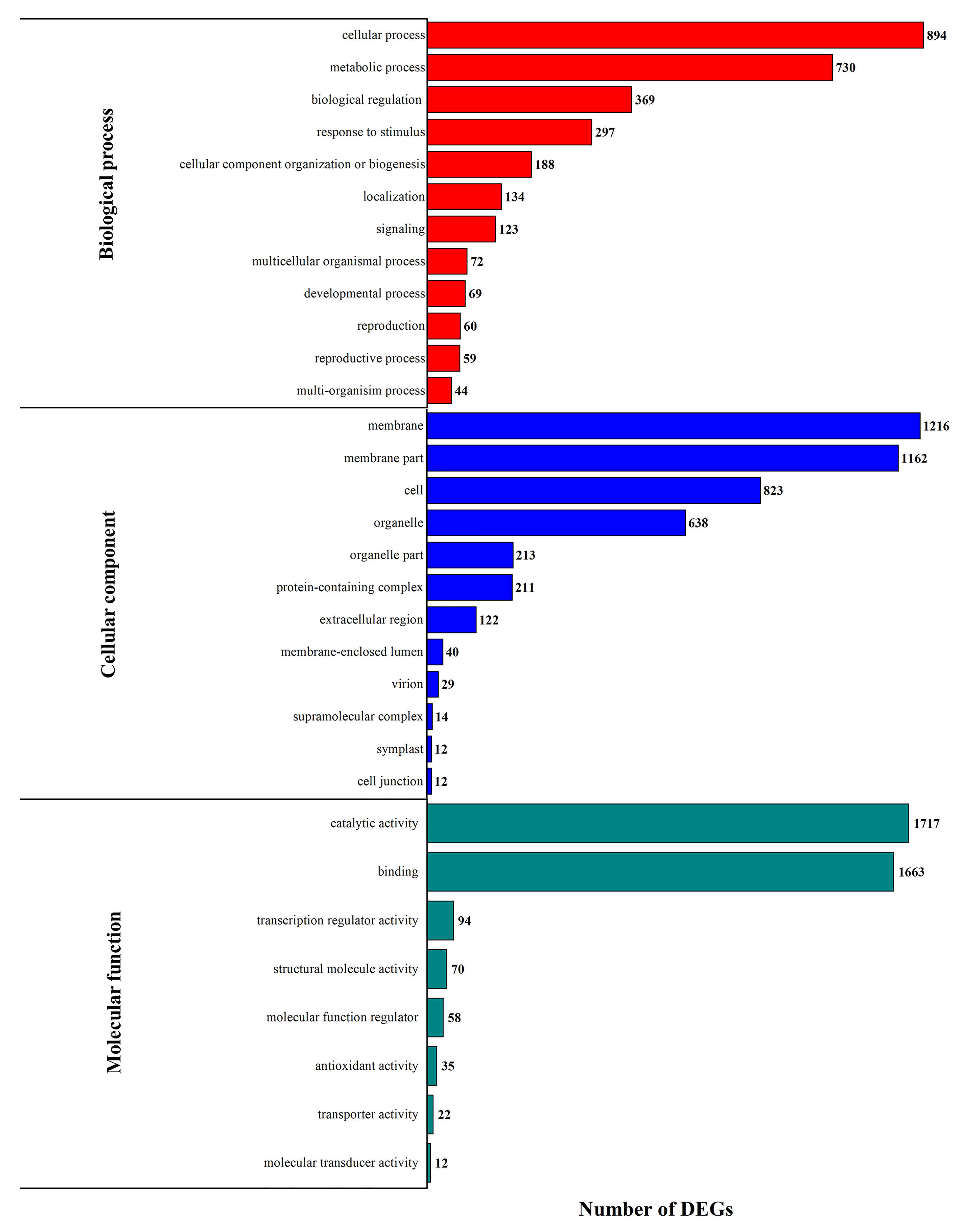
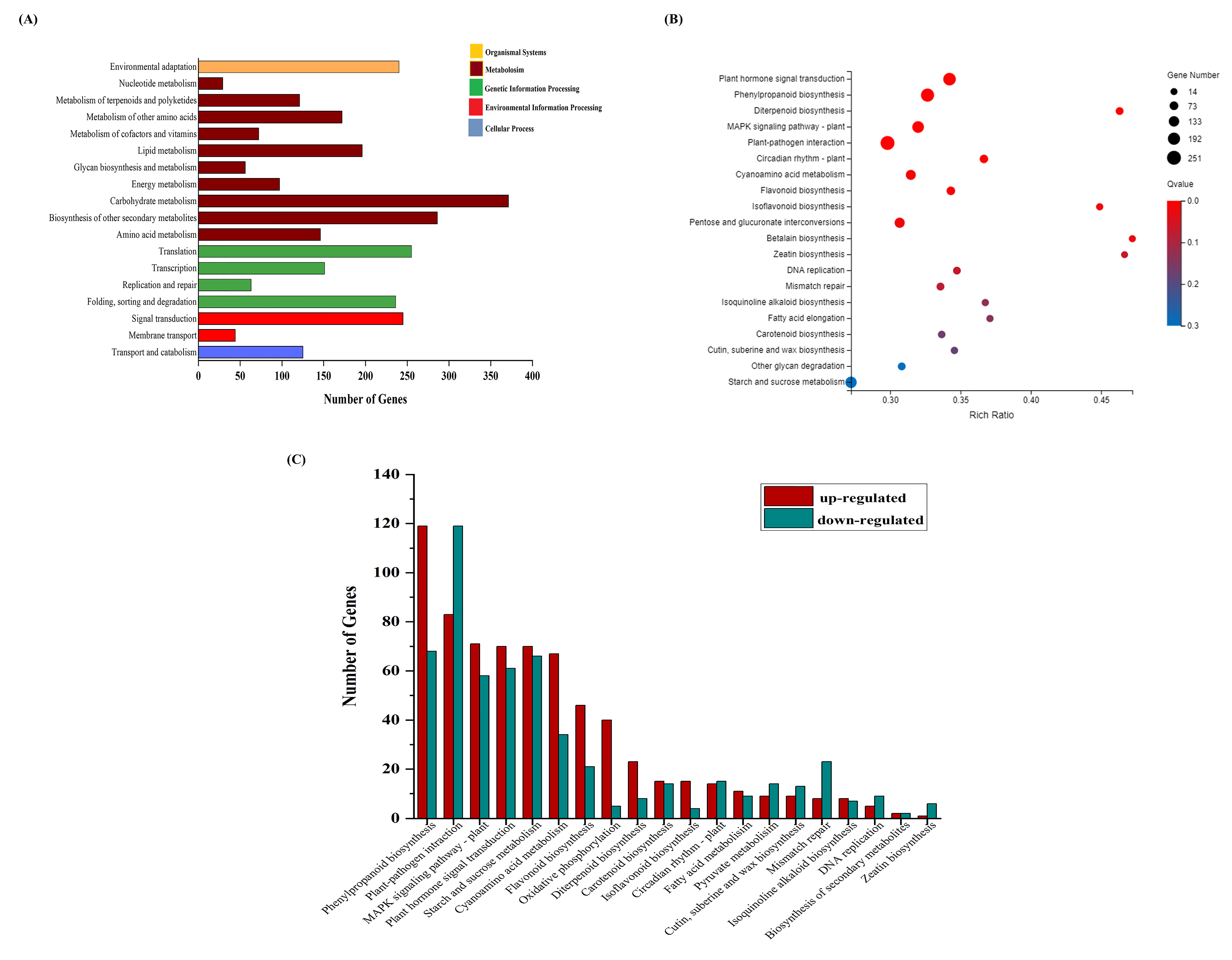
3.3. DEG Functional Enrichment Analysis
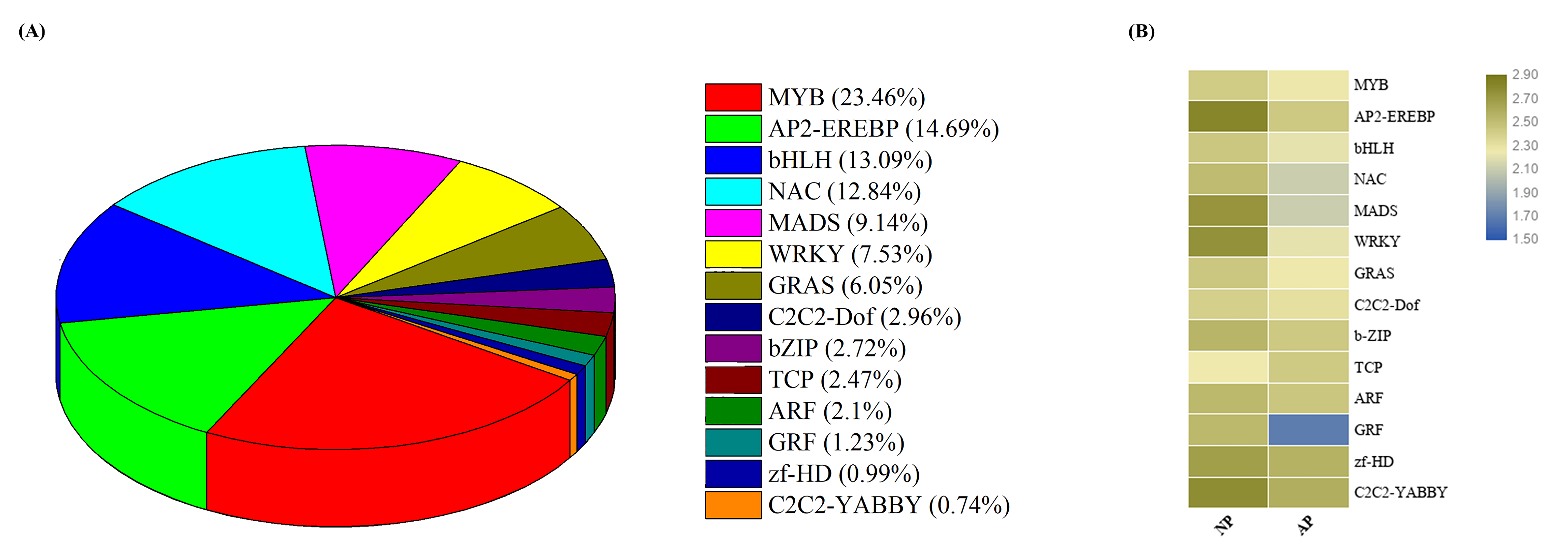
3.4. Expression Profiling of Transcription Factor Encoding Genes
3.5. DEGs in Response to Plant Hormone Signaling Transduction Pathway
3.6. DEGs in Response to Different Metabolic Pathways
3.7. Validation of Genes Through RT-qPCR
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Adachi, M.; Suzuki, Y.; Mizuta, T.; Osawa, T.; Adachi, T.; Osaka, K.; Suzuki, K.; Shiojima, K.; Arai, Y.; Masuda, K.; et al. The “Prunus mume Sieb. et Zucc” (Ume) is a Rich Natural Source of Novel Anti-Cancer Substance. Int. J. Food Prop. 2007, 10, 375–384. [Google Scholar] [CrossRef]
- Chu, M. Chinese Fruit Tree: Prunus mume; China Forestry Publishing House: Beijing, China, 1999. [Google Scholar]
- Huang, Y.; Liu, L.; Huang, J.; Wang, Z.; Chen, F.-F.; Zhang, Q.; Zheng, B.; Chen, M. Use of transcriptome sequencing to understand the pistillate flowering in hickory (Carya cathayensis Sarg.). BMC Genom. 2013, 14, 691. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Zhang, J.; Li, H.; Niu, J.; Xue, H.; Liu, B.; Wang, Q.; Luo, X.; Zhang, F.; Zhao, D.; et al. Transcriptomic Analysis Reveals Candidate Genes for Female Sterility in Pomegranate Flowers. Front. Plant Sci. 2017, 8, 8. [Google Scholar] [CrossRef] [PubMed]
- Gao, S.M.; Ma, K.; Du, X.H.; Li, F.L. Advances in research on Xanthoceras sorbifolia. Chin. Bull Bot 2002, 19, 296–301. [Google Scholar]
- Reale, L.; Sgromo, C.; Ederli, L.; Pasqualini, S.; Orlandi, F.; Fornaciari, M.; Ferranti, F.; Romano, B. Morphological and cytological development and starch accumulation in hermaphrodite and staminate flowers of olive (Olea europaea L.). Sex. Plant Reprod. 2009, 22, 109–119. [Google Scholar] [CrossRef] [PubMed]
- Hou, J.-H.; Gao, Z.-H.; Zhang, Z.; Chen, S.-M.; Ando, T.; Zhang, J.-Y.; Wang, X. Isolation and Characterization of an AGAMOUS Homologue PmAG from the Japanese Apricot (Prunus mume Sieb. et Zucc.). Plant Mol. Boil. Rep. 2010, 29, 473–480. [Google Scholar] [CrossRef]
