Using Micro- and Macro-Level Network Metrics Unveils Top Communicative Gene Modules in Psoriasis



Abstract
1. Introduction
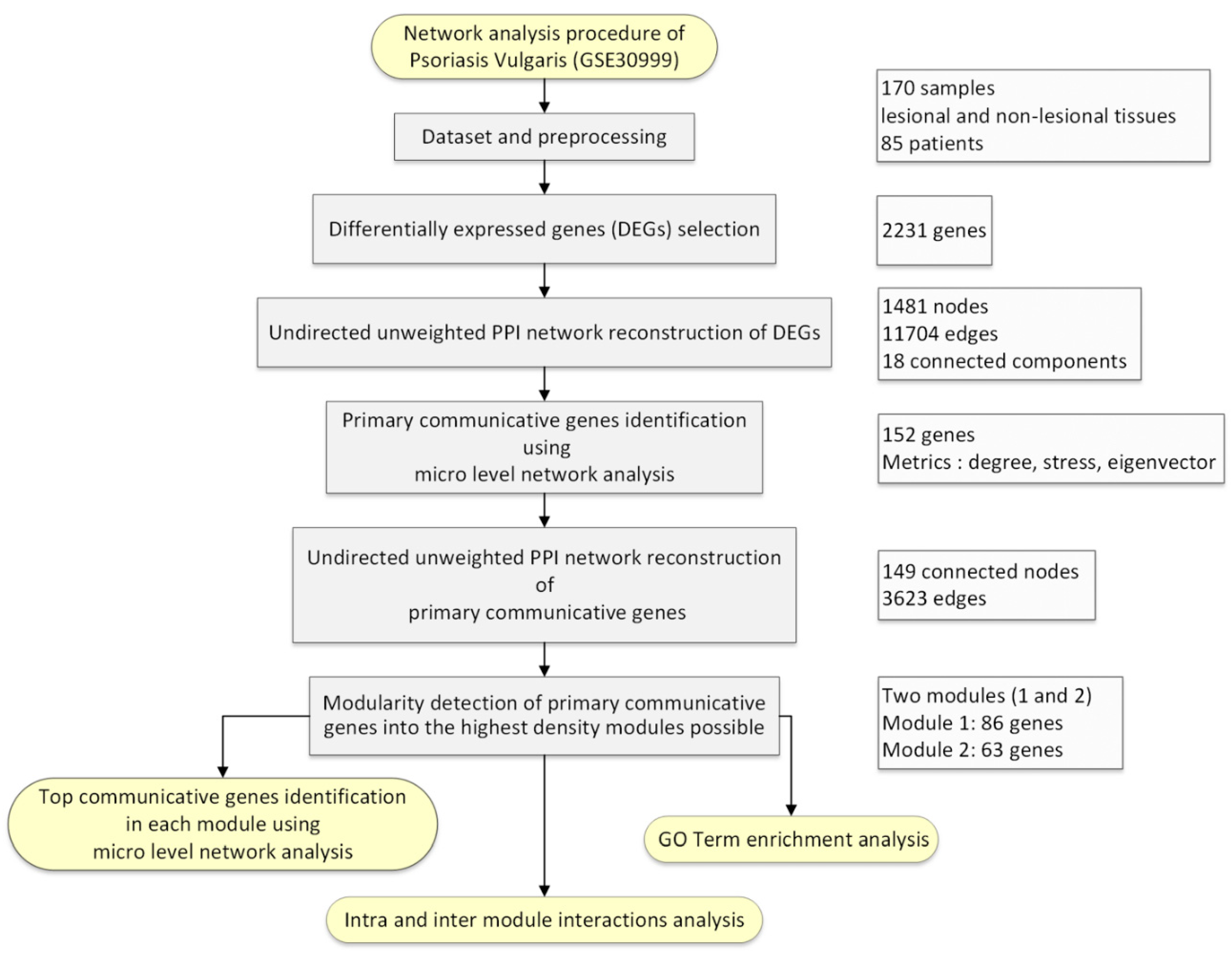
2. Materials and Methods
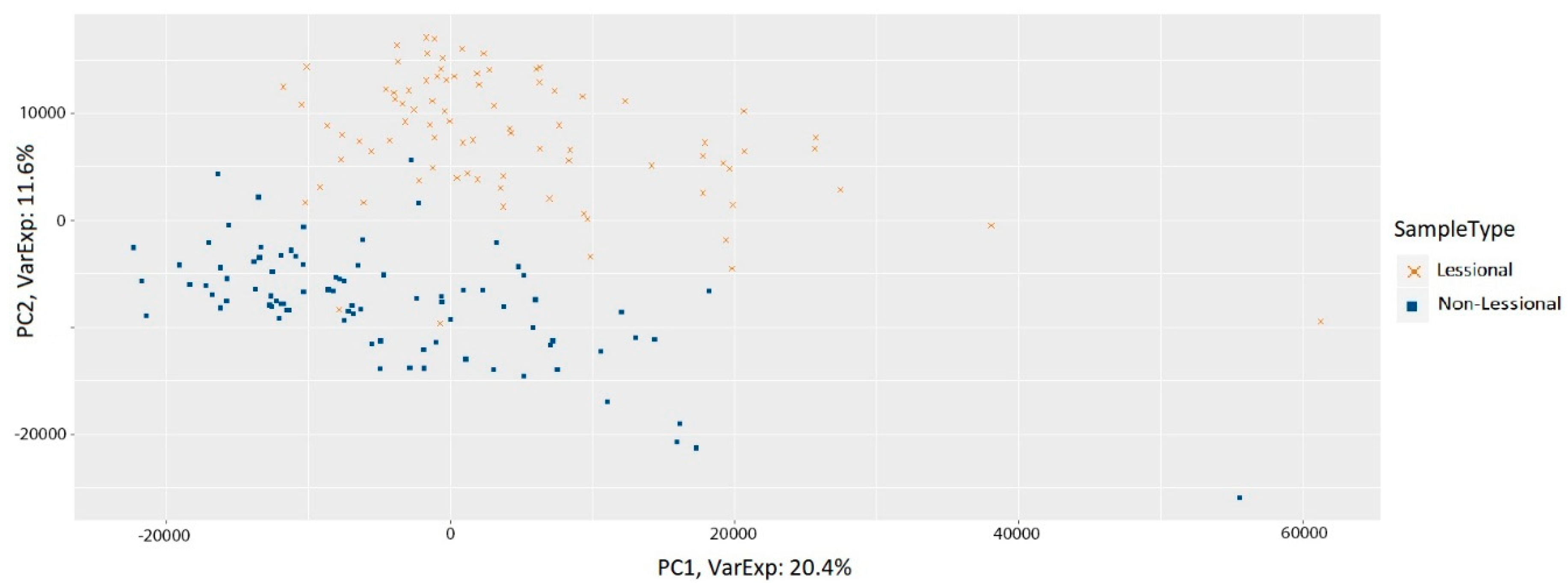
2.1. Dataset
2.2. Preprocessing
2.3. Selection of Differentially Expressed Genes (DEGs) in Psoriasis Vulgaris Patients
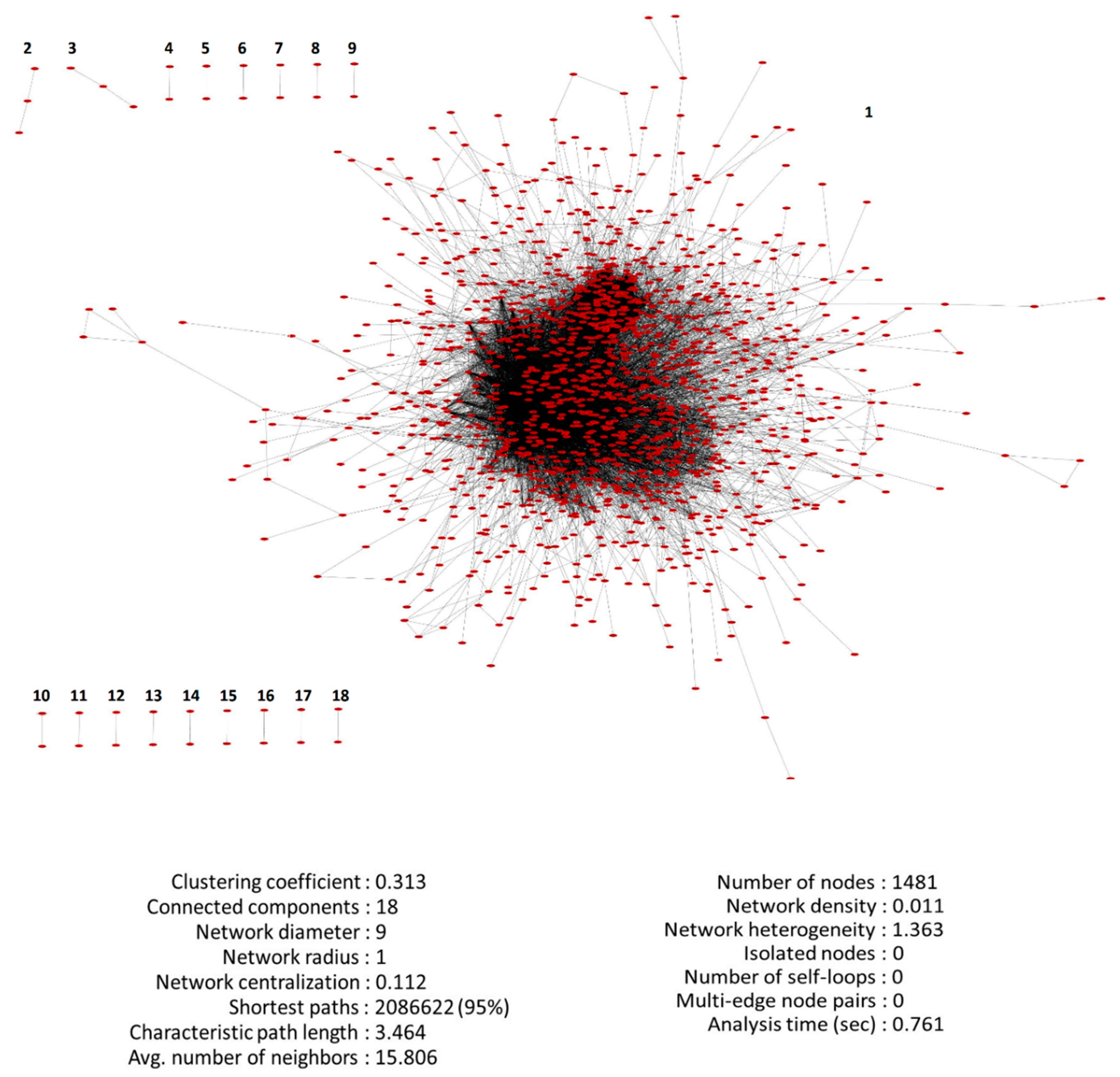
2.4. PPI Undirected Unweighted Network Reconstruction of DEGs
2.5. Primary Communicative Genes Identification
2.6. PPI Network Reconstruction and Analysis of Primary Communicative Genes
2.7. Macro-Level Network Analysis Followed by Top Communicative Genes Identification
3. Results
3.1. Preprocessing of Samples
3.2. DEGs in Psoriasis Vulgaris Patients
3.3. PPI Network of DEGs Reconstruction and Analysis
3.4. PPI Network of Primary Communicative Gene Reconstruction and Analysis
3.5. Macro-Level Network Analysis
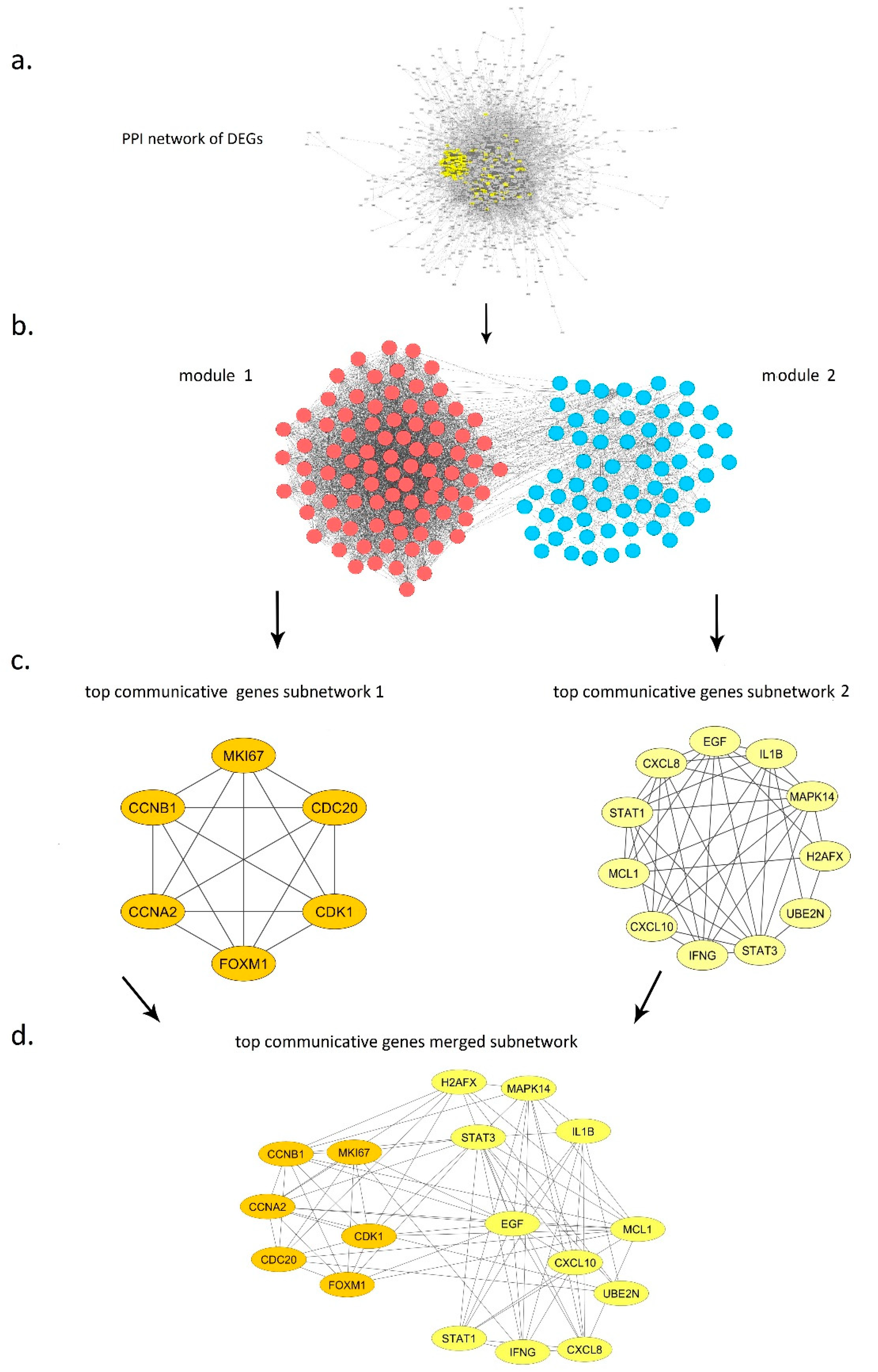
3.6. Top Communicative Genes Selection
3.6.1. Module 1
- Enriched pathway analysis (module 1)
3.6.2. Module 2
- Enriched pathway analysis (module 2)
3.7. Identification of Pathways Associated with Genes in Both Modules 1 and 2
3.8. Inter Top Communicative Genes Subnetwork Interactions
4. Discussion
4.1. Module 1 (Cell Cycle)
4.2. Module 2 (Immune System)
4.3. Pathway Enrichment
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Ayala-Fontánez, N.; Soler, D.C.; McCormick, T.S. Current knowledge on psoriasis and autoimmune diseases. Psoriasis 2016, 6, 7–32. [Google Scholar] [CrossRef] [PubMed]
- Schleicher, S.M. Psoriasis: Pathogenesis, Assessment, and Therapeutic Update. Clin. Podiatr. Med. Surg. 2016, 33, 355–366. [Google Scholar] [CrossRef] [PubMed]
