The Characterization of chIFITMs in Avian Coronavirus Infection In Vivo, Ex Vivo and In Vitro


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Ethics Statement
2.2. Viruses and Cells
2.3. Virus Infection In Vivo
2.4. Virus Infection In Vitro
2.5. Virus Infection in Ex Vivo Tracheal Organ Cultures (TOCs)
2.6. Total RNA Extraction and Two-Step Reverse Transcription
2.7. Quantification of Virus Subgenomic RNA (sgRNA) in Infected Tissues, Cells and TOCs
2.8. IFITM Gene Expression Analysis
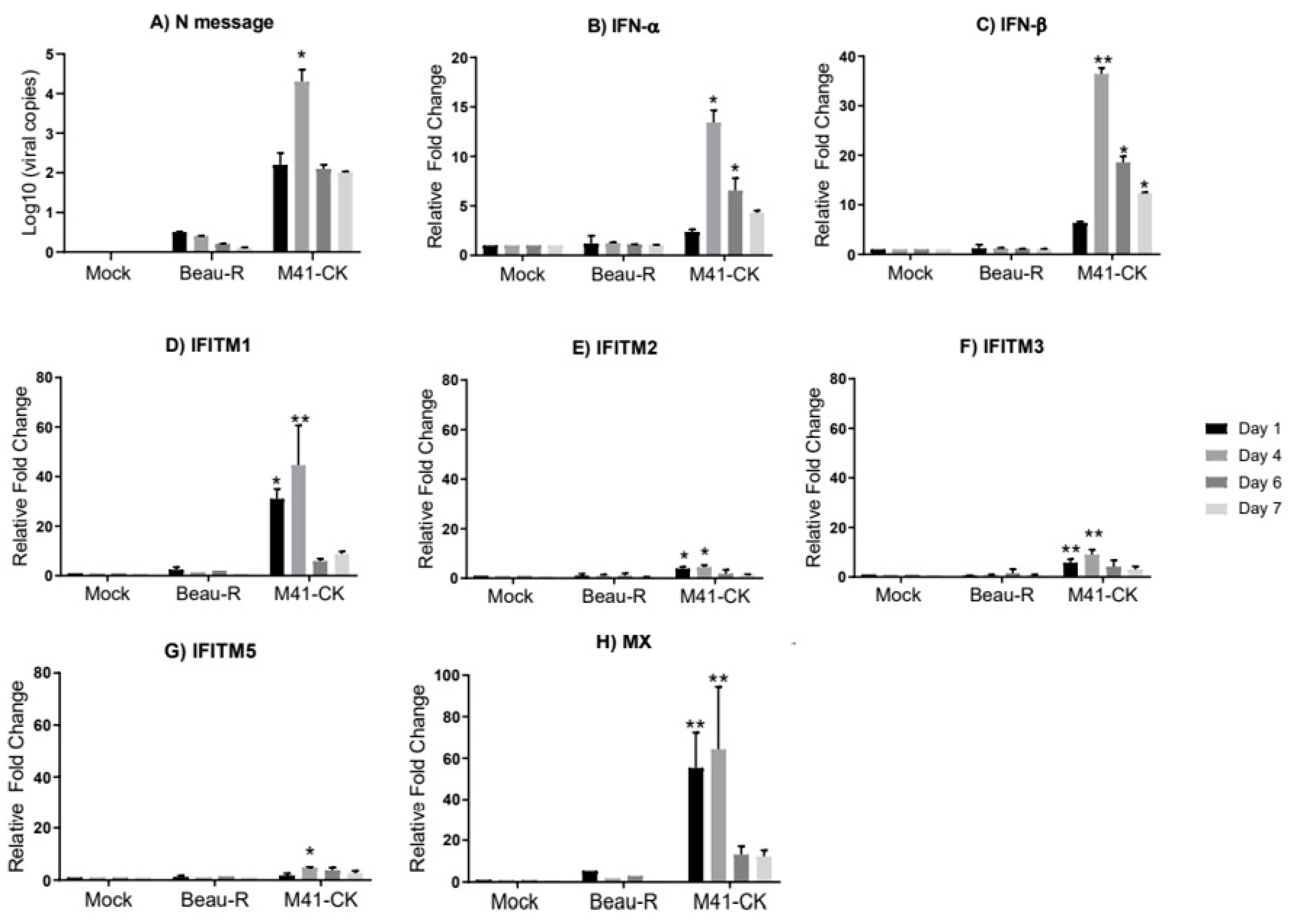
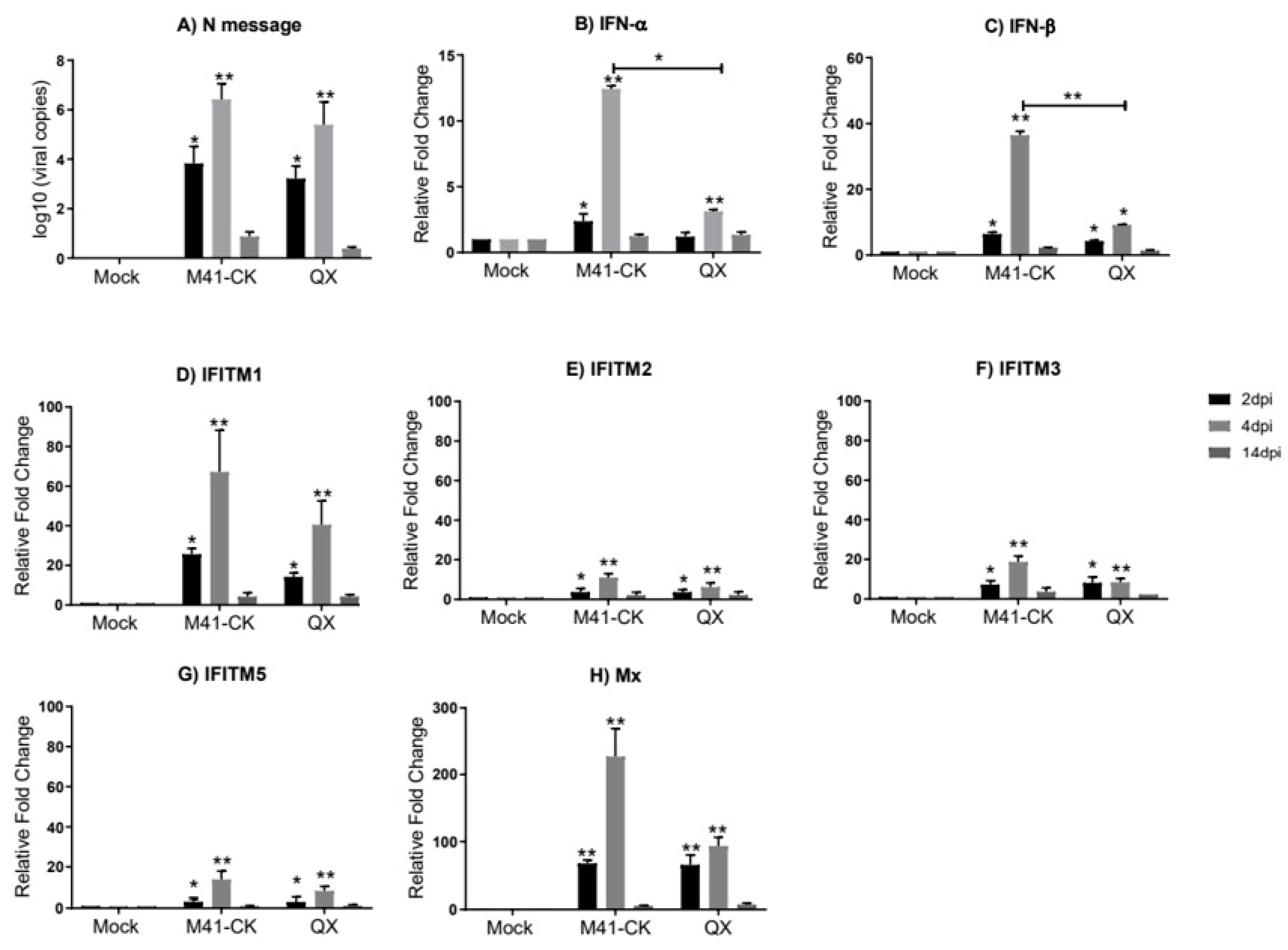
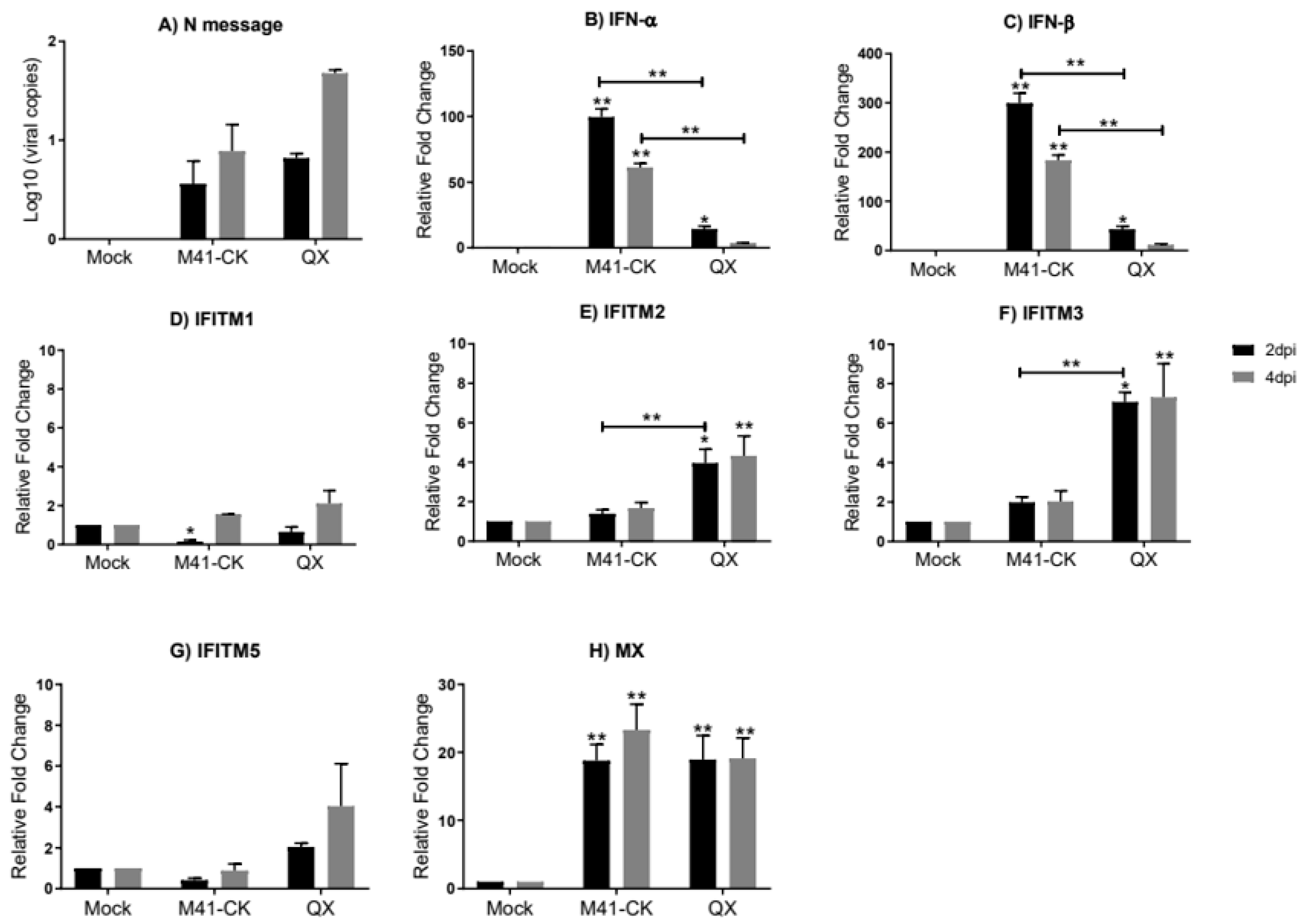
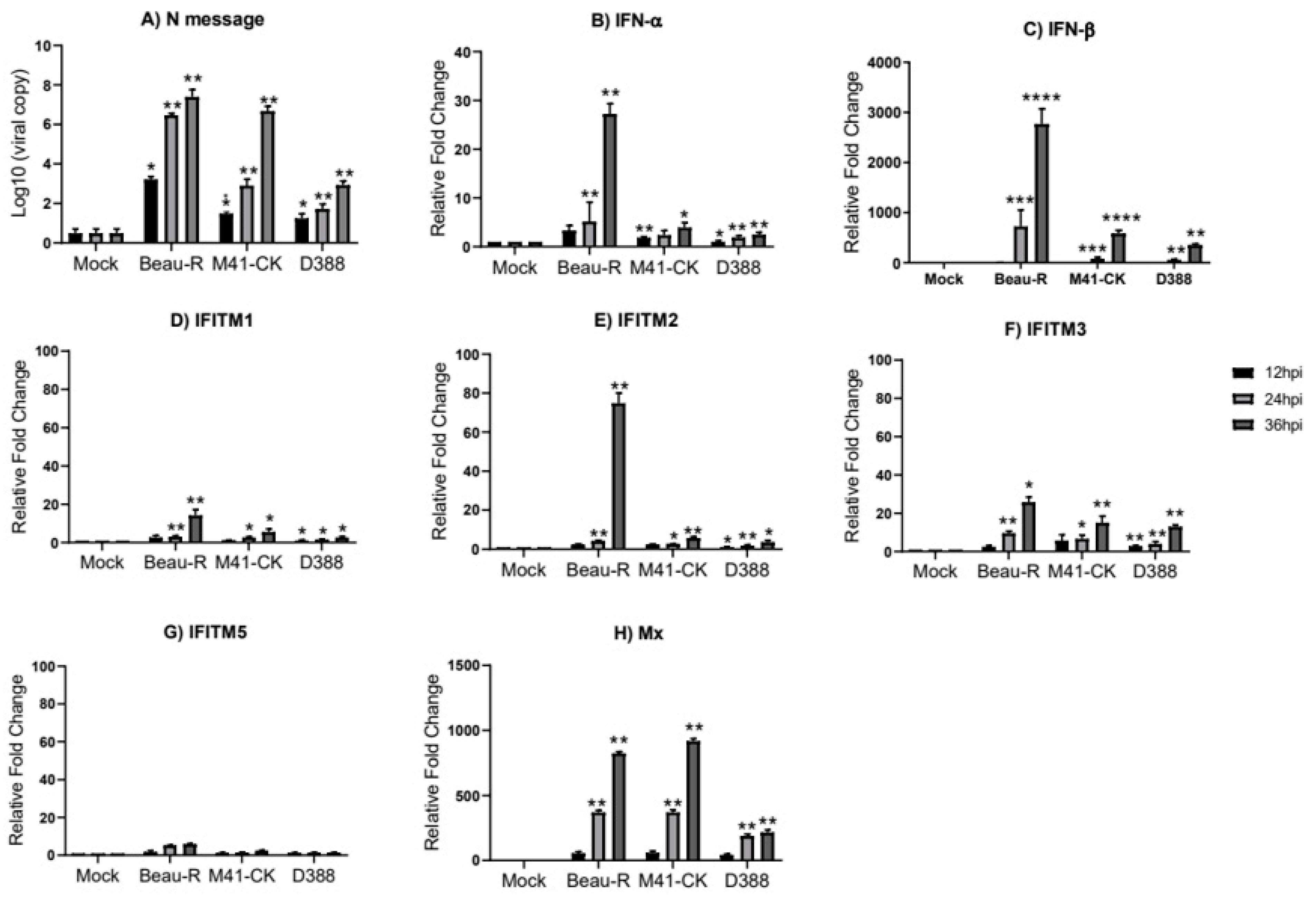
3. Results
3.1. Clinical Observation
3.2. Transcriptional Profile of chIFITMs In Vivo
3.3. Transcriptional Profiles of chIFITMs in Ex Vivo TOCs
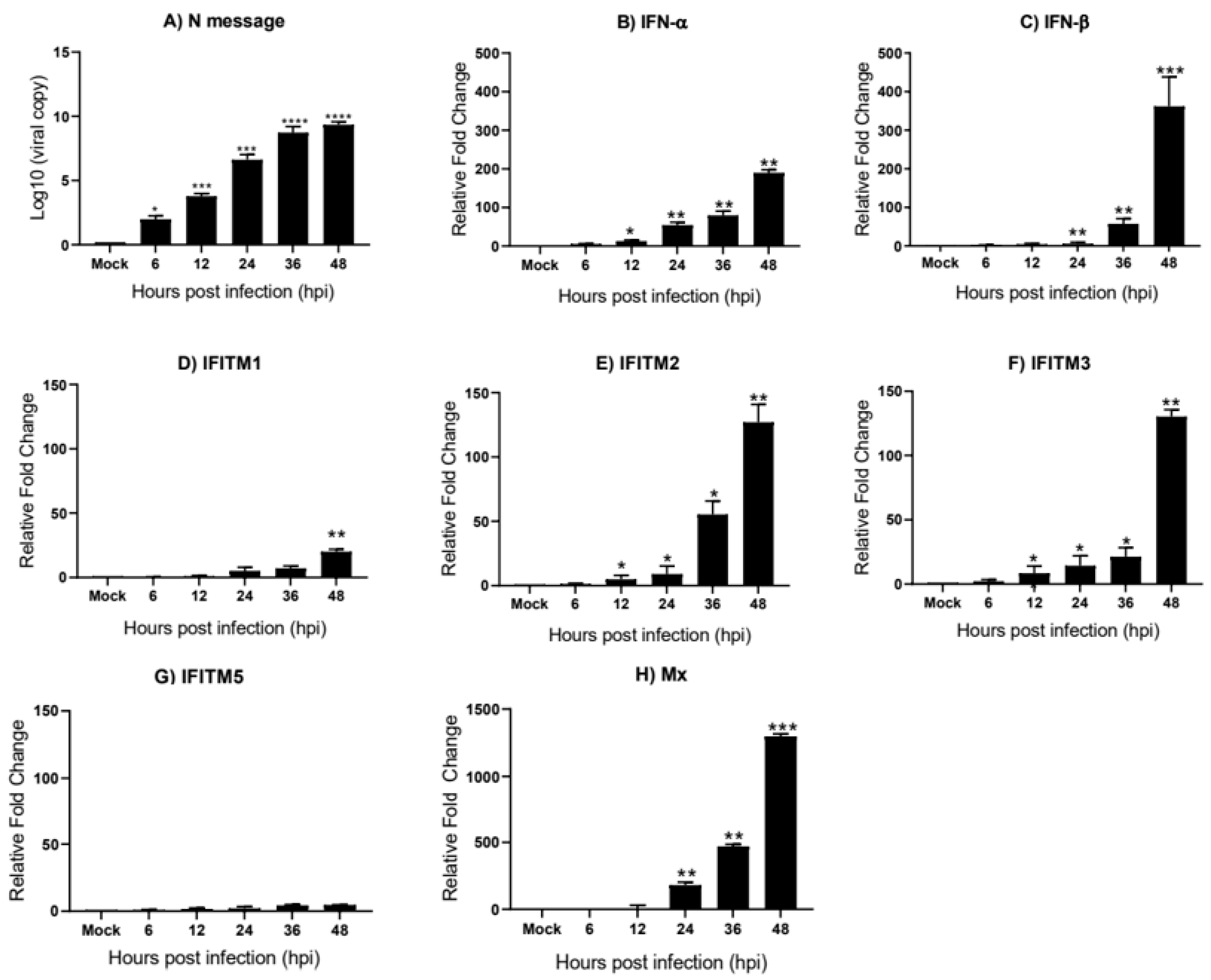
3.4. Infectious Bronchitis Virus Infection Induces chIFITM Expression In Vitro
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Fehr, A.R.; Perlman, S. Coronaviruses: An overview of their replication and pathogenesis. Methods Mol. Biol. 2015, 1282, 1–23. [Google Scholar]
- Rota, P.A.; Oberste, M.S.; Monroe, S.S.; Nix, W.A.; Campagnoli, R.; Icenogle, J.P.; Penaranda, S.; Bankamp, B.; Maher, K.; Chen, M.H.; et al. Characterization of a novel coronavirus associated with severe acute respiratory syndrome. Science 2003, 300, 1394–1399. [Google Scholar] [CrossRef]
- Zhu, N.; Zhang, D.; Wang, W.; Li, X.; Yang, B.; Song, J.; Zhao, X.; Huang, B.; Shi, W.; Lu, R.; et al. A novel coronavirus from patients with pneumonia in China, 2019. N. Engl. J. Med. 2020, 382, 727–733. [Google Scholar] [CrossRef]
- Kin, N.; Miszczak, F.; Lin, W.; Gouilh, M.A.; Vabret, A.; Epicorem Consortium. Genomic analysis of 15 human coronaviruses OC43 (HCoV-OC43s) circulating in France from 2001 to 2013 reveals a high intra-specific diversity with new recombinant genotypes. Viruses 2015, 7, 2358–2377. [Google Scholar] [CrossRef]
- Casais, R.; Thiel, V.; Siddell, S.G.; Cavanagh, D.; Britton, P. Reverse genetics system for the avian coronavirus infectious bronchitis virus. J. Virol. 2001, 75, 12359–12369. [Google Scholar] [CrossRef]
- Bande, F.; Arshad, S.S.; Omar, A.R.; Bejo, M.H.; Abubakar, M.S.; Abba, Y. Pathogenesis and diagnostic approaches of avian infectious bronchitis. Adv. Virol. 2016, 2016, 4621659. [Google Scholar] [CrossRef]
- Okino, C.H.; Mores, M.A.; Trevisol, I.M.; Coldebella, A.; Montassier, H.J.; Brentano, L. Early immune responses and development of pathogenesis of avian infectious bronchitis viruses with different virulence profiles. PLoS ONE 2017, 12, e0172275. [Google Scholar] [CrossRef]
- Ignjatovic, J.; Sapats, S. Avian infectious bronchitis virus. Rev. Sci. Tech. 2000, 19, 493–508. [Google Scholar] [CrossRef]