- Wang, S. Preliminary Studies on Differences of Related Characteristics between Perfect Flower and Imperfect Flower and Protemics in Japanese Apricot; Nanjing Agricultural University: Nanjing, China, 2008. [Google Scholar]
- Wetzstein, H.Y.; Ravid, N.; Wilkins, E.; Martinelli, A.P. A Morphological and Histological Characterization of Bisexual and Male Flower Types in Pomegranate. J. Am. Soc. Hortic. Sci. 2011, 136, 83–92. [Google Scholar] [CrossRef]
- Shi, T.; Zhang, Q.; Gao, Z.; Zhang, Z.; Zhuang, W. Analyses on pistil differentiation process and related biochemical indexes of two cultivars of Prunus mume. J. Plant Resour. Environ. 2011, 20, 35–41. [Google Scholar]
- Bai, Z.; Feng, J.; Li, W.; Hu, Y.; Cao, X.; Sun, J. Investigation of pistil abortion rate of five apricot cultivars in Xinjiang. Xinjiang Agric. Sci. 2012, 49, 1805–1809. [Google Scholar]
- Dokoozlian, N.K. Grape berry growth and development. Raisin Prod. Man. 2000, 3393, 30. [Google Scholar]
- Davis, S.J. Integrating hormones into the floral-transition pathway ofArabidopsis thaliana. Plant Cell Environ. 2009, 32, 1201–1210. [Google Scholar] [CrossRef]
- Zhang, D.; Ren, L.; Yue, J.-H.; Wang, L.; Zhuo, L.-H.; Shen, X. A comprehensive analysis of flowering transition in Agapanthus praecox ssp. orientalis (Leighton) Leighton by using transcriptomic and proteomic techniques. J. Proteom. 2013, 80, 1–25. [Google Scholar] [CrossRef]
- Fu, C.; Huang, N.; Li, S.; Zhao, Z.; Huang, Z.; Shi, Y.; Tang, F. Endogenous hormones contents and growth development in South Feng-shui pear. Southwest China J. Agric. Sci. 2014, 27, 276–279. [Google Scholar]
- Zhu, Y.; Li, Y.; Xin, D.; Chen, W.; Shao, X.; Wang, Y.; Guo, W. RNA-Seq-based transcriptome analysis of dormant flower buds of Chinese cherry (Prunus pseudocerasus). Gene 2015, 555, 362–376. [Google Scholar] [CrossRef]
- Jacobsen, J.V.; Pearce, D.W.; Poole, A.T.; Pharis, R.P.; Mander, L.N. Abscisic acid, phaseic acid and gibberellin contents associated with dormancy and germination in barley. Physiol. Plant 2002, 115, 428–441. [Google Scholar] [CrossRef]