- Ashtiani, M.; Salehzadehyazdi, A.; Razaghimoghadam, Z.; Hennig, H.; Wolkenhauer, O.; Mirzaie, M.; Jafari, M. A systematic survey of centrality measures for protein-protein interaction networks. BMC Syst. Biol. 2018, 12, 80. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Yang, C.; Zhang, J.; Zhong, W.; Zhu, L.; Chen, Y. Identification of potential key mRNAs and LncRNAs for psoriasis by bioinformatic analysis using weighted gene co-expression network analysis. Mol. Genet. Genom. 2020, 295, 741–749. [Google Scholar] [CrossRef]
- Zhang, Y.J.; Sun, Y.Z.; Gao, X.H.; Qi, R.Q. Integrated bioinformatic analysis of differentially expressed genes and signaling pathways in plaque psoriasis. Mol. Med. Rep. 2019, 20, 225–235. [Google Scholar] [CrossRef] [PubMed]
- Mei, R.; Mei, X. Screening of Skin Lesion-Associated Genes in Patients with Psoriasis by Meta-Integration Analysis. Dermatology 2018, 233, 277–288. [Google Scholar] [CrossRef]
- Sundarrajan, S.; Arumugam, M. Comorbidities of psoriasis—Exploring the links by network approach. PLoS ONE 2016, 11. [Google Scholar] [CrossRef]
- Sevimoglu, T.; Arga, K.Y. Computational Systems Biology of Psoriasis: Are We Ready for the Age of Omics and Systems Biomarkers. Omics A J. Integr. Biol. 2015, 19, 669–687. [Google Scholar] [CrossRef]
- Piruzian, E.S.; Bruskin, S.A.; Ishkin, A.; Abdeev, R.M.; Moshkovskii, S.; Melnik, S.; Nikolsky, Y.; Nikolskaya, T. Integrated network analysis of transcriptomic and proteomic data in psoriasis. BMC Syst. Biol. 2010, 4. [Google Scholar] [CrossRef]