- Wang, Y.; Wang, Y.; Zhang, Z.; Fan, G.; Jiang, Y.; Liu, X.; Ding, J.; Wang, S. Isolation and identification of glandular stomach type IBV (QX IBV) in chickens. Chin. J. Anim. Quar. 1998, 15, 1–3. [Google Scholar]
- Jackwood, M.W. Review of infectious bronchitis virus around the world. Avian Dis. 2012, 56, 634–641. [Google Scholar] [CrossRef]
- Boursnell, M.E.; Brown, T.D.; Foulds, I.J.; Green, P.F.; Tomley, F.M.; Binns, M.M. Completion of the sequence of the genome of the coronavirus avian infectious bronchitis virus. J. Gen. Virol. 1987, 68, 57–77. [Google Scholar] [CrossRef]
- Boursnell, M.E.; Brown, T.D.; Foulds, I.J.; Green, P.F.; Tomley, F.M.; Binns, M.M. The complete nucleotide sequence of avian infectious bronchitis virus: Analysis of the polymerase-coding region. Adv. Exp. Med. Biol. 1987, 218, 15–29. [Google Scholar]
- Brierley, I.; Boursnell, M.E.; Binns, M.M.; Bilimoria, B.; Blok, V.C.; Brown, T.D.; Inglis, S.C. An efficient ribosomal frame-shifting signal in the polymerase-encoding region of the coronavirus IBV. EMBO J. 1987, 6, 3779–3785. [Google Scholar] [CrossRef]
- Brierley, I.; Digard, P.; Inglis, S.C. Characterization of an efficient coronavirus ribosomal frameshifting signal: Requirement for an RNA pseudoknot. Cell 1989, 57, 537–547. [Google Scholar] [CrossRef]
- Lai, M.M.; Cavanagh, D. The molecular biology of coronaviruses. Adv. Virus Res. 1997, 48, 1–100. [Google Scholar]
- Shang, J.; Zheng, Y.; Yang, Y.; Liu, C.; Geng, Q.; Luo, C.; Zhang, W.; Li, F. Cryo-EM structure of infectious bronchitis coronavirus spike protein reveals structural and functional evolution of coronavirus spike proteins. PLoS Pathog. 2018, 14, e1007009. [Google Scholar] [CrossRef]
- Bickerton, E.; Maier, H.J.; Stevenson-Leggett, P.; Armesto, M.; Britton, P. The S2 subunit of infectious bronchitis virus beaudette is a determinant of cellular tropism. J. Virol. 2018, 92, e01044-18. [Google Scholar] [CrossRef]
- Casais, R.; Dove, B.; Cavanagh, D.; Britton, P. Recombinant avian infectious bronchitis virus expressing a heterologous spike gene demonstrates that the spike protein is a determinant of cell tropism. J. Virol. 2003, 77, 9084–9089. [Google Scholar] [CrossRef]
- Wickramasinghe, I.N.; de Vries, R.P.; Grone, A.; de Haan, C.A.; Verheije, M.H. Binding of avian coronavirus spike proteins to host factors reflects virus tropism and pathogenicity. J. Virol. 2011, 85, 8903–8912. [Google Scholar] [CrossRef]
- Hodgson, T.; Casais, R.; Dove, B.; Britton, P.; Cavanagh, D. Recombinant infectious bronchitis coronavirus Beaudette with the spike protein gene of the pathogenic M41 strain remains attenuated but induces protective immunity. J. Virol. 2004, 78, 13804–13811. [Google Scholar] [CrossRef]