- Bartrina, I.; Otto, E.; Strnad, M.; Werner, T.; Schmülling, T. Cytokinin Regulates the Activity of Reproductive Meristems, Flower Organ Size, Ovule Formation, and Thus Seed Yield in Arabidopsis thaliana. Plant Cell 2011, 23, 69–80. [Google Scholar] [CrossRef]
- Corbesier, L.; Prinsen, E.; Jacqmard, A.; Lejeune, P.; Van Onckelen, H.; Perilleux, C.; Bernier, G. Cytokinin levels in leaves, leaf exudate and shoot apical meristem of Arabidopsis thaliana during floral transition. J. Exp. Bot. 2003, 54, 2511–2517. [Google Scholar] [CrossRef]
- Debeaujon, I. Gibberellin Requirement for Arabidopsis Seed Germination Is Determined Both by Testa Characteristics and Embryonic Abscisic Acid. Plant Physiol. 2000, 122, 415–424. [Google Scholar] [CrossRef]
- Bencivenga, S.; Simonini, S.; Benková, E.; Colombo, L. The Transcription Factors BEL1 and SPL Are Required for Cytokinin and Auxin Signaling During Ovule Development in Arabidopsis. Plant Cell 2012, 24, 2886–2897. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Wang, L.; Yang, Y.; Cui, J.; Chang, F.; Wang, Y.; Zhang, X. Analysis of Arabidopsis floral transcriptome: Detection of new florally expressed genes and expansion of Brassicaceae-specific gene families. Front. Plant Sci. 2015, 5, 802. [Google Scholar] [CrossRef]
- Liu, K.; Feng, S.; Pan, Y.; Zhong, J.; Chen, Y.; Yuan, C.; Li, H. Transcriptome Analysis and Identification of Genes Associated with Floral Transition and Flower Development in Sugar Apple (Annona squamosa L.). Front. Plant Sci. 2016, 7. [Google Scholar] [CrossRef] [PubMed]
- Alagna, F.; Cirilli, M.; Galla, G.; Carbone, F.; Daddiego, L.; Facella, P.; Lopez, L.; Colao, C.; Mariotti, R.; Cultrera, N.G.M.; et al. Transcript Analysis and Regulative Events during Flower Development in Olive (Olea europaea L.). PLoS ONE 2016, 11, e0152943. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.; Langmead, B.; Salzberg, S.L. HISAT: A fast spliced aligner with low memory requirements. Nat. Methods 2015, 12, 357–360. [Google Scholar] [CrossRef]