- Suárez-Farinas, M.; Li, K.; Fuentes-Duculan, J.; Hayden, K.; Brodmerkel, C.; Krueger, J.G. Expanding the Psoriasis Disease Profile: Interrogation of the Skin and Serum of Patients with Moderate-to-Severe Psoriasis. J. Investig. Dermatol. 2012, 132, 2552–2564. [Google Scholar] [CrossRef]
- Hubbell, E.; Liu, W.M.; Mei, R. Robust estimators for expression analysis. Bioinformatics 2002, 18, 1585–1592. [Google Scholar] [CrossRef] [PubMed]
- Smyth, G.K. Linear Models and Empirical Bayes Methods for Assessing Differential Expression in Microarray Experiments. Stat. Appl. Genet. Mol. Biol. 2004, 3, 1–25. [Google Scholar] [CrossRef] [PubMed]
- Szklarczyk, D.; Franceschini, A.; Wyder, S.; Forslund, K.; Heller, D.; Huertacepas, J.; Simonovic, M.; Roth, A.; Santos, A.; Tsafou, K.; et al. STRING v10: Protein–Protein interaction networks, integrated over the tree of life. Nucleic Acids Res. 2015, 43, 447–452. [Google Scholar] [CrossRef] [PubMed]
- Carlson, M. hgu133plus2.db: Affymetrix Human Genome U133 Plus 2.0 Array Annotation Data (chip hgu133plus2); 2016. [Google Scholar]
- Assenov, Y.; Ramírez, F.; Schelhorn, S.E.S.E.; Lengauer, T.; Albrecht, M. Computing topological parameters of biological networks. Bioinformatics 2008, 24, 282–284. [Google Scholar] [CrossRef]
- Shannon, P.; Markiel, A.; Ozier, O.; Baliga, N.S.; Wang, J.T.; Ramage, D.; Amin, N.; Schwikowski, B.; Ideker, T. Cytoscape: A Software Environment for Integrated Models of Biomolecular Interaction Networks. Genome Res. 1971, 13, 426. [Google Scholar] [CrossRef]
- Das, K.; Samanta, S.; Pal, M. Study on centrality measures in social networks: A survey. Soc. Netw. Anal. Min. 2018, 8. [Google Scholar] [CrossRef]