- Winter, C.; Schwegmann-Wessels, C.; Cavanagh, D.; Neumann, U.; Herrler, G. Sialic acid is a receptor determinant for infection of cells by avian Infectious bronchitis virus. J. Gen. Virol. 2006, 87, 1209–1216. [Google Scholar] [CrossRef]
- Abd El Rahman, S.; El-Kenawy, A.A.; Neumann, U.; Herrler, G.; Winter, C. Comparative analysis of the sialic acid binding activity and the tropism for the respiratory epithelium of four different strains of avian infectious bronchitis virus. Avian Pathol. 2009, 38, 41–45. [Google Scholar] [CrossRef]
- Chhabra, R.; Ball, C.; Chantrey, J.; Ganapathy, K. Differential innate immune responses induced by classical and variant infectious bronchitis viruses in specific pathogen free chicks. Dev. Comp. Immunol. 2018, 87, 16–23. [Google Scholar] [CrossRef]
- Chhabra, R.; Kuchipudi, S.V.; Chantrey, J.; Ganapathy, K. Pathogenicity and tissue tropism of infectious bronchitis virus is associated with elevated apoptosis and innate immune responses. Virology 2016, 488, 232–241. [Google Scholar] [CrossRef]
- Cavanagh, D. Severe acute respiratory syndrome vaccine development: Experiences of vaccination against avian infectious bronchitis coronavirus. Avian Pathol. 2003, 32, 567–582. [Google Scholar] [CrossRef]
- De Wit, J.J. Detection of infectious bronchitis virus. Avian Pathol. 2000, 29, 71–93. [Google Scholar] [CrossRef]
- Jordan, B. Vaccination against infectious bronchitis virus: A continuous challenge. Vet. Microbiol. 2017, 206, 137–143. [Google Scholar] [CrossRef]
- De Wit, J.J.; Cook, J.K.; Van der Heijden, H.M. Infectious bronchitis virus variants: A review of the history, current situation and control measures. Avian Pathol. 2011, 40, 223–235. [Google Scholar] [CrossRef]
- Shimazaki, Y.; Watanabe, Y.; Harada, M.; Seki, Y.; Kuroda, Y.; Fukuda, M.; Honda, E.; Suzuki, S.; Nakamura, S. Genetic analysis of the S1 gene of 4/91 type infectious bronchitis virus isolated in Japan. J. Vet. Med. Sci. 2009, 71, 583–588. [Google Scholar] [CrossRef]
- Xu, C.; Zhao, J.; Hu, X.; Zhang, G. Isolation and identification of four infectious bronchitis virus strains in China and analyses of their S1 glycoprotein gene. Vet. Microbiol. 2007, 122, 61–71. [Google Scholar] [CrossRef]
- Kameka, A.M.; Haddadi, S.; Kim, D.S.; Cork, S.C.; Abdul-Careem, M.F. Induction of innate immune response following infectious bronchitis corona virus infection in the respiratory tract of chickens. Virology 2014, 450, 114–121. [Google Scholar] [CrossRef]
- Bailey, C.C.; Zhong, G.; Huang, I.C.; Farzan, M. IFITM-family proteins: The cell’s first line of antiviral defense. Annu. Rev. Virol. 2014, 1, 261–283. [Google Scholar] [CrossRef]
- Munoz-Moreno, R.; Cuesta-Geijo, M.A.; Martinez-Romero, C.; Barrado-Gil, L.; Galindo, I.; Garcia-Sastre, A.; Alonso, C. Antiviral Role of IFITM Proteins in African Swine Fever Virus Infection. PLoS ONE 2016, 11, e0154366. [Google Scholar] [CrossRef]
