Genomic Islands in Mycoplasmas

 ,
,  ,
,
Abstract
1. Common and Specific Features of Mycoplasma Genomes
2. Genomic Islands and the Mycoplasma Flexible Gene Pool
3. Mycoplasma Integrative Conjugative Elements
4. Mycoplasma Viruses and Prophages
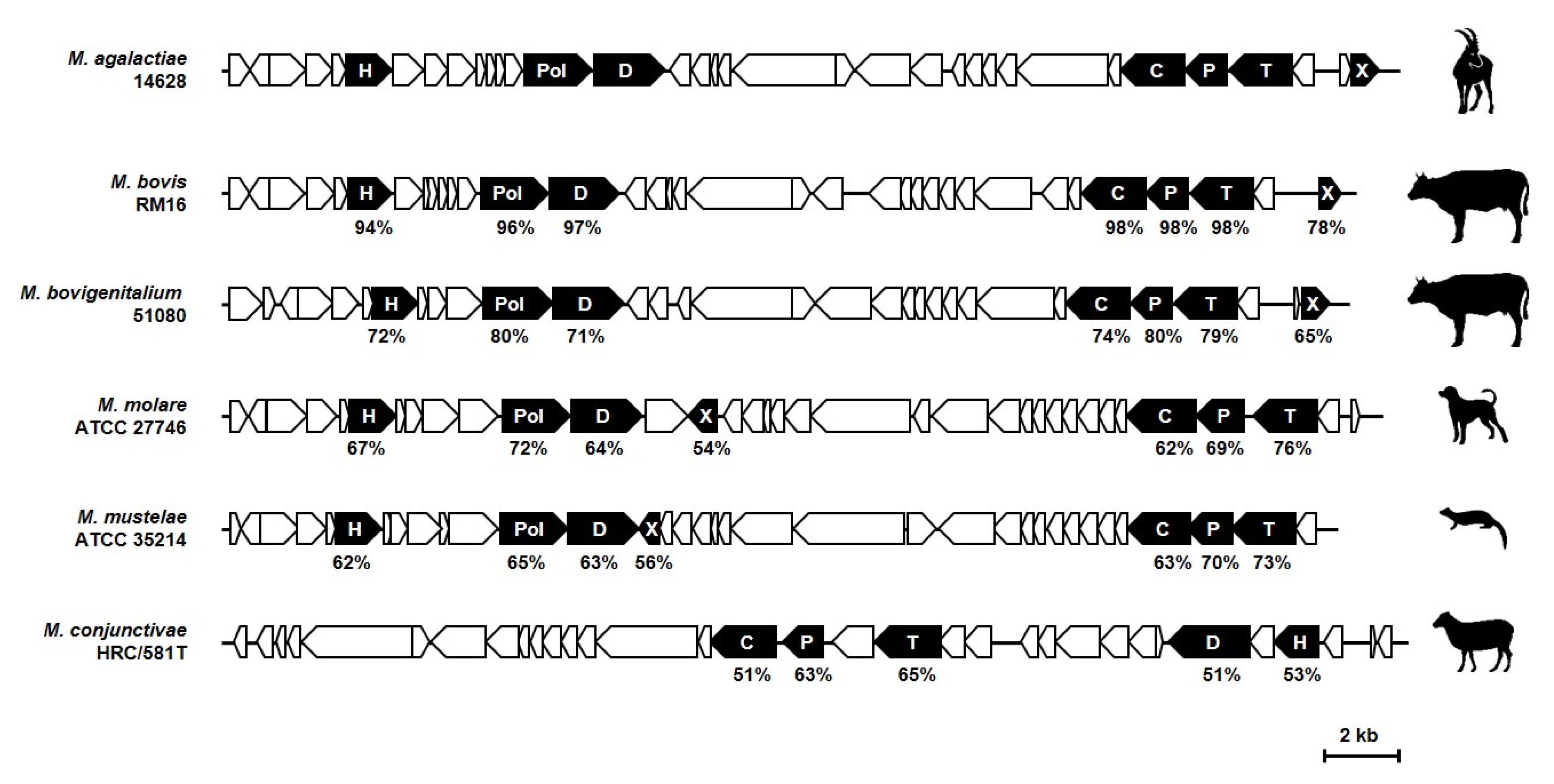
5. Distribution of MICE and Prophages in Mycoplasma Species
6. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Citti, C.; Blanchard, A. Mycoplasmas and their host: Emerging and re-emerging minimal pathogens. Trends Microbiol. 2013, 21, 196–203. [Google Scholar] [CrossRef]
- Woese, C.R.; Maniloff, J.; Zablen, L.B. Phylogenetic analysis of the mycoplasmas. Proc. Natl. Acad. Sci. USA 1980, 77, 494–498. [Google Scholar] [CrossRef] [PubMed]
- Ciccarelli, F.D.; Doerks, T.; von Mering, C.; Creevey, C.J.; Snel, B.; Bork, P. Toward automatic reconstruction of a highly resolved tree of life. Science 2006, 311, 1283–1287. [Google Scholar] [CrossRef] [PubMed]
- Sirand-Pugnet, P.; Citti, C.; Barré, A.; Blanchard, A. Evolution of mollicutes: Down a bumpy road with twists and turns. Res. Microbiol. 2007, 158, 754–766. [Google Scholar] [CrossRef]
- Fraser, C.M.; Gocayne, J.D.; White, O.; Adams, M.D.; Clayton, R.A.; Fleischmann, R.D.; Bult, C.J.; Kerlavage, A.R.; Sutton, G.; Kelley, J.M.; et al. The minimal gene complement of Mycoplasma genitalium. Science 1995, 270, 397–403. [Google Scholar] [CrossRef] [PubMed]
- Mushegian, A.R.; Koonin, E.V. A minimal gene set for cellular life derived by comparison of complete bacterial genomes. Proc. Natl. Acad. Sci. USA 1996, 93, 10268–10273. [Google Scholar] [CrossRef]
- Hutchison, C.A.; Peterson, S.N.; Gill, S.R.; Cline, R.T.; White, O.; Fraser, C.M.; Smith, H.O.; Venter, J.C. Global transposon mutagenesis and a minimal mycoplasma genome. Science 1999, 286, 2165–2169. [Google Scholar] [CrossRef]
- Dybvig, K.; Lao, P.; Jordan, D.S.; Simmons, W.L. Fewer essential genes in mycoplasmas than previous studies suggest. FEMS Microbiol. Lett. 2010, 311, 51–55. [Google Scholar] [CrossRef][Green Version]
- Sirand-Pugnet, P.; Lartigue, C.; Marenda, M.; Jacob, D.; Barré, A.; Barbe, V.; Schenowitz, C.; Mangenot, S.; Couloux, A.; Segurens, B.; et al. Being pathogenic, plastic, and sexual while living with a nearly minimal bacterial genome. PLoS Genet. 2007, 3, e75. [Google Scholar] [CrossRef]
- Dordet-Frisoni, E.; Sagné, E.; Baranowski, E.; Breton, M.; Nouvel, L.X.; Blanchard, A.; Marenda, M.S.; Tardy, F.; Sirand-Pugnet, P.; Citti, C. Chromosomal transfers in mycoplasmas: When minimal genomes go mobile. mBio 2014, 5, e01958. [Google Scholar] [CrossRef]
- Dordet-Frisoni, E.; Faucher, M.; Sagné, E.; Baranowski, E.; Tardy, F.; Nouvel, L.X.; Citti, C. Mycoplasma chromosomal transfer: A distributive, conjugative process creating an infinite variety of mosaic genomes. Front. Microbiol. 2019, 10, 2441. [Google Scholar] [CrossRef] [PubMed]