- Scardoni, G.; Petterlini, M.; Laudanna, C. Analyzing biological network parameters with CentiScaPe. Bioinformatics 2009, 25, 2857–2859. [Google Scholar] [CrossRef]
- Blondel, V.D.; Guillaume, J.L.; Lambiotte, R.; Lefebvre, E. Fast unfolding of communities in large networks. J. Stat. Mech. Theory Exp. 2008, 2008, 10008. [Google Scholar] [CrossRef]
- Chen, E.Y.; Tan, C.M.; Kou, Y.; Duan, Q.; Wang, Z.; Meirelles, G.V.; Clark, N.R.; Ma’ayan, A. Enrichr: Interactive and collaborative HTML5 gene list enrichment analysis tool. BMC Bioinform. 2013, 14, 128. [Google Scholar] [CrossRef]
- Slenter, D.; Kutmon, M.; Hanspers, K.; Riutta, A.; Windsor, J.; Nunes, N.; Melius, J.; Cirillo, E.; Coort, S.L.; Digles, D.; et al. WikiPathways: A multifaceted pathway database bridging metabolomics to other omics research. Nucleic Acids Res. 2018. Available online: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC5753270/ (accessed on 15 June 2020). [CrossRef]
- Ogata, H.; Goto, S.; Sato, K.; Fujibuchi, W.; Bono, H.; Kanehisa, M. KEGG: Kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 1999, 27, 29–34. [Google Scholar] [CrossRef] [PubMed]
- Leonard, M.; Graham, S.; Bonacum, D. The human factor: The critical importance of effective teamwork and communication in providing safe care. Qual. Saf. Heal. Care 2004, 13, 361–362. [Google Scholar] [CrossRef]
- MacDonald, A.; Burden, A.D. Psoriasis: Advances in pathophysiology and management. Postgrad. Med. J. 2007, 83, 690–697. [Google Scholar] [CrossRef] [PubMed]
- Greenberg, E.N.; Marshall, M.E.; Jin, S.; Venkatesh, S.; Dragan, M.; Tsoi, L.C.; Andersen, B. Circadian control of interferon-sensitive gene expression in murine skin. Proc. Natl. Acad. Sci. USA 2020, 117, 5761–5771. [Google Scholar] [CrossRef] [PubMed]