- Blyth, G.A.; Chan, W.F.; Webster, R.G.; Magor, K.E. Duck interferon-inducible transmembrane protein 3 mediates restriction of influenza viruses. J. Virol. 2016, 90, 103–116. [Google Scholar] [CrossRef]
- Chen, S.; Luo, G.; Yang, Z.; Lin, S.; Chen, S.; Wang, S.; Goraya, M.U.; Chi, X.; Zeng, X.; Chen, J.L. Avian Tembusu virus infection effectively triggers host innate immune response through MDA5 and TLR3-dependent signaling pathways. Vet. Res. 2016, 47, 74. [Google Scholar] [CrossRef]
- Diamond, M.S.; Farzan, M. The broad-spectrum antiviral functions of IFIT and IFITM proteins. Nat. Rev. Immunol. 2013, 13, 46–57. [Google Scholar] [CrossRef]
- Brass, A.L.; Huang, I.C.; Benita, Y.; John, S.P.; Krishnan, M.N.; Feeley, E.M.; Ryan, B.J.; Weyer, J.L.; van der Weyden, L.; Fikrig, E.; et al. The IFITM proteins mediate cellular resistance to influenza A H1N1 virus, West Nile virus, and dengue virus. Cell 2009, 139, 1243–1254. [Google Scholar] [CrossRef]
- Huang, I.C.; Bailey, C.C.; Weyer, J.L.; Radoshitzky, S.R.; Becker, M.M.; Chiang, J.J.; Brass, A.L.; Ahmed, A.A.; Chi, X.; Dong, L.; et al. Distinct patterns of IFITM-mediated restriction of filoviruses, SARS coronavirus, and influenza A virus. PLoS Pathog. 2011, 7, e1001258. [Google Scholar] [CrossRef]
- Lu, J.; Pan, Q.; Rong, L.; He, W.; Liu, S.L.; Liang, C. The IFITM proteins inhibit HIV-1 infection. J. Virol. 2011, 85, 2126–2137. [Google Scholar] [CrossRef]
- Perreira, J.M.; Chin, C.R.; Feeley, E.M.; Brass, A.L. IFITMs restrict the replication of multiple pathogenic viruses. J. Mol. Biol. 2013, 425, 4937–4955. [Google Scholar] [CrossRef]
- Smith, J.; Smith, N.; Yu, L.; Paton, I.R.; Gutowska, M.W.; Forrest, H.L.; Danner, A.F.; Seiler, J.P.; Digard, P.; Webster, R.G.; et al. A comparative analysis of host responses to avian influenza infection in ducks and chickens highlights a role for the interferon-induced transmembrane proteins in viral resistance. BMC Genom. 2015, 16, 574. [Google Scholar] [CrossRef]
- Smith, S.; Weston, S.; Kellam, P.; Marsh, M. IFITM proteins-cellular inhibitors of viral entry. Curr. Opin. Virol. 2014, 4, 71–77. [Google Scholar] [CrossRef]
- Wilkins, C.; Woodward, J.; Lau, D.T.; Barnes, A.; Joyce, M.; McFarlane, N.; McKeating, J.A.; Tyrrell, D.L.; Gale, M., Jr. IFITM1 is a tight junction protein that inhibits hepatitis C virus entry. Hepatology 2013, 57, 461–469. [Google Scholar] [CrossRef]
- Wilkins, J.; Zheng, Y.M.; Yu, J.; Liang, C.; Liu, S.L. Nonhuman Primate IFITM Proteins Are Potent Inhibitors of HIV and SIV. PLoS ONE 2016, 11, e0156739. [Google Scholar] [CrossRef]