- Langmead, B.; Salzberg, S.L. Fast gapped-read alignment with Bowtie 2. Nat. Methods 2012, 9, 357–359. [Google Scholar] [CrossRef]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 002832. [Google Scholar] [CrossRef] [PubMed]
- Kanehisa, M.; Goto, S. KEGG: Kyoto Encyclopedia of Genes and Genomes. Nucleic Acids Res. 2000, 28, 27–30. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.-T.; Li, Y.-F.; Luan, T.-G.; Lan, C.-Y.; Shu, W.-S. Simultaneous Determination of Phytohormones in Plant Extracts using SPME and HPLC. Chromatographia 2007, 66, 515–520. [Google Scholar] [CrossRef]
- Guo, S.; Iqbal, S.; Ma, R.; Song, J.; Yu, M.; Gao, Z. High-density genetic map construction and quantitative trait loci analysis of the stony hard phenotype in peach based on restriction-site associated DNA sequencing. BMC Genom. 2018, 19, 612. [Google Scholar] [CrossRef]
- Iqbal, S.; Ni, X.; Bilal, M.S.; Shi, T.; Khalil-Ur-Rehman, M.; Zhenpeng, P.; Jie, G.; Usman, M.; Gao, Z. Identification and expression profiling of sugar transporter genes during sugar accumulation at different stages of fruit development in apricot. Gene 2020, 742, 144584. [Google Scholar] [CrossRef]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2−ΔΔCT method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef]
- Ali, M.A.; Azeem, F.; Nawaz, M.A.; Acet, T.; Abbas, A.; Imran, Q.M.; Shah, K.H.; Rehman, H.M.; Chung, G.; Yang, S.H.; et al. Transcription factors WRKY11 and WRKY17 are involved in abiotic stress responses in Arabidopsis. J. Plant Physiol. 2018, 226, 12–21. [Google Scholar] [CrossRef] [PubMed]
- Stewart, D.; Graciet, E.; Wellmer, F. Molecular and regulatory mechanisms controlling floral organ development. FEBS J. 2016, 283, 1823–1830. [Google Scholar] [CrossRef] [PubMed]
- Heisler, M.G.; Ohno, C.; Das, P.; Sieber, P.; Reddy, G.V.; Long, J.A.; Meyerowitz, E.M. Patterns of Auxin Transport and Gene Expression during Primordium Development Revealed by Live Imaging of the Arabidopsis Inflorescence Meristem. Curr. Boil. 2005, 15, 1899–1911. [Google Scholar] [CrossRef]