- Gavet, O.; Pines, J. Progressive Activation of CyclinB1-Cdk1 Coordinates Entry to Mitosis. Dev. Cell 2010, 18, 533–543. [Google Scholar] [CrossRef]
- Katsuno, Y.; Suzuki, A.; Sugimura, K.; Okumura, K.; Zineldeen, D.H.; Shimada, M.; Niida, H.; Mizuno, T.; Hanaoka, F.; Nakanishi, M. Cyclin A-Cdk1 regulates the origin firing program in mammalian cells. Proc. Natl. Acad. Sci. USA 2009, 106, 3184–3189. [Google Scholar] [CrossRef]
- Kotani, T.; Yoshida, N.; Mita, K.; Yamashita, M. Requirement of cyclin B2, but not cyclin B1, for bipolar spindle formation in frog (Rana japonica) oocytes. Mol. Reprod. Dev. 2001, 59, 199–208. [Google Scholar] [CrossRef]
- Mee, J.B.; Johnson, C.M.; Morar, N.; Burslem, F.; Groves, R.W. The psoriatic transcriptome closely resembles that induced by interleukin-1 in cultured keratinocytes: Dominance of innate immune responses in psoriasis. Am. J. Pathol. 2007, 171, 32–42. [Google Scholar] [CrossRef]
- Zolotarenko, A.; Chekalin, E.; Mesentsev, A.; Kiseleva, L.; Gribanova, E.; Mehta, R.; Baranova, A.; Tatarinova, T.V.; Piruzian, E.S.; Bruskin, S. Integrated computational approach to the analysis of RNA-seq data reveals new transcriptional regulators of psoriasis. Exp. Mol. Med. 2016, 48. [Google Scholar] [CrossRef]
- Xue, L.; Chiang, L.; He, B.; Zhao, Y.Y.; Winoto, A. FoxM1, a Forkhead Transcription Factor Is a Master Cell Cycle Regulator for Mouse Mature T Cells but Not Double Positive Thymocytes. PLoS ONE 2010, 5, e9229. [Google Scholar] [CrossRef]
- Levine, M.S.; Holland, A.J. The impact of mitotic errors on cell proliferation and tumorigenesis. Genes Dev. 2018, 32, 620–638. [Google Scholar] [CrossRef] [PubMed]