- Desai, T.M.; Marin, M.; Chin, C.R.; Savidis, G.; Brass, A.L.; Melikyan, G.B. IFITM3 restricts influenza A virus entry by blocking the formation of fusion pores following virus-endosome hemifusion. PLoS Pathog. 2014, 10, e1004048. [Google Scholar] [CrossRef]
- Li, K.; Markosyan, R.M.; Zheng, Y.M.; Golfetto, O.; Bungart, B.; Li, M.; Ding, S.; He, Y.; Liang, C.; Lee, J.C. IFITM proteins restrict viral membrane hemifusion. PLoS Pathog. 2013, 9, e1003124. [Google Scholar] [CrossRef]
- Weston, S.; Czieso, S.; White, I.J.; Smith, S.E.; Wash, R.S.; Diaz-Soria, C.; Kellam, P.; Marsh, M. Alphavirus Restriction by IFITM Proteins. Traffic 2016, 17, 997–1013. [Google Scholar] [CrossRef]
- Zhao, X.; Li, J.; Winkler, C.A.; An, P.; Guo, J.T. IFITM Genes, Variants, and Their Roles in the Control and Pathogenesis of Viral Infections. Front. Microbiol. 2018, 9, 3228. [Google Scholar] [CrossRef]
- Amini-Bavil-Olyaee, S.; Choi, Y.J.; Lee, J.H.; Shi, M.; Huang, I.C.; Farzan, M.; Jung, J.U. The antiviral effector IFITM3 disrupts intracellular cholesterol homeostasis to block viral entry. Cell Host Microbe 2013, 13, 452–464. [Google Scholar] [CrossRef]
- Smith, S.E.; Gibson, M.S.; Wash, R.S.; Ferrara, F.; Wright, E.; Temperton, N.; Kellam, P.; Fife, M. Chicken interferon-inducible transmembrane protein 3 restricts influenza viruses and lyssaviruses in vitro. J. Virol. 2013, 87, 12957–12966. [Google Scholar] [CrossRef]
- Bassano, I.; Ong, S.H.; Lawless, N.; Whitehead, T.; Fife, M.; Kellam, P. Accurate characterization of the IFITM locus using MiSeq and PacBio sequencing shows genetic variation in Galliformes. BMC Genom. 2017, 18, 419. [Google Scholar] [CrossRef]
- Yu, J.; Li, M.; Wilkins, J.; Ding, S.; Swartz, T.H.; Esposito, A.M.; Zheng, Y.M.; Freed, E.O.; Liang, C.; Chen, B.K. IFITM Proteins Restrict HIV-1 Infection by Antagonizing the Envelope Glycoprotein. Cell Rep. 2015, 13, 145–156. [Google Scholar] [CrossRef]
- Yu, Y.Y.; Shi, G.C. The antivirual function of ifitm 3 in influenza A virus infection. Chin. J. Tuberc. Respir. Dis. 2017, 40, 294–297. [Google Scholar]
- Ellis, S.; Keep, S.; Britton, P.; de Wit, S.; Bickerton, E.; Vervelde, L. Recombinant infectious bronchitis viruses expressing chimeric spike glycoproteins induce partial protective immunity against homologous challenge despite limited replication in vivo. J. Virol. 2018, 92, e01473-18. [Google Scholar] [CrossRef]
- Keep, S.; Stevenson-Leggett, P.; Steyn, A.; Oade, M.S.; Webb, I.; Stuart, J.; Vervelde, L.; Britton, P.; Maier, H.J.; Bickerton, E. Temperature sensitivity: A potential method for the generation of vaccines against the avian coronavirus infectious bronchitis virus. Viruses 2020, 12, 754. [Google Scholar] [CrossRef]
- Van Roekel, H.C.M.; Bullis, K.L.; Olesiuk, O.M.; Sperling, F.G. Infectious bronchitis. Am. J. Vet. Res. 1951, 12, 140–146. [Google Scholar]