- Dobrindt, U.; Hochhut, B.; Hentschel, U.; Hacker, J. Genomic islands in pathogenic and environmental microorganisms. Nat. Rev. Microbiol. 2004, 2, 414–424. [Google Scholar] [CrossRef] [PubMed]
- Razin, S.; Yogev, D.; Naot, Y. Molecular biology and pathogenicity of mycoplasmas. Microbiol. Mol. Biol. Rev. 1998, 62, 1094–1156. [Google Scholar] [CrossRef] [PubMed]
- Citti, C.; Dordet-Frisoni, E.; Nouvel, L.X.; Kuo, C.H.; Baranowski, E. Horizontal gene transfers in mycoplasmas (Mollicutes). Curr. Issues Mol. Biol. 2018, 29, 3–22. [Google Scholar] [CrossRef]
- Lo, W.-S.; Gasparich, G.E.; Kuo, C.-H. Convergent evolution among ruminant-pathogenic mycoplasma involved extensive gene content changes. Genome Biol. Evol. 2018, 10, 2130–2139. [Google Scholar] [CrossRef]
- Bertelli, C.; Laird, M.R.; Williams, K.P.; Lau, B.Y.; Hoad, G.; Winsor, G.L.; Brinkman, F.S. IslandViewer 4: Expanded prediction of genomic islands for larger-scale datasets. Nucleic Acids Res. 2017, 45, W30–W35. [Google Scholar] [CrossRef]
- Nouvel, L.X.; Sirand-Pugnet, P.; Marenda, M.S.; Sagné, E.; Barbe, V.; Mangenot, S.; Schenowitz, C.; Jacob, D.; Barré, A.; Claverol, S.; et al. Comparative genomic and proteomic analyses of two Mycoplasma agalactiae strains: Clues to the macro- and micro-events that are shaping mycoplasma diversity. BMC Genom. 2010, 11, 86. [Google Scholar] [CrossRef]
- Nouvel, L.-X.; Marenda, M.; Sirand-Pugnet, P.; Sagné, E.; Glew, M.; Mangenot, S.; Barbe, V.; Barré, A.; Claverol, S.; Citti, C. Occurrence, plasticity, and evolution of the vpma gene family, a genetic system devoted to high-frequency surface variation in Mycoplasma agalactiae. J. Bacteriol. 2009, 191, 4111–4121. [Google Scholar] [CrossRef]
- Nouvel, L.-X.; Marenda, M.S.; Glew, M.D.; Sagné, E.; Giammarinaro, P.; Tardy, F.; Poumarat, F.; Rosengarten, R.; Citti, C. Molecular typing of Mycoplasma agalactiae: Tracing European-wide genetic diversity and an endemic clonal population. Comp. Immunol. Microbiol. Infect. Dis. 2012, 35, 487–496. [Google Scholar] [CrossRef]
- Glew, M.D.; Marenda, M.; Rosengarten, R.; Citti, C. Surface diversity in Mycoplasma agalactiae is driven by site-specific DNA inversions within the vpma multigene locus. J. Bacteriol. 2002, 184, 5987–5998. [Google Scholar] [CrossRef]
- Chopra-Dewasthaly, R.; Spergser, J.; Zimmermann, M.; Citti, C.; Jechlinger, W.; Rosengarten, R. Vpma phase variation is important for survival and persistence of Mycoplasma agalactiae in the immunocompetent host. PLoS Pathog. 2017, 13, e1006656. [Google Scholar] [CrossRef]