- Kapanidou, M.; Curtis, N.L.; Bolanos-Garcia, V.M. Cdc20: At the Crossroads between Chromosome Segregation and Mitotic Exit. Trends Biochem. Sci. 2017, 42, 193–205. [Google Scholar] [CrossRef] [PubMed]
- Zwicker, S.; Hattinger, E.; Bureik, D.; Batyckabaran, A.; Schmidt, A.; Gerber, P.; Rothenfusser, S.; Gilliet, M.; Ruzicka, T.; Wolf, R. Th17 micro-milieu regulates NLRP1-dependent caspase-5 activity in skin autoinflammation. PLoS ONE 2017, 12. [Google Scholar] [CrossRef] [PubMed]
- King, L.E.; Gates, R.E.; Stoscheck, C.M.; Nanney, L.B. The EGF/TGF α receptor in skin. J. Investig. Dermatol. 1990, 94, 164S–170S. Available online: http://www.ncbi.nlm.nih.gov/pubmed/2191051 (accessed on 29 December 2019). [CrossRef]
- Manczinger, M.; Kemény, L. Novel factors in the pathogenesis of psoriasis and potential drug candidates are found with systems biology approach. PLoS ONE 2013, 8. [Google Scholar] [CrossRef]
- Chiricozzi, A.; Romanelli, P.; Volpe, E.; Borsellino, G.; Romanelli, M. Scanning the immunopathogenesis of psoriasis. Int. J. Mol. Sci. 2018, 19, 179. [Google Scholar] [CrossRef]
- Chowdhari, S.; Saini, N. hsa-miR-4516 Mediated Downregulation of STAT3/CDK6/UBE2N Plays a Role in PUVA Induced Apoptosis in Keratinocytes. J. Cell. Physiol. 2014, 229, 1630–1638. [Google Scholar] [CrossRef]
- Pollard, K.M.; Cauvi, D.M.; Toomey, C.B.; Morris, K.V.; Kono, D.H. Interferon-γ and systemic autoimmunity. Discov. Med. 2013, 16, 123–131. Available online: http://www.ncbi.nlm.nih.gov/pubmed/23998448%0Ahttp://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=PMC3934799 (accessed on 11 January 2020).