- De Wit, J.J.; Nieuwenhuisen-van Wilgen, J.; Hoogkamer, A.; van de Sande, H.; Zuidam, G.J.; Fabri, T.H. Induction of cystic oviducts and protection against early challenge with infectious bronchitis virus serotype D388 (genotype QX) by maternally derived antibodies and by early vaccination. Avian Pathol. 2011, 40, 463–471. [Google Scholar] [CrossRef]
- Keep, S.; Bickerton, E.; Armesto, M.; Britton, P. The ADRP domain from a virulent strain of infectious bronchitis virus is not sufficient to confer a pathogenic phenotype to the attenuated Beaudette strain. J. Gen. Virol. 2018, 99, 1097–1102. [Google Scholar] [CrossRef]
- Bickerton, E.; Dowgier, G.; Britton, P. Recombinant infectious bronchitis viruses expressing heterologous S1 subunits: Potential for a new generation of vaccines that replicate in Vero cells. J. Gen. Virol. 2018, 99, 1681–1685. [Google Scholar] [CrossRef]
- Penzes, Z.; Tibbles, K.; Shaw, K.; Britton, P.; Brown, T.D.; Cavanagh, D. Characterization of a replicating and packaged defective RNA of avian coronavirus infectious bronchitis virus. Virology 1994, 203, 286–293. [Google Scholar] [CrossRef]
- Penzes, Z.; Wroe, C.; Brown, T.D.; Britton, P.; Cavanagh, D. Replication and packaging of coronavirus infectious bronchitis virus defective RNAs lacking a long open reading frame. J. Virol. 1996, 70, 8660–8668. [Google Scholar] [CrossRef] [PubMed]
- Stirrups, K.; Shaw, K.; Evans, S.; Dalton, K.; Cavanagh, D.; Britton, P. Leader switching occurs during the rescue of defective RNAs by heterologous strains of the coronavirus infectious bronchitis virus. J. Gen. Virol. 2000, 81, 791–801. [Google Scholar] [CrossRef] [PubMed]
- Hennion, R.M.; Hill, G. The preparation of chicken kidney cell cultures for virus propagation. Methods Mol. Biol. 2015, 1282, 57–62. [Google Scholar] [PubMed]
- Hennion, R.M. The preparation of chicken tracheal organ cultures for virus isolation, propagation, and titration. Methods Mol. Biol. 2015, 1282, 51–56. [Google Scholar] [PubMed]
- Cook, J.K.; Orbell, S.J.; Woods, M.A.; Huggins, M.B. Breadth of protection of the respiratory tract provided by different live-attenuated infectious bronchitis vaccines against challenge with infectious bronchitis viruses of heterologous serotypes. Avian Pathol. 1999, 28, 477–485. [Google Scholar] [CrossRef] [PubMed]
- Keep, S.M.; Bickerton, E.; Britton, P. Transient dominant selection for the modification and generation of recombinant infectious bronchitis coronaviruses. Methods Mol. Biol. 2015, 1282, 115–133. [Google Scholar]
- Reemers, S.S.; Groot Koerkamp, M.J.; Holstege, F.C.; van Eden, W.; Vervelde, L. Cellular host transcriptional responses to influenza A virus in chicken tracheal organ cultures differ from responses in in vivo infected trachea. Vet. Immunol. Immunopathol. 2009, 132, 91–100. [Google Scholar] [CrossRef]