- Glew, M.D.; Papazisi, L.; Poumarat, F.; Bergonier, D.; Rosengarten, R.; Citti, C. Characterization of a multigene family undergoing high-frequency DNA rearrangements and coding for abundant variable surface proteins in Mycoplasma agalactiae. Inf. Immun. 2000, 68, 4539–4548. [Google Scholar] [CrossRef] [PubMed]
- Fleury, B.; Bergonier, D.; Berthelot, X.; Peterhans, E.; Frey, J.; Vilei, E.M. Characterization of P40, a cytadhesin of Mycoplasma agalactiae. Infect. Immun. 2002, 70, 5612–5621. [Google Scholar] [CrossRef] [PubMed]
- Lysnyansky, I.; Sachse, K.; Rosenbusch, R.; Levisohn, S.; Yogev, D. The vsp locus of Mycoplasma bovis: Gene organization and structural features. J. Bacteriol. 1999, 181, 5734–5741. [Google Scholar] [CrossRef] [PubMed]
- Marenda, M.; Barbe, V.; Gourgues, G.; Mangenot, S.; Sagne, E.; Citti, C. A new integrative conjugative element occurs in Mycoplasma agalactiae as chromosomal and free circular forms. J. Bacteriol. 2006, 188, 4137–4141. [Google Scholar] [CrossRef] [PubMed]
- Calcutt, M.J.; Lewis, M.S.; Wise, K.S. Molecular genetic analysis of ICEF, an integrative conjugal element that is present as a repetitive sequence in the chromosome of Mycoplasma fermentans PG18. J. Bacteriol. 2002, 184, 6929–6941. [Google Scholar] [CrossRef]
- Alvarez-Martinez, C.E.; Christie, P.J. Biological diversity of prokaryotic type IV secretion systems. Microbiol. Mol. Biol. Rev. 2009, 73, 775–808. [Google Scholar] [CrossRef]
- Liu, M.; Li, X.; Xie, Y.; Bi, D.; Sun, J.; Li, J.; Tai, C.; Deng, Z.; Ou, H.-Y. ICEberg 2.0: An updated database of bacterial integrative and conjugative elements. Nucleic Acids Res. 2019, 47, D660–D665. [Google Scholar] [CrossRef]
- Guérillot, R.; Siguier, P.; Gourbeyre, E.; Chandler, M.; Glaser, P. The diversity of prokaryotic DDE transposases of the mutator superfamily, insertion specificity, and association with conjugation machineries. Genome Biol. Evol. 2014, 6, 260–272. [Google Scholar] [CrossRef]
- Tardy, F.; Mick, V.; Dordet-Frisoni, E.; Marenda, M.S.; Sirand-Pugnet, P.; Blanchard, A.; Citti, C. Integrative conjugative elements are widespread in field isolates of Mycoplasma species pathogenic for ruminants. Appl. Environ. Microbiol. 2015, 81, 1634–1643. [Google Scholar] [CrossRef]
- Dordet Frisoni, E.; Marenda, M.S.; Sagné, E.; Nouvel, L.X.; Guérillot, R.; Glaser, P.; Blanchard, A.; Tardy, F.; Sirand-Pugnet, P.; Baranowski, E.; et al. ICEA of Mycoplasma agalactiae: A new family of self-transmissible integrative elements that confers conjugative properties to the recipient strain. Mol. Microbiol. 2013, 89, 1226–1239. [Google Scholar] [CrossRef] [PubMed]
- Calderon-Copete, S.P.; Wigger, G.; Wunderlin, C.; Schmidheini, T.; Frey, J.; Quail, M.A.; Falquet, L. The Mycoplasma conjunctivae genome sequencing, annotation and analysis. BMC Bioinform. 2009, 10, S7. [Google Scholar] [CrossRef] [PubMed]