- Fuentesduculan, J.; Suarezfarinas, M.; Zaba, L.C.; Nograles, K.E.; Pierson, K.C.; Mitsui, H.; Pensabene, C.A.; Kzhyshkowska, J.; Krueger, J.G.; Lowes, M.A. A subpopulation of CD163-positive macrophages is classically activated in psoriasis. J. Investig. Dermatol. 2010, 130, 2412–2422. [Google Scholar] [CrossRef]
- Duan, H.; Koga, T.; Kohda, F.; Hara, H.; Urabe, K.; Furue, M. Interleukin-8-positive neutrophils in psoriasis. J. Dermatol. Sci. 2001, 26, 119–124. [Google Scholar] [CrossRef]
- Russo, R.C.; Garcia, C.C.; Teixeira, M.M.; Amaral, F.A. The CXCL8/IL-8 chemokine family and its receptors in inflammatory diseases. Expert Rev. Clin. Immunol. 2014, 10, 593–619. [Google Scholar] [CrossRef] [PubMed]
- Yucel-Lindberg, T.; Brunius, G. Epidermal growth factor synergistically enhances interleukin-8 production in human gingival fibroblasts stimulated with interleukin-1β. Arch. Oral Biol. 2006, 51, 892–898. [Google Scholar] [CrossRef] [PubMed]
- Johnston, A.; Xing, X.; Swindell, W.R.; Kochkodan, J.J.; Riblett, M.; Nair, R.P.; Stuart, P.E.; Ding, J.; Voorhees, J.J.; Elder, J.T.; et al. Susceptibility-associated genetic variation at IL12B enhances Th1 polarization in psoriasis. Hum. Mol. Genet. 2013, 22, 1807–1815. [Google Scholar] [CrossRef] [PubMed]
- Chang, J.H.; Xiao, Y.; Hu, H.; Jin, J.; Yu, J.; Zhou, X.; Wu, X.; Johnson, H.M.; Akira, S.; Pasparakis, M.; et al. Ubc13 maintains the suppressive function of regulatory T cells and prevents their conversion into effector-like T cells HHS Public Access. Nat. Immunol. 2012, 13, 481–490. [Google Scholar] [CrossRef] [PubMed]
- Kawai, T.; Akira, S. Toll-like receptor downstream signaling. Arthritis Res. Ther. 2005, 7, 12–19. [Google Scholar] [CrossRef]
- Cretu, D.; Prassas, I.; Saraon, P.; Batruch, I.; Gandhi, R.; Diamandis, E.P.; Chandran, V. Identification of psoriatic arthritis mediators in synovial fluid by quantitative mass spectrometry. Clin. Proteom. 2014. [Google Scholar] [CrossRef]
- Hald, A.; Andrés, R.M.; Salskov-Iversen, M.L.; Kjellerup, R.B.; Iversen, L.; Johansen, C. STAT1 expression and activation is increased in lesional psoriatic skin. Br. J. Dermatol. 2013, 168, 302–310. [Google Scholar] [CrossRef]
- Park, Y.K.; Jang, B.C. UVB-induced anti-survival and pro-apoptotic effects on HaCaT human keratinocytes via caspase- and PKC-dependent downregulation of PKB, HIAP-1, Mcl-1, XIAP and ER stress. Int. J. Mol. Med. 2014, 33, 695–702. [Google Scholar] [CrossRef]
- Sundarrajan, S.; Lulu, S.; Arumugam, M. Insights into protein interaction networks reveal non-receptor kinases as significant druggable targets for psoriasis. Gene 2015, 566, 138–147. [Google Scholar] [CrossRef]