- Armesto, M.; Cavanagh, D.; Britton, P. The replicase gene of avian coronavirus infectious bronchitis virus is a determinant of pathogenicity. PLoS ONE 2009, 4, e7384. [Google Scholar] [CrossRef]
- Maier, H.J.; Hawes, P.C.; Cottam, E.M.; Mantell, J.; Verkade, P.; Monaghan, P.; Wileman, T.; Britton, P. Infectious bronchitis virus generates spherules from zippered endoplasmic reticulum membranes. MBio 2013, 4, e00801–e00813. [Google Scholar] [CrossRef]
- Staines, K.; Batra, A.; Mwangi, W.; Maier, H.J.; Van Borm, S.; Young, J.R.; Fife, M.; Butter, C. A versatile panel of reference gene assays for the measurement of chicken mRNA by quantitative PCR. PLoS ONE 2016, 11, e0160173. [Google Scholar] [CrossRef]
- Reddy, V.R.; Trus, I.; Desmarets, L.M.; Li, Y.; Theuns, S.; Nauwynck, H.J. Productive replication of nephropathogenic infectious bronchitis virus in peripheral blood monocytic cells, a strategy for viral dissemination and kidney infection in chickens. Vet. Res. 2016, 47, 70. [Google Scholar] [CrossRef] [PubMed]
- Bailey, C.C.; Huang, I.C.; Kam, C.; Farzan, M. Ifitm3 limits the severity of acute influenza in mice. PLoS Pathog. 2012, 8, e1002909. [Google Scholar] [CrossRef] [PubMed]
- Everitt, A.R.; Clare, S.; Pertel, T.; John, S.P.; Wash, R.S.; Smith, S.E.; Chin, C.R.; Feeley, E.M.; Sims, J.S.; Adams, D.J.; et al. IFITM3 restricts the morbidity and mortality associated with influenza. Nature 2012, 484, 519–523. [Google Scholar] [CrossRef] [PubMed]
- Everitt, A.R.; Clare, S.; McDonald, J.U.; Kane, L.; Harcourt, K.; Ahras, M.; Lall, A.; Hale, C.; Rodgers, A.; Young, D.B.; et al. Defining the range of pathogens susceptible to Ifitm3 restriction using a knockout mouse model. PLoS ONE 2013, 8, e80723. [Google Scholar] [CrossRef]
- Wrensch, F.; Winkler, M.; Pohlmann, S. IFITM proteins inhibit entry driven by the MERS-coronavirus spike protein: Evidence for cholesterol-independent mechanisms. Viruses 2014, 6, 3683–3698. [Google Scholar] [CrossRef]
- Zhao, X.; Guo, F.; Liu, F.; Cuconati, A.; Chang, J.; Block, T.M.; Guo, J.T. Interferon induction of IFITM proteins promotes infection by human coronavirus OC43. Proc. Natl. Acad. Sci. USA 2014, 111, 6756–6761. [Google Scholar] [CrossRef]
- Chen, B.Y.; Hosi, S.; Nunoya, T.; Itakura, C. Histopathology and immunohistochemistry of renal lesions due to infectious bronchitis virus in chicks. Avian Pathol. 1996, 25, 269–283. [Google Scholar] [CrossRef]
- Owen, R.L.; Cowen, B.S.; Hattel, A.L.; Naqi, S.A.; Wilson, R.A. Detection of viral antigen following exposure of one-day-old chickens to the Holland 52 strain of infectious bronchitis virus. Avian Pathol. 1991, 20, 663–673. [Google Scholar] [CrossRef]