- Vasconcelos, A.T.R.; Ferreira, H.B.; Bizarro, C.V.; Bonatto, S.L.; Carvalho, M.O.; Pinto, P.M.; Almeida, D.F.; Almeida, L.G.P.; Almeida, R.; Alves-Filho, L.; et al. Swine and poultry pathogens: The complete genome sequences of two strains of Mycoplasma hyopneumoniae and a strain of Mycoplasma synoviae. J. Bacteriol. 2005, 187, 5568–5577. [Google Scholar] [CrossRef] [PubMed]
- Liu, W.; Feng, Z.; Fang, L.; Zhou, Z.; Li, Q.; Li, S.; Luo, R.; Wang, L.; Chen, H.; Shao, G.; et al. Complete genome sequence of Mycoplasma hyopneumoniae strain 168. J. Bacteriol. 2011, 193, 1016–1017. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Minion, F.C.; Lefkowitz, E.J.; Madsen, M.L.; Cleary, B.J.; Swartzell, S.M.; Mahairas, G.G. The genome sequence of Mycoplasma hyopneumoniae strain 232, the agent of swine Mycoplasmosis. J. Bacteriol. 2004, 186, 7123–7133. [Google Scholar] [CrossRef]
- Rechnitzer, H.; Brzuszkiewicz, E.; Strittmatter, A.; Liesegang, H.; Lysnyansky, I.; Daniel, R.; Gottschalk, G.; Rottem, S. Genomic features and insights into the biology of Mycoplasma fermentans. Microbiology 2011, 157, 760–773. [Google Scholar] [CrossRef]
- Shu, H.-W.; Liu, T.-T.; Chan, H.-I.; Liu, Y.-M.; Wu, K.-M.; Shu, H.-Y.; Tsai, S.-F.; Hsiao, K.-J.; Hu, W.S.; Ng, W.V. Genome sequence of the repetitive-sequence-rich Mycoplasma fermentans strain M64. J. Bacteriol. 2011, 193, 4302–4303. [Google Scholar] [CrossRef]
- Wise, K.S.; Calcutt, M.J.; Foecking, M.F.; Röske, K.; Madupu, R.; Methé, B.A. Complete genome sequence of Mycoplasma bovis type strain PG45 (ATCC 25523). Inf. Immun. 2011, 79, 982–983. [Google Scholar] [CrossRef]
- Li, Y.; Zheng, H.; Liu, Y.; Jiang, Y.; Xin, J.; Chen, W.; Song, Z. The complete genome sequence of Mycoplasma bovis strain Hubei-1. PLoS ONE 2011, 6, e20999. [Google Scholar] [CrossRef]
- Qi, J.; Guo, A.; Cui, P.; Chen, Y.; Mustafa, R.; Ba, X.; Hu, C.; Bai, Z.; Chen, X.; Shi, L.; et al. Comparative geno-plasticity analysis of Mycoplasma bovis HB0801 (Chinese Isolate). PLoS ONE 2012, 7, e38239. [Google Scholar] [CrossRef]
- Manso-Silván, L.; Tardy, F.; Baranowski, E.; Barré, A.; Blanchard, A.; Breton, M.; Couture, C.; Citti, C.; Dordet-Frisoni, E.; Dupuy, V.; et al. Draft genome sequences of Mycoplasma alkalescens, Mycoplasma arginini, and Mycoplasma bovigenitalium, three species with equivocal pathogenic status for cattle. Genome Announc. 2013, 1, e00348-13. [Google Scholar] [CrossRef]
- Meygret, A.; Peuchant, O.; Dordet-Frisoni, E.; Sirand-Pugnet, P.; Citti, C.; Bébéar, C.; Béven, L.; Pereyre, S. High prevalence of integrative and conjugative elements encoding transcription activator-like effector repeats in Mycoplasma hominis. Front. Microbiol. 2019, 10, 2385. [Google Scholar] [CrossRef] [PubMed]