- Fargnoli, A.S.; Mu, A.; Katz, M.G.; Williams, R.D.; Margulies, K.B.; Weiner, D.B.; Yang, S.; Bridges, C.R. Anti-inflammatory loaded poly-lactic glycolic acid nanoparticle formulations to enhance myocardial gene transfer: An in-vitro assessment of a drug/gene combination therapeutic approach for direct injection. J. Transl. Med. 2014, 12, 171. [Google Scholar] [CrossRef]
- Weber, F.; Shen, L.; Aldred, M.A.; Morrison, C.; Frilling, A.; Saji, M.; Schuppert, F.; Broelsch, C.E.; Ringel, M.D.; Eng, C. Genetic Classification of Benign and Malignant Thyroid Follicular Neoplasia Based on a Three-Gene Combination. J. Clin. Endocrinol. Metab. 2005, 90, 2512–2521. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Micro-Level Network Metrics | Min Value (All Genes) | Max Value (All Genes) | Min Value (Extracted Genes) | Max Value (Extracted Genes) | No. of Extracted Genes |
|---|---|---|---|---|---|
| Degree | 1 | 182 | 50 | 182 | 113 |
| Stress | 0 | 2,118,086 | 200,000 | 2,118,086 | 69 |
| Eigenvector | 0 | 0.13 | 0.05 | 0.13 | 86 |
| a. | b. | c. | ||||
|---|---|---|---|---|---|---|
| Rank | Gene Symbol | Degree | Gene Symbol | Betweenness | Gene Symbol | Stress |
| 1 | CCNB1 | 92 | CCNB1 | 0.05 | CCNB1 | 15,636 |
| 2 | CDC20 | 91 | CDC20 | 0.04 | MKI67 | 13,412 |
| 3 | CDK1 | 90 | MKI67 | 0.04 | CDK1 | 10,020 |
| 4 | CCNA2 | 87 | FOXM1 | 0.03 | CCNA2 | 9618 |
| 5 | FOXM1 | 8920 | ||||
| 6 | CDC20 | 8732 | ||||
| a. | b. | c. | d. | |||||
|---|---|---|---|---|---|---|---|---|
| Rank | Gene Symbol | Degree | Gene Symbol | Eigenvector | Gene Symbol | Stress | Gene Symbol | Betweenness |
| 1 | STAT3 | 43 | H2AFX | 0.05 | STAT3 | 29,876 | EGF | 0.07913 |
| 2 | H2AFX | 43 | MCL1 | 0.018 | EGF | 27,610 | STAT3 | 0.06636 |
| 3 | EGF | 40 | STAT3 | 0.015 | MAPK14 | 10,910 | MAPK14 | 0.044914 |
| 4 | IL1B | 36 | EGF | 0.014 | IL1B | 8518 | ||
| 5 | STAT1 | 34 | UBE2N | 0.012 | ||||
| 6 | IFNG | 31 | MAPK14 | 0.007 | ||||
| 7 | CXCL8 | 31 | ||||||
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Naderi, R.; Saadati Mollaei, H.; Elofsson, A.; Hosseini Ashtiani, S. Using Micro- and Macro-Level Network Metrics Unveils Top Communicative Gene Modules in Psoriasis. Genes 2020, 11, 914. https://doi.org/10.3390/genes11080914
Naderi R, Saadati Mollaei H, Elofsson A, Hosseini Ashtiani S. Using Micro- and Macro-Level Network Metrics Unveils Top Communicative Gene Modules in Psoriasis. Genes. 2020; 11(8):914. https://doi.org/10.3390/genes11080914
Chicago/Turabian StyleNaderi, Reyhaneh, Homa Saadati Mollaei, Arne Elofsson, and Saman Hosseini Ashtiani. 2020. "Using Micro- and Macro-Level Network Metrics Unveils Top Communicative Gene Modules in Psoriasis" Genes 11, no. 8: 914. https://doi.org/10.3390/genes11080914
APA StyleNaderi, R., Saadati Mollaei, H., Elofsson, A., & Hosseini Ashtiani, S. (2020). Using Micro- and Macro-Level Network Metrics Unveils Top Communicative Gene Modules in Psoriasis. Genes, 11(8), 914. https://doi.org/10.3390/genes11080914