- Cong, F.; Liu, X.; Han, Z.; Shao, Y.; Kong, X.; Liu, S. Transcriptome analysis of chicken kidney tissues following coronavirus avian infectious bronchitis virus infection. BMC Genom. 2013, 14, 743. [Google Scholar] [CrossRef]
- Wang, H.N.; Wu, Q.Z.; Huang, Y.; Liu, P. Isolation and identification of infectious bronchitis virus from chickens in Sichuan, China. Avian Dis. 1997, 41, 279–282. [Google Scholar] [CrossRef]
- Winter, C.; Herrler, G.; Neumann, U. Infection of the tracheal epithelium by infectious bronchitis virus is sialic acid dependent. Microbes Infect. 2008, 10, 367–373. [Google Scholar] [CrossRef] [PubMed]
- Kint, J.; Fernandez-Gutierrez, M.; Maier, H.J.; Britton, P.; Langereis, M.A.; Koumans, J.; Wiegertjes, G.F.; Forlenza, M. Activation of the chicken type I interferon response by infectious bronchitis coronavirus. J. Virol. 2015, 89, 1156–1167. [Google Scholar] [CrossRef] [PubMed]
- Kint, J.; Dickhout, A.; Kutter, J.; Maier, H.J.; Britton, P.; Koumans, J.; Pijlman, G.P.; Fros, J.J.; Wiegertjes, G.F.; Forlenza, M. Infectious bronchitis coronavirus inhibits STAT1 signaling and requires accessory proteins for resistance to type I interferon activity. J. Virol. 2015, 89, 12047–12057. [Google Scholar] [CrossRef] [PubMed]
- Kint, J.; Langereis, M.A.; Maier, H.J.; Britton, P.; van Kuppeveld, F.J.; Koumans, J.; Wiegertjes, G.F.; Forlenza, M. Infectious bronchitis coronavirus limits interferon production by inducing a host shutoff that requires accessory protein 5b. J. Virol. 2016, 90, 7519–7528. [Google Scholar] [CrossRef] [PubMed]
- Laconi, A.; van Beurden, S.J.; Berends, A.J.; Kramer-Kuhl, A.; Jansen, C.A.; Spekreijse, D.; Chenard, G.; Philipp, H.C.; Mundt, E.; Rottier, P.J.M.; et al. Deletion of accessory genes 3a, 3b, 5a or 5b from avian coronavirus infectious bronchitis virus induces an attenuated phenotype both in vitro and in vivo. J. Gen. Virol. 2018, 99, 1381. [Google Scholar] [CrossRef] [PubMed]





© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Steyn, A.; Keep, S.; Bickerton, E.; Fife, M. The Characterization of chIFITMs in Avian Coronavirus Infection In Vivo, Ex Vivo and In Vitro. Genes 2020, 11, 918. https://doi.org/10.3390/genes11080918
Steyn A, Keep S, Bickerton E, Fife M. The Characterization of chIFITMs in Avian Coronavirus Infection In Vivo, Ex Vivo and In Vitro. Genes. 2020; 11(8):918. https://doi.org/10.3390/genes11080918
Chicago/Turabian StyleSteyn, Angela, Sarah Keep, Erica Bickerton, and Mark Fife. 2020. "The Characterization of chIFITMs in Avian Coronavirus Infection In Vivo, Ex Vivo and In Vitro" Genes 11, no. 8: 918. https://doi.org/10.3390/genes11080918
APA StyleSteyn, A., Keep, S., Bickerton, E., & Fife, M. (2020). The Characterization of chIFITMs in Avian Coronavirus Infection In Vivo, Ex Vivo and In Vitro. Genes, 11(8), 918. https://doi.org/10.3390/genes11080918