The Potential of HTS Approaches for Accurate Genotyping in Grapevine (Vitis vinifera L.)




Abstract
1. Introduction
2. Materials and Methods
2.1. SSRs and Cultivars
2.2. DNA Extraction
2.3. PCR Amplification
2.3.1. PCR for Locus-Specific Amplification
2.3.2. PCR for Barcode Integration
2.4. Pooling and Sequencing
2.5. Bioinformatics Analysis
3. Results and Discussion
3.1. Sequencing Analysis
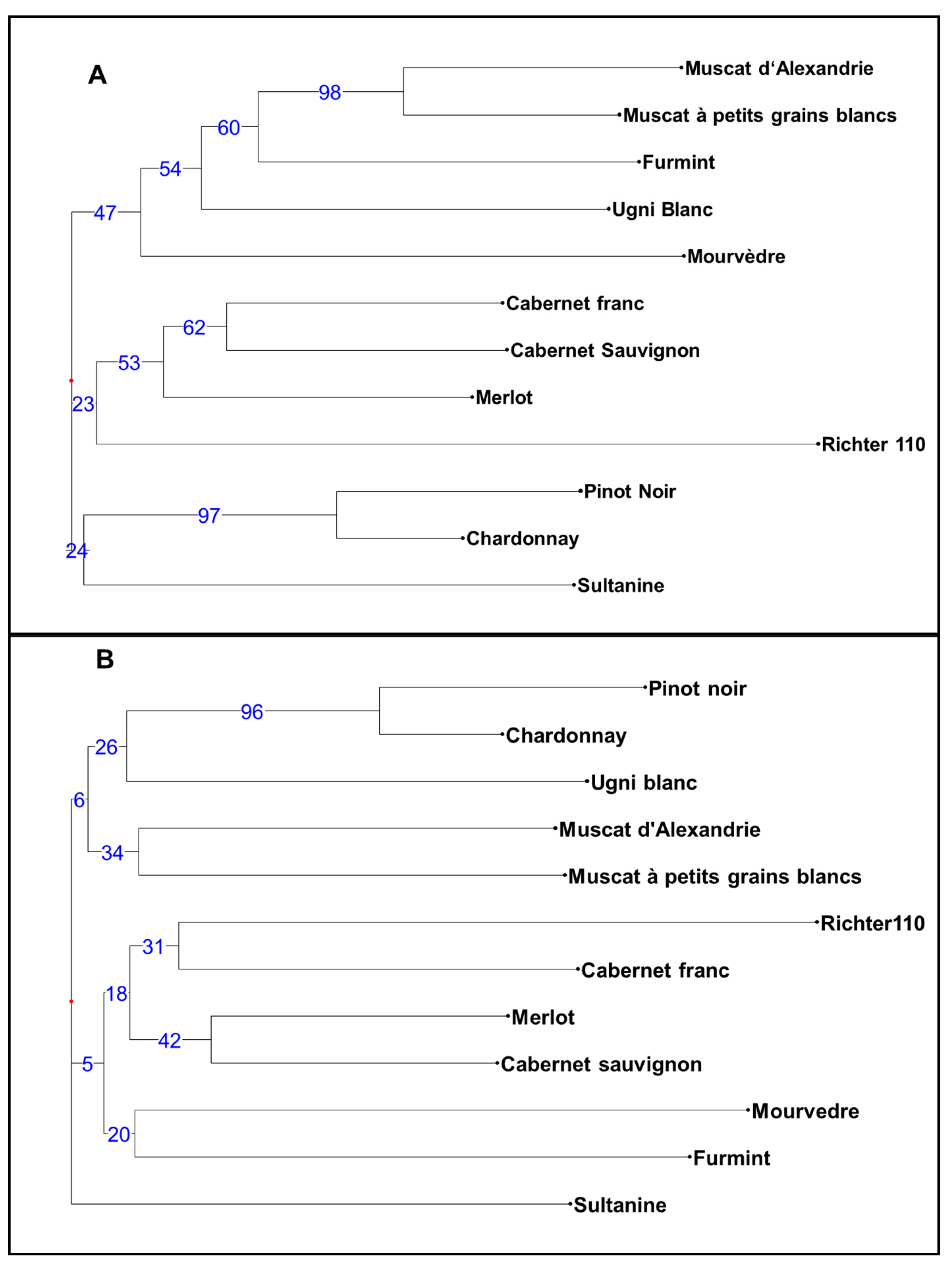
3.2. Comparison of CE and HTS Approaches
3.3. The HTS Approach Creates a Bias in Calling True Alleles for Some Loci
3.4. Analyses of 96 V. vinifera Samples
3.5. HTS Genotyping Economy
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Southern, E.M. Detection of specific sequences among DNA fragments separated by gel electrophoresis. JMB 1975, 98, 503–517. [Google Scholar] [CrossRef]
- Saiki, R.K.; Scharf, S.; Faloona, F.; Mullis, K.B.; Horn, G.T.; Erlich, H.A.; Arnheim, N. Enzymatic amplification of beta-globin genomic sequences and restriction site analysis for diagnosis of sickle cell anemia. Science 1985, 230, 1350–1354. [Google Scholar] [CrossRef] [PubMed]
- Williams, J.G.; Kubelik, A.R.; Livak, K.J.; Rafalski, J.A.; Tingey, S.V. DNA polymorphisms amplified by arbitrary primers are useful as genetic markers. Nucleic Acids Res. 1990, 18, 6531–6535. [Google Scholar] [CrossRef] [PubMed]
- Semagn, K.; Babu, R.; Hearne, S.; Olsen, M. Single nucleotide polymorphism genotyping using Kompetitive Allele Specific PCR (KASP): Overview of the technology and its application in crop improvement. Mol. Breed. 2014, 33, 1–14. [Google Scholar] [CrossRef]
- Ganal, M.W.; Polley, A.; Graner, E.M.; Plieske, J.; Wieseke, R.; Luerssen, H.; Durstewitz, G. Large SNP arrays for genotyping in crop plants. J. Biosci. 2012, 37, 821–828. [Google Scholar] [CrossRef] [PubMed]
- He, J.; Zhao, X.; Laroche, A.; Lu, Z.X.; Liu, H.; Li, Z. Genotyping-by-sequencing (GBS), an ultimate marker-assisted selection (MAS) tool to accelerate plant breeding. Front. Plant Sci. 2014, 5, 484. [Google Scholar] [CrossRef]
- Deokar, A.A.; Ramsay, L.; Sharpe, A.G.; Diapari, M.; Sindhu, A.; Bett, K.; Warkentin, T.D.; Tar’an, B. Genome wide SNP identification in chickpea for use in development of a high density genetic map and improvement of chickpea reference genome assembly. BMC Genom. 2014, 15, 708. [Google Scholar] [CrossRef]
- Su, Q.; Zhang, X.; Zhang, W.; Zhang, N.; Song, L.; Liu, L.; Xue, X.; Liu, G.; Liu, J.; Meng, D.; et al. QTL detection for kernel size and weight in bread wheat (Triticum aestivum L.) using a high-density SNP and SSR-based linkage map. Front. Plant Sci. 2018, 9, 1484. [Google Scholar] [CrossRef]
- Cipriani, G.; Spadotto, A.; Jurman, I.; Di Gaspero, G.; Crespan, M.; Meneghetti, S.; Frare, E.; Vignani, R.; Cresti, M.; Morgante, M.; et al. The SSR-based molecular profile of 1005 grapevine (Vitis vinifera L.) accessions uncovers new synonymy and parentages, and reveals a large admixture amongst varieties of different geographic origin. Theor. App. Genet. 2010, 121, 1569–1585. [Google Scholar] [CrossRef]