- Krizek, B.A. AINTEGUMENTA-LIKE genes have partly overlapping functions with AINTEGUMENTA but make distinct contributions to Arabidopsis thaliana flower development. J. Exp. Bot. 2015, 66, 4537–4549. [Google Scholar] [CrossRef]
- Sharma, R.; Agarwal, P.; Ray, S.; Deveshwar, P.; Sharma, P.; Sharma, N.; Nijhawan, A.; Jain, M.; Singh, A.K.; Singh, V.P.; et al. Expression dynamics of metabolic and regulatory components across stages of panicle and seed development in indica rice. Funct. Integr. Genom. 2012, 12, 229–248. [Google Scholar] [CrossRef]
- Wang, L.; Xie, W.; Chen, Y.; Tang, W.; Yang, J.; Ye, R.; Liu, L.; Lin, Y.; Xu, C.; Xiao, J.; et al. A dynamic gene expression atlas covering the entire life cycle of rice. Plant J. 2010, 61, 752–766. [Google Scholar] [CrossRef]
- Schwechheimer, C.; Zourelidou, M.; Bevan, M. Plant transcription factor studies. Annu. Rev. Plant Boil. 1998, 49, 127–150. [Google Scholar] [CrossRef]
- Zhang, W.; Sun, Y.; Timofejeva, L.; Chen, C.; Grossniklaus, U.; Ma, H. Regulation of Arabidopsis tapetum development and function by DYSFUNCTIONAL TAPETUM1 (DYT1) encoding a putative bHLH transcription factor. Development 2006, 133, 3085–3095. [Google Scholar] [CrossRef]
- Uno, Y.; Furihata, T.; Abe, H.; Yoshida, R.; Shinozaki, K.; Yamaguchi-Shinozaki, K. Arabidopsis Basic Leucine Zipper Transcription Factors Involved in an Abscisic Acid-Dependent Signal Transduction Pathway under Drought and High-Salinity Conditions; National Academy of Sciences: Washington, DC, USA, 2000; Volume 97, pp. 11632–11637. [Google Scholar]
- Liang, G.; He, H.; Li, Y.; Wang, F.; Yu, D. Molecular Mechanism of microRNA396 Mediating Pistil Development in Arabidopsis1. Plant Physiol. 2013, 164, 249–258. [Google Scholar] [CrossRef]
- Liscum, E.; Reed, J.W. Genetics of Aux/IAA and ARF action in plant growth and development. Plant Mol. Boil. 2002, 49, 387–400. [Google Scholar] [CrossRef]
- Kariali, E.; Mohapatra, P.K. Hormonal regulation of tiller dynamics in differentially-tillering rice cultivars. Plant Growth Regul. 2007, 53, 215–223. [Google Scholar] [CrossRef]
- Sorce, C.; Montanaro, G.; Bottega, S.; Spano’, C. Indole-3-acetic acid metabolism and growth in young kiwifruit berry. Plant Growth Regul. 2017, 82, 505–515. [Google Scholar] [CrossRef]
- Nagpal, P.; Ellis, C.M.; Weber, H.; Ploense, S.E.; Barkawi, L.S.; Guilfoyle, T.J.; Hagen, G.; Alonso, J.M.; Cohen, J.D.; Farmer, E.E.; et al. Auxin response factors ARF6 and ARF8 promote jasmonic acid production and flower maturation. Development 2005, 132, 4107–4118. [Google Scholar] [CrossRef]
- Li, G.; Ma, J.; Tan, M.; Mao, J.; An, N.; Sha, G.; Zhang, D.; Zhao, C.; Han, M. Transcriptome analysis reveals the effects of sugar metabolism and auxin and cytokinin signaling pathways on root growth and development of grafted apple. BMC Genom. 2016, 17, 150. [Google Scholar] [CrossRef] [PubMed]
- Liu, G.; Li, W.; Zheng, P.; Xu, T.; Chen, L.; Liu, D.; Hussain, S.; Teng, Y. Transcriptomic analysis of ‘Suli’ pear (Pyrus pyrifolia white pear group) buds during the dormancy by RNA-Seq. BMC Genom. 2012, 13, 700. [Google Scholar] [CrossRef]
- Chen, D.; Wang, F.; Gao, A.; Wang, Q.; Ding, Q. Effects of GA_ (3) and ethylene on abortion of apricot flower. Acta Agric. Boreali Occident. Sinica 2001, 10, 52–55. [Google Scholar]
- Pearce, S.; Huttly, A.K.; Prosser, I.M.; Li, Y.D.; Vaughan, S.P.; Gallova, B.; Patil, A.; Coghill, J.; Dubcovsky, J.; Hedden, P.; et al. Heterologous expression and transcript analysis of gibberellin biosynthetic genes of grasses reveals novel functionality in the GA3ox family. BMC Plant Boil. 2015, 15, 130. [Google Scholar] [CrossRef]
- Li, L.; Zhang, W.; Zhang, L.; Li, N.; Peng, J.; Wang, Y.; Zhong, C.; Yang, Y.; Sun, S.; Liang, S.; et al. Transcriptomic insights into antagonistic effects of gibberellin and abscisic acid on petal growth in Gerbera hybrida. Front. Plant Sci. 2015, 6. [Google Scholar] [CrossRef]
- Riboni, M.; Test, A.R.; Galbiati, M.; Tonelli, C.; Conti, L. ABA-dependent control of GIGANTEA signalling enables drought escape via up-regulation of FLOWERING LOCUS T in Arabidopsis thaliana. J. Exp. Bot. 2016, 67, 6309–6322. [Google Scholar] [CrossRef]
- Meng, Y.; Ma, S.; Shao, J.; Sun, J.; Ma, B.; Wang, H. Effects of spraying 6-BA on axillary bud growth and the dynamic changes of endogenous hormones in ’Tianhong 2′ Fuji nursery apple trees. Acta Hortic. Sinica 2012, 39, 837–844. [Google Scholar]
- McLoughlin, F.; Galvan-Ampudia, C.S.; Julkowska, M.M.; Caarls, L.; Van Der Does, D.; Laurière, C.; Munnik, T.; Haring, M.A.; Testerink, C. The Snf1-related protein kinases SnRK2.4 and SnRK2.10 are involved in maintenance of root system architecture during salt stress. Plant J. 2012, 72, 436–449. [Google Scholar] [CrossRef] [PubMed]
- Yoshida, T.; Fujita, Y.; Maruyama, K.; Mogami, J.; Todaka, D.; Shinozaki, K.; Yamaguchi-Shinozaki, K. Four A rabidopsis AREB/ABF transcription factors function predominantly in gene expression downstream of SnRK2 kinases in abscisic acid signalling in response to osmotic stress. Plant Cell Environ. 2015, 38, 35–49. [Google Scholar] [CrossRef]
- Kaur-Sawhney, R.; Tiburcio, A.F.; Galston, A.W. Spermidine and flower-bud differentiation in thin-layer explants of tobacco. Planta 1988, 173, 282–284. [Google Scholar] [CrossRef]
- Hennig, L.; Gruissem, W.; Grossniklaus, U.; Köhler, C. Transcriptional Programs of Early Reproductive Stages in Arabidopsis1. Plant Physiol. 2004, 135, 1765–1775. [Google Scholar] [CrossRef]
- Laitinen, R.A.; Immanen, J.; Auvinen, P.; Rudd, S.; Alatalo, E.; Paulin, L.; Ainasoja, M.; Kotilainen, M.; Koskela, S.; Teeri, T.H.; et al. Analysis of the floral transcriptome uncovers new regulators of organ determination and gene families related to flower organ differentiation in Gerbera hybrida (Asteraceae). Genome Res. 2005, 15, 475–486. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Xie, D.-Y.; Sharma, S.B.; Paiva, N.L.; Ferreira, D.; Dixon, R.A. Role of Anthocyanidin Reductase, Encoded by BANYULS in Plant Flavonoid Biosynthesis. Science 2003, 299, 396–399. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample Name | Total Raw Reads (Mb) | Total Clean Reads (Mb) | Clean Reads Q20 (%) | Clean Reads Q30 (%) | Clean Reads Ratio (%) | Total Mapping (%) | Uniquely Mapping (%) | Accession Number |
|---|---|---|---|---|---|---|---|---|
| NP-1 | 62.47 | 61.66 | 97.21 | 92.63 | 98.71 | 86.19 | 61.90 | SRR12234382 |
| NP-2 | 62.52 | 61.72 | 97.22 | 92.67 | 98.80 | 84.95 | 61.07 | SRR12234384 |
| NP-3 | 62.47 | 61.74 | 97.12 | 92.41 | 98.83 | 88.24 | 62.97 | SRR12234380 |
| AP-1 | 69.96 | 65.16 | 97.48 | 89.81 | 93.13 | 86.42 | 65.53 | SRR12234385 |
| AP-2 | 67.47 | 63.54 | 97.59 | 90.16 | 94.18 | 87.77 | 66.69 | SRR12234383 |
| AP-3 | 69.96 | 65.83 | 97.61 | 90.22 | 94.08 | 86.28 | 65.53 | SRR12234381 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Shi, T.; Iqbal, S.; Ayaz, A.; Bai, Y.; Pan, Z.; Ni, X.; Hayat, F.; Saqib Bilal, M.; Khuram Razzaq, M.; Gao, Z. Analyzing Differentially Expressed Genes and Pathways Associated with Pistil Abortion in Japanese Apricot via RNA-Seq. Genes 2020, 11, 1079. https://doi.org/10.3390/genes11091079
Shi T, Iqbal S, Ayaz A, Bai Y, Pan Z, Ni X, Hayat F, Saqib Bilal M, Khuram Razzaq M, Gao Z. Analyzing Differentially Expressed Genes and Pathways Associated with Pistil Abortion in Japanese Apricot via RNA-Seq. Genes. 2020; 11(9):1079. https://doi.org/10.3390/genes11091079
Chicago/Turabian StyleShi, Ting, Shahid Iqbal, Aliya Ayaz, Yang Bai, Zhenpeng Pan, Xiaopeng Ni, Faisal Hayat, Muhammad Saqib Bilal, Muhammad Khuram Razzaq, and Zhihong Gao. 2020. "Analyzing Differentially Expressed Genes and Pathways Associated with Pistil Abortion in Japanese Apricot via RNA-Seq" Genes 11, no. 9: 1079. https://doi.org/10.3390/genes11091079
APA StyleShi, T., Iqbal, S., Ayaz, A., Bai, Y., Pan, Z., Ni, X., Hayat, F., Saqib Bilal, M., Khuram Razzaq, M., & Gao, Z. (2020). Analyzing Differentially Expressed Genes and Pathways Associated with Pistil Abortion in Japanese Apricot via RNA-Seq. Genes, 11(9), 1079. https://doi.org/10.3390/genes11091079