- Calcutt, M.J.; Foecking, M.F. An Excision-Competent and Exogenous Mosaic Transposon Harbors the tetM Gene in Multiple Mycoplasma hominis Lineages. Antimicrob. Agents Chemother. 2015, 59, 6665–6666. [Google Scholar] [CrossRef] [PubMed]
- Thiaucourt, F.; Manso-Silvan, L.; Salah, W.; Barbe, V.; Vacherie, B.; Jacob, D.; Breton, M.; Dupuy, V.; Lomenech, A.M.; Blanchard, A.; et al. Mycoplasma mycoides, from “mycoides Small Colony” to “capri”. A microevolutionary perspective. BMC Genom. 2011, 12, 114. [Google Scholar] [CrossRef]
- Dupuy, V.; Sirand-Pugnet, P.; Baranowski, E.; Barré, A.; Breton, M.; Couture, C.; Dordet-Frisoni, E.; Gaurivaud, P.; Jacob, D.; Lemaitre, C.; et al. Complete genome sequence of Mycoplasma putrefaciens strain 9231, one of the agents of contagious Agalactia in goats. Genome Announc. 2013, 1, e00354-13. [Google Scholar] [CrossRef]
- Baranowski, E.; Dordet-Frisoni, E.; Sagné, E.; Hygonenq, M.-C.; Pretre, G.; Claverol, S.; Fernandez, L.; Nouvel, L.X.; Citti, C. The Integrative Conjugative Element (ICE) of Mycoplasma agalactiae: Key elements involved in horizontal dissemination and influence of coresident ICEs. mBio 2018, 9, e00873-18. [Google Scholar] [CrossRef]
- Faucher, M.; Nouvel, L.-X.; Dordet-Frisoni, E.; Sagné, E.; Baranowski, E.; Hygonenq, M.-C.; Marenda, M.-S.; Tardy, F.; Citti, C. Mycoplasmas under experimental antimicrobial selection: The unpredicted contribution of horizontal chromosomal transfer. PLoS Genet. 2019, 15, e1007910. [Google Scholar] [CrossRef]
- Jores, J.; Ma, L.; Ssajjakambwe, P.; Schieck, E.; Liljander, A.; Chandran, S.; Stoffel, M.H.; Cippa, V.; Arfi, Y.; Assad-Garcia, N.; et al. Removal of a subset of non-essential genes fully attenuates a highly virulent Mycoplasma strain. Front. Microbiol. 2019, 10, 664. [Google Scholar] [CrossRef]
- Gourlay, R.N. Isolation of a virus infecting a strain of Mycoplasma laidlawii. Nature 1970, 225, 1165. [Google Scholar] [CrossRef]
- Gourlay, R.N. Mycoplasma viruses: Isolation, physicochemical, and biological properties. CRC Crit. Rev. Microbiol. 1974, 3, 315–331. [Google Scholar] [CrossRef]
- Maniloff, J.; Haberer, K.; Gourlay, R.N.; Das, J.; Cole, R. Mycoplasma viruses. Intervirology 1982, 18, 177–188. [Google Scholar] [CrossRef]
- Maniloff, J. Mycoplasma viruses. Crit. Rev. Microbiol. 1988, 15, 339–389. [Google Scholar] [CrossRef] [PubMed]
- Dybvig, K.; Tu, A.-H.; Clapper, B. Mycoplasma phages. Phages 2005, 223–237. [Google Scholar] [CrossRef]
- Howard, C.J.; Gourlay, R.N.; Wyld, S.G. Isolation of a virus, MVBr1, from Mycoplasma bovirhinis. FEMS Microbiol. Lett. 1980, 7, 163–165. [Google Scholar] [CrossRef]
- Gourlay, R.N.; Wyld, S.G.; Garwes, D.J. Some properties of mycoplasma virus Br 1. Arch. Virol. 1983, 75, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Tardy, F.; Baranowski, E.; Nouvel, L.-X.; Mick, V