- Morgante, M.; Olivieri, A.M. PCR-amplified microsatellites as markers in plant genetics. Plant J. 1993, 3, 175–182. [Google Scholar] [CrossRef]
- Stępień, Ł.; Mohler, V.; Bocianowski, J.; Koczyk, G. Assessing genetic diversity of Polish wheat (Triticum aestivum) varieties using microsatellite markers. Genet. Resour. Crop Evol. 2007, 54, 1499–1506. [Google Scholar] [CrossRef]
- Jacob, H.J.; Lindpaintner, K.; Lincoln, S.E.; Kusumi, K.; Bunker, R.K.; Mao, Y.P.; Ganten, D.; Dzau, V.J.; Lander, E.S. Genetic mapping of a gene causing hypertension in the stroke-prone spontaneously hypertensive rat. Cell 1991, 67, 213–224. [Google Scholar] [CrossRef]
- Edwards, A.; Civitello, A.; Hammond, H.A.; Caskey, C.T. DNA typing and genetic mapping with trimeric and tetrameric tandem repeats. Am. J. Hum. Genet. 1991, 49, 746. [Google Scholar] [PubMed]
- Parida, S.K.; Dalal, V.; Singh, A.K.; Singh, N.K.; Mohapatra, T. Genic non-coding microsatellites in the rice genome: Characterization, marker design and use in assessing genetic and evolutionary relationships among domesticated groups. BMC Genom. 2009, 10, 140. [Google Scholar] [CrossRef] [PubMed]
- Powell, W.; Machray, G.C.; Provan, J. Polymorphism revealed by simple sequence repeats. Trends Plant Sci. 1996, 1, 215–222. [Google Scholar] [CrossRef]
- Richard, G.F.; Hennequin, C.; Thierry, A.; Dujon, B. Trinucleotide repeats and other microsatellites in yeasts. Res. Microbiol. 1999, 150, 589–602. [Google Scholar] [CrossRef]
- Richard, G.F.; Pâques, F. Mini- and microsatellite expansions: The recombination connection. EMBO Rep. 2000, 1, 122–126. [Google Scholar] [CrossRef]
- Van Dijk, E.L.; Auger, H.; Jaszczyszyn, Y.; Thermes, C. Ten years of next-generation sequencing technology. Trends Genet. 2014, 30, 418–426. [Google Scholar] [CrossRef]
- Bornman, D.M.; Hester, M.E.; Schuetter, J.M.; Kasoji, M.D.; Minard-Smith, A.; Barden, C.A.; Nelson, S.C.; Godbold, G.D.; Baker, C.H.; Yang, B.; et al. Short-read, high-throughput sequencing technology for STR genotyping. BioTech. Rapid Dispatches 2012, 2012, 1–6. [Google Scholar] [CrossRef]
- Fordyce, S.L.; Ávila-Arcos, M.C.; Rockenbauer, E.; Børsting, C.; Frank-Hansen, R.; Petersen, F.T.; Willerslev, E.; Hansen, A.J.; Morling, N.; Gilbert, M.T. High-throughput sequencing of core STR loci for forensic genetic investigations using the Roche Genome Sequencer FLX platform. Biotechniques 2011, 51, 127–133. [Google Scholar] [CrossRef]
- Darby, B.J.; Erickson, S.F.; Hervey, S.D.; Ellis-Felege, S.N. Digital fragment analysis of short tandem repeats by high-throughput amplicon sequencing. Ecol. Evol. 2016, 6, 4502–4512. [Google Scholar] [CrossRef] [PubMed]
- Lepais, O.; Chancerel, E.; Boury, C.; Salin, F.; Manicki, A.; Taillebois, L.; Dutech, C.; Aissi, A.; Bacles, C.F.E.; Daverat, F.; et al. Fast sequence-based microsatellite genotyping development workflow. PeerJ 2020, 8, e9085. [Google Scholar] [CrossRef] [PubMed]
- Albarghouthi, M.N.; Buchholz, B.A.; Doherty, E.A.; Bogdan, F.M.; Zhou, H.; Barron, A.E. Impact of polymer hydrophobicity on the properties and performance of DNA sequencing matrices for capillary electrophoresis. Electrophoresis 2001, 22, 737–747. [Google Scholar] [CrossRef]
- Tu, O.; Knott, T.; Marsh, M.; Bechtol, K.; Harris, D.; Barker, D.; Bashkin, J. The influence of fluorescent dye structure on the electrophoretic mobility of end-labeled DNA. Nucleic Acids Res. 1998, 26, 2797–2802. [Google Scholar] [CrossRef] [PubMed]
- Estoup, A.; Jarne, P.; Cornuet, J.M. Homoplasy and mutation model at microsatellite loci and their consequences for population genetic analysis. Mol. Ecol. 2002, 11, 1591–1604. [Google Scholar] [CrossRef]
- Koressaar, T.; Lepamets, M.; Kaplinski, L.; Raime, K.; Andreson, R.; Remm, M. Primer3_masker: Integrating masking of template sequence with primer design software. Bioinformatics 2018, 34, 1937–1938. [Google Scholar] [CrossRef] [PubMed]
- Laucou, V.; Lacombe, T.; Dechesne, F.; Siret, R.; Bruno, J.P.; Dessup, M.; Dessup, T.; Ortigosa, P.; Parra, P.; Roux, C.; et al. High throughput analysis of grape genetic diversity as a tool for germplasm collection management. Theor. Appl. Genet. 2011, 122, 1233–1245. [Google Scholar] [CrossRef]
- Kump, B.; Javornik, B. Evaluation of genetic variability among common buckwheat (Fagopyrum esculentum Moench) populations by RAPD markers. Plant Sci. 1996, 114, 149–158. [Google Scholar] [CrossRef]
- Gohl, D.M.; MacLean, A.; Hauge, A.; Becker, A.; Walek, D.; Beckman, K.B. An optimized protocol for high-throughput amplicon-based microbiome profiling. Protoc. Exch. 2016. [Google Scholar] [CrossRef]
- Vartia, S.; Villanueva-Cañas, J.L.; Finarelli, J.; Farrell, E.D.; Collins, P.C.; Graham, H.M.; Carlsson, J.E.L.; Gauthier, D.T.; McGinnity, P.; Cross, T.F.; et al. A novel method of microsatellite genotyping by sequencing using individual combinatorial barcoding. R. Soc. Open Sci. 2016, 3, 150565. [Google Scholar] [CrossRef]
- Sefc, K.M.; Regner, F.; Turetschek, E.; Glossl, J.; Steinkellner, H. Identification of microsatellite sequences in Vitis riparia and their applicability for genotyping of different Vitis species. Genome 1999, 42, 367–373. [Google Scholar] [CrossRef] [PubMed]
- Merdinoglu, D.; Butterlin, G.; Bevilacqua, L.; Chiquet, V.; Adam-Blondon, A.F.; Decroocq, S. Development of a large set of microsatellite markers in grapevine (Vitis vinifera L.) suitable for multiplex PCR. Mol. Breed. 2005, 15, 349–366. [Google Scholar] [CrossRef]
- Browers, J.E.; Dangl, G.S.; Vignani, R.; Meredith, C.P. Isolation and characterization of new polymorphic simple sequence repeat loci in grape (Vitis vinifera L.). Genome 1996, 39, 628–633. [Google Scholar] [CrossRef] [PubMed]
- Aronesty, E. Comparison of sequencing utility programs. Open Bioinforma. J. 2013, 7, 1–8. [Google Scholar] [CrossRef]
- Martin, M. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet. J. 2011, 17. [Google Scholar] [CrossRef]
- Beier, S.; Thiel, T.; Munch, T.; Scholz, U.; Mascher, M. MISA-web: A web server for microsatellite prediction. Bioinformatics 2017, 33, 2583–2585. [Google Scholar] [CrossRef] [PubMed]
- Rice, P.; Longden, I.; Bleassby, A. EMBOSS: The European molecular biology open software suite. Trends Genet. 2000, 508, 276–277. [Google Scholar] [CrossRef]
- Kedzierska, K.Z.; Gerber, L.; Cagnazzi, D.; Krutzen, M.; Ratan, A.; Kistler, L. SONiCS: PCR stutter noise correction in genome-scale microsatellites. Bioinformatics 2018, 34, 4115–4117. [Google Scholar] [CrossRef]
- Gymrek, M.; Golan, D.; Rosset, S.; Erlich, Y. lobSTR: A short tandem repeat profiler for personal genomes. Genome Res. 2012, 22, 1154–1162. [Google Scholar] [CrossRef]
- Highnam, G.; Franck, C.; Martin, A.; Stephens, C.; Puthige, A.; Mittelman, D. Accurate human microsatellite genotypes from high-throughput resequencing data using informed error profiles. Nucleic Acids Res. 2012, 41, e32. [Google Scholar] [CrossRef]
- Cao, M.D.; Tasker, E.; Willadsen, K.; Imelfort, M.; Vishwanathan, S.; Sureshkumar, S.; Balasubramanian, S.; Bodén, M. Inferring short tandem repeat variation from paired-end short reads. Nucleic Acids Res. 2013, 42, e16. [Google Scholar] [CrossRef] [PubMed]
- Fungtammasan, A.; Ananda, G.; Hile, S.E.; Su, M.S.-W.; Sun, C.; Harris, R.; Medvedev, P.; Eckert, K.; Makova, K.D. Accurate typing of short tandem repeats from genome-wide sequencing data and its applications. Genome Res. 2015, 25, 736–749. [Google Scholar] [CrossRef] [PubMed]
- Cantarella, C.; D’Agostino, N. PSR: Polymorphic SSR retrieval. BMC Res. Notes 2015, 8, 525. [Google Scholar] [CrossRef] [PubMed]
- Buckler, E.S.; Ilut, D.C.; Wang, X.; Kretzschmar, T.; Gore, M.A.; Mitchell, S.E. rAmpSeq: Using repetitive sequences for robust genotyping. BioRxiv 2016. [Google Scholar] [CrossRef]
- Tang, H.; Nzabarushimana, E. STRScan: Targeted profiling of short tandem repeats in whole-genome sequencing data. BMC Bioinforma. 2017, 18, 398. [Google Scholar] [CrossRef]
- Riaz, S.; Garrison, K.E.; Dangl, G.S.; Boursiqot, J.-M.; Meredith, C.P. Genetic divergance and chimerism within ancient asexually propagated winegrape cultivars. J. Am. Soc. Hortic. Sci. 2002, 127, 508–514. [Google Scholar] [CrossRef]
- Crespan, M. Evidence on the evolution of polymorphism of microsatellite markers in varieties of Vitis vinifera L. Theor. Appl. Genet. 2004, 108, 231–237. [Google Scholar] [CrossRef]
- Ibanez, J.; De Andres, M.T.; Borrego, J. Allelic variation observed at one microsatellite locus between the two synonym grape cultivars Black Currant and Mavri Corinthiaki. Vitis 2000, 39, 173–174. [Google Scholar]
- Hocquigny, S.; Pelsy, F.; Dumas, V.; Kindt, S.; Heloir, M.C.; Merdinoglu, D. Diversification within grapevine cultivars goes through chimeric states. Genome 2004, 47, 579–589. [Google Scholar] [CrossRef]
- Štajner, N.; Rusjan, D.; Korosec-Koruza, Z.; Javornik, B. Genetic Characterization of Old Slovenian Grapevine Varieties of Vitis vinifera L. by Microsatellite Genotyping. Am. J. Enol. Viticult. 2011, 62, 250–255. [Google Scholar] [CrossRef]
- Koncilja, K. Intravarietal Variability analysis of Grapevine Variety ‘Merlot’ (Vitis vinifera L.) with Microsatelites Markers. Master’s Thesis, University of Ljubljana, Ljubljana, Slovenia, 2010. [Google Scholar]
- Vélez, M.D.; Ibáñez, J. Assessment of the uniformity and stability of grapevine cultivars using a set of microsatellite markers. Euphytica 2012, 186, 419–432. [Google Scholar] [CrossRef]
- Thompson, M.M.; Olmo, H.P. Cytohistological studies of cytochimeric and tetraploid grapes. Am. J. Bot. 1963, 50, 901–906. Available online: https://www.jstor.org/stable/2439777 (accessed on 9 August 2020). [CrossRef]
- Deschamps, S.; Llaca, V.; May, G.D. Genotyping-by-sequencing in plants. Biology 2012, 1, 460–483. [Google Scholar] [CrossRef] [PubMed]
- Mertes, F.; Elsharawy, A.; Sauer, S.; van Helvoort, J.M.; van der Zaag, P.J.; Franke, A.; Nilsson, M.; Lehrach, H.; Brookes, A.J. Targeted enrichment of genomic DNA regions for next-generation sequencing. Brief. Funct. Genom. 2011, 10, 374–386. [Google Scholar] [CrossRef] [PubMed]
- Zhou, L.; Holliday, J.A. Targeted enrichment of the black cottonwood (Populus trichocarpa) gene space using sequence capture. BMC Genom. 2012, 13, 703. [Google Scholar] [CrossRef] [PubMed]
- Hill, C.B.; Wong, D.; Tibbits, J.; Forrest, K.; Hayden, M.; Zhang, X.Q.; Westcott, S.; Angessa, T.T.; Li, C. Targeted enrichment by solution-based hybrid capture to identify genetic sequence variants in barley. Sci. Data 2019, 6, 12. [Google Scholar] [CrossRef] [PubMed]
- Lachagari, V.; Gupta, R.; Lekkala, S.P.; Mahadevan, L.; Kuriakose, B.; Chakravartty, N.; Katta, A.M.; Santhosh, S.; Reddy, A.R.; Thomas, G. Whole genome sequencing and comparative genomic analysis reveal allelic variations unique to a purple colored rice landrace (Oryza sativa ssp. indica cv. Purpleputtu). Front. Plant Sci. 2019, 10, 513. [Google Scholar] [CrossRef]
- Šarhanová, P.; Pfanzelt, S.; Brandt, R.; Himmelbach, A.; Blattner, F.R. SSE-R-seq: Genotyping of microsatellites using next-generation sequencing reveals higher level of polymorphism as compared to traditional fragment sizes coring. Ecol. Evol. 2018, 8, 10817–10833. [Google Scholar] [CrossRef]
- Curto, M.; Winter, S.; Seiter, A.; Schmid, L.; Scheicher, K.; Barthel, L.M.F.; Plass, J.; Meimberg, H. Application of a SSR-GBS marker system on investigation of European Hedgehog species and their hybrid zone dynamics. Ecol. Evol. 2019, 9, 2814–2832. [Google Scholar] [CrossRef]
- Farrell, E.D.; Carlsson, J.E.L.; Carlsson, J. Next Gen Pop Gen: Implementing a high-throughput approach to population genetics in boarfish (Capros aper). R. Soc. Open Sci. 2016, 3, 160651. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Barcode | Cultivar | Country of Origin | Barcode | Cultivar | Country of Origin |
|---|---|---|---|---|---|
| F1-R1 | Kratošija | North Macedonia | F7-R1 | GnetKras | Slovenia |
| F1-R2 | Neznana Bela | Slovenia | F7-R2 | Kratošija I | Montenegro |
| F1-R3 | Rebula | Slovenia | F7-R3 | Muštoš Feher | Serbia |
| F1-R4 | Sremska Zelenika | Serbia | F7-R4 | Rebula Portalis | Slovenia |
| F1-R5 | Zimsko Belo | Serbia | F7-R5 | Smederevka | Bosnia and Herzegovina |
| F1-R6 | Manastirsko Belo | North Macedonia | F7-R6 | Vranac | Bosnia and Herzegovina |
| F1-R7 | Dobrogostina | Bosnia and Herzegovina | F7-R7 | Belovina | North Macedonia |
| F1-R8 | Godominka | Serbia | F7-R8 | Gnjet | Slovenia |
| F2-R1 | Bagrina | Serbia | F8-R1 | Kreaca | Serbia |
| F2-R2 | DrenakCrni | Serbia | F8-R2 | Refosco | Slovenia |
| F2-R3 | Kadarka Bela | Serbia | F8-R3 | Stanušina | North Macedonia |
| F2-R4 | Krkošija Šupljica | Serbia | F8-R4 | Žametovka | Bosnia and Herzegovina |
| F2-R5 | Prokupac | Bosnia and Herzegovina | F8-R5 | Ohridsko Belo | North Macedonia |
| F2-R6 | Ružica | Serbia | F8-R6 | Refošk | Slovenia |
| F2-R7 | Bela Zgodnja | Slovenia | F8-R7 | DolgiGrozdi | Slovenia |
| F2-R8 | Chardonnay | Slovenia | F8-R8 | Gročanka | Serbia |
| F3-R1 | Drenak | Bosnia and Herzegovina | F9-R1 | PlovdinaCrna | Serbia |
| F3-R2 | Kadarka | Serbia | F9-R2 | Rezaklija | Bosnia and Herzegovina |
| F3-R3 | Kujundžuša | Bosnia and Herzegovina | F9-R3 | Stari Rizling VI | Montenegro |
| F3-R4 | Prokupac | Serbia | F9-R4 | Žlozder | Bosnia and Herzegovina |
| F3-R5 | Ružica V | Montenegro | F9-R5 | Debinë e Zezë | Albania |
| F3-R6 | TamjanikaCrna | Serbia | F9-R6 | Kallmet | Albania |
| F3-R7 | Merlot | Slovenia | F9-R7 | Potek e Zezë | Albania |
| F3-R8 | Beli Medenac | Serbia | F9-R8 | Shesh i Zi | Albania |
| F4-R1 | Ružica VI | Montenegro | F10-R1 | Stambolleshë | Albania |
| F4-R2 | TrbljanBeli | Serbia | F10-R2 | Sheshi Bardhë | Albania |
| F4-R3 | Pinot noir | Slovenia | F10-R3 | Kosinjot | Albania |
| F4-R4 | Bena | Bosnia and Herzegovina | F10-R4 | Vlosh | Albania |
| F4-R5 | Elezovka | Bosnia and Herzegovina | F10-R5 | Tajgë e Zezë | Albania |
| F4-R6 | Kavčina | Serbia | F10-R6 | Meresnik | Albania |
| F4-R7 | Marburger | Slovenia | F10-R7 | Korith i Bardhë | Albania |
| F4-R8 | Prošip | Bosnia and Herzegovina | F10-R8 | Tajgë e Bardhë | Albania |
| F5-R1 | Sipa | Slovenia | F11-R1 | Pulëz | Albania |
| F5-R2 | Trnjak | Bosnia and Herzegovina | F11-R2 | Razaki e Kuqe | Albania |
| F5-R3 | Cabernet Sauvignon | Slovenia | F11-R3 | Serinë e Bardhë | Albania |
| F5-R4 | BlatinaI | Bosnia and Herzegovina | F11-R4 | Debinë e Bardhë | Albania |
| F5-R5 | Furmint | Serbia | F11-R5 | Furmint | France |
| F5-R6 | Menigovka | Bosnia and Herzegovina | F11-R6 | Chardonnay | France |
| F5-R7 | Radovača VII | Montenegro | F11-R7 | Pinot Noir | France |
| F5-R8 | Sipon | Slovenia | F11-R8 | Mourvedre | France |
| F6-R1 | Sultanine | Slovenia | F12-R1 | Ugni B/Trebbianotoscano | France |
| F6-R2 | CrnValandovskiDrenok | North Macedonia | F12-R2 | Muscat a petit grains | France |
| F6-R3 | Gavran | Serbia | F12-R3 | Muscat d’Alexandrie | France |
| F6-R4 | Končanka | North Macedonia | F12-R4 | Merlot | France |
| F6-R5 | Muskat Ruža | Serbia | F12-R5 | Cabernet Sauvignon | France |
| F6-R6 | Slankamenka Crvena | Serbia | F12-R6 | Cabernet franc | France |
| F6-R7 | Touriga Nacional | Slovenia | F12-R7 | Sultanine | France |
| F6-R8 | Čauš Bel | North Macedonia | F12-R8 | Richter110 | France |
| Locus | Reference Sequence | Microsatellite Core Repeat | Reference Length |
|---|---|---|---|
| VMC1b11-NGS | GACCTAAGTTTCTGAGGCTTTGAAAATTACCTTCCGGGTTTCTAGAGAGGGAGAGAGAGAGAGAGAGAGAGAGAGAGGAAGGTTCGGCAACACAAAATGAGAGGCA | (GA)n | 106 |
| VrZAG79-NGS | TTAGCCGAAGCCATCTCTGTTCTCAAGCAGAATGGAAGTGAGAGAGAGAGAGAGAGARGAGAGAGAGAGAGATAAAGGTGGTGAGGTGCTTGTGTTTCTTGA | (CT)n | 102 |
| VVIb01-NGS | CCTGTGAAACCACCACTATCCTCAGAGAAGCTCTCTCTCTCTCTCTCTCTCTTCACACTCACATCACTCGTTTACCTTGTGCAACCA | (CT)n | 87 |
| VVIn73-NGS | AGGCTTCAAAGCCCTCTCATCTTAATTCGTGTGTGTGTGTGTGTGTGTTGGGGCCTTTGGGGCTCCACTGACACCCACAAGGGTGT | (CA)n | 86 |
| VVIp31-NGS | TTGGGAAACCACAGAAGTGACAATTTATAGAGAGAGAGAGAGAGAGAGAGAGAGAGAGAGGCATATCCATTAGAATGATCACATTCCAGGAACAACCCATT | (GA)n | 101 |
| VVIq52-NGS | CAGGAAAGTGTTCAATGGTTACAAAACAGGAGAGAGAGAGAGAGAGAGTGTGTCACTGGTTCTGTCATCTACCATCCTT | (CT)n | 79 |
| VVIv37-NGS | ACCAGTATTAAGAACGCAGTCACTGCCCACAGAGAGAGAGAGAGAGAGAGAGAGAGAGAGAGAGAGAGAGAGATGGGGTGAGTGGGAAGTTAAGAGTAGGG | (TC)n(GT)n | 101 |
| VVMD24-NGS | AGAAGACTTGTCTCTCTCAATCAAATTGTGGTCCTCCTCTCTCTCTCTCTCTCTCTCTCTCTACTACTGCATATCATTGATAGTCCTTGTCTCAATTTCTTTGCG | (CT)n | 105 |
| VVMD25-NGS | TGAAAAGTGTAGTGACCCTTTGACTAGGCCTCCCTTCTCTCTCTCTCTCTCTCTCATGTTTATGTTATTTATTGTTTTTTTCCTTGAAACCACAAGACAAGCCTCCA | (CT)n | 107 |
| VVMD27-NGS | CCTCTCTCTCCGGCGGTATTCTCAATCTCCCTCCTCCTTCCGCCCAAGTTGAGGTCTCTCTCTCTCTCTCTCTCTCTCTATTTATATACTTACGGATGTATTCAGATCTGGT | (CT)n | 112 |
| VVMD32-NGS | TGAAACGTCTCGCCATTACCCCTCCCTCTCTCTCTCTCTCTCTCTCTCTCTCTCTCTCTCTCTCTCTCTCTCTCTCTCAAGCCAGGCGTCAAAACATGAACTGTTTGTC | (CT)n | 109 |
| VVMD7-NGS | CCTCAAGCAGCGTATCCATAGCGAGTGGAGGAGAGAGAGAGAGAGAGAGAGAGAGAGAGAGTGAGCGCCAAAGAGAGAGGGAGGAGGG | (CT)n | 88 |
| SSR Name | Linkage | Locus Specific Forward Primer with Universal Tail | Locus Specific Reverse Primer with Universal Tail | Reference |
|---|---|---|---|---|
| VMC1b11-NGS | 8 | AATTAACCCTGACCTAAGTTTCTGAGGCTTTGA | CAGTCGGGCGTGCCTCTCATTTTGTGTTGC | BV681754 |
| VrZAG79-NGS | 5 | AATTAACCCTTTAGCCGAAGCCATCTCTGT | CAGTCGGGCGTCAAGAAACACAAGCACCTCA | [31] |
| VVIb01-NGS | 2 | AATTAACCCTCCTGTGAAACCACCACTATCC | CAGTCGGGCGTGGTTGCACAAGGTAAACGA | [32] |
| VVIn73-NGS | 17 | AATTAACCCTAGGCTTCAAAGCCCTCTCAT | CAGTCGGGCGACACCCTTGTGGGTGTCAGT | [32] |
| VVIp31-NGS | 19 | AATTAACCCTTTGGGAAACCACAGAAGTGA | CAGTCGGGCGAATGGGTTGTTCCTGGAATG | [32] |
| VVIq52-NGS | 9 | AATTAACCCTCAGGAAAGTGTTCAATGGTTAC | CAGTCGGGCGAAGGATGGTAGATGACAGAACCA | [32] |
| VVIv37-NGS | 10 | AATTAACCCTACCAGTATTAAGAACGCAGTCAC | CAGTCGGGCGCCCTACTCTTAACTTCCCACTCA | [32] |
| VVMD24-NGS | 14 | AATTAACCCTAGAAGACTTGTCTCTCTCAATCAAA | CAGTCGGGCGCGCAAAGAAATTGAGACAAGG | [33] |
| VVMD25-NGS | 11 | AATTAACCCTTGAAAAGTGTAGTGACCCTTTGA | CAGTCGGGCGTGGAGGCTTGTCTTGTGGTT | [33] |
| VVMD27-NGS | 5 | AATTAACCCTCCTCTCTCTCCGGCGGTA | CAGTCGGGCGACCAGATCTGAATACATCCGTAA | [33] |
| VVMD32-NGS | 4 | AATTAACCCTTGAAACGTCTCGCCATTACC | CAGTCGGGCGGACAAACAGTTCATGTTTTGACG | [33] |
| VVMD7-NGS | 7 | AATTAACCCTCCTCAAGCAGCGTATCCATAG | CAGTCGGGCGCCCTCCTCCCTCTCTCTTTG | [33] |
| Locus | Reference Allele Length | Mapped Reads 1 | Amount of Data [bp] after Mapping 2 | Average Coverage after Mapping | No. of Sequences after Filtering 3 | Amount of Data (bp) after Demultiplexing | Average Coverage after Filtering |
|---|---|---|---|---|---|---|---|
| VMC1b11 | 106 | 3,649,804 | 551,120,404 | 2,888,215 | 2,927,098 | 305,691,052 | 2,883,878 |
| VrZAG79 | 102 | 1,192,832 | 180,117,632 | 1,148,842 | 1,694,202 | 156,444,090 | 1,533,766 |
| VVIb01 | 87 | 2,713,933 | 409,803,883 | 2,712,447 | 2,444,143 | 223,916,517 | 2,573,753 |
| VVIn73 | 86 | 1,085,003 | 163,835,453 | 1,084,374 | 957,723 | 81,434,445 | 946,912 |
| VVIp31 | 101 | 1,963,722 | 296,522,022 | 1,819,017 | 1,447,397 | 149,040,267 | 1,475,646 |
| VVIq52 | 79 | 792,078 | 119,603,778 | 791,626 | 704,921 | 56,007,493 | 708,956 |
| VVIv37 | 101 | 1,119,484 | 169,042,084 | 1,022,452 | 1,264,019 | 115,785,796 | 1,146,394 |
| VVMD24 | 105 | 1,813,906 | 273,899,806 | 1,810,404 | 1,588,685 | 162,085,558 | 1,543,672 |
| VVMD25 | 107 | 2,348,776 | 354,665,176 | 2,192,870 | 2,000,291 | 221,586,577 | 2,070,903 |
| VVMD27 | 112 | 2,911,241 | 439,597,391 | 2,895,057 | 2,468,380 | 277,195,183 | 2,474,957 |
| VVMD32 | 109 | 1,556,828 | 235,081,028 | 1,163,432 | 1,037,861 | 92,983,922 | 853,064 |
| VVMD7 | 88 | 949.509 | 143,375,859 | 937,902 | 899,408 | 75,335,819 | 856,089 |
| Total | 22,097,116 | 3,336,664,516 | 19,434,128 | 1.917,506,719 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kunej, U.; Dervishi, A.; Laucou, V.; Jakše, J.; Štajner, N. The Potential of HTS Approaches for Accurate Genotyping in Grapevine (Vitis vinifera L.). Genes 2020, 11, 917. https://doi.org/10.3390/genes11080917
Kunej U, Dervishi A, Laucou V, Jakše J, Štajner N. The Potential of HTS Approaches for Accurate Genotyping in Grapevine (Vitis vinifera L.). Genes. 2020; 11(8):917. https://doi.org/10.3390/genes11080917
Chicago/Turabian StyleKunej, Urban, Aida Dervishi, Valérie Laucou, Jernej Jakše, and Nataša Štajner. 2020. "The Potential of HTS Approaches for Accurate Genotyping in Grapevine (Vitis vinifera L.)" Genes 11, no. 8: 917. https://doi.org/10.3390/genes11080917
APA StyleKunej, U., Dervishi, A., Laucou, V., Jakše, J., & Štajner, N. (2020). The Potential of HTS Approaches for Accurate Genotyping in Grapevine (Vitis vinifera L.). Genes, 11(8), 917. https://doi.org/10.3390/genes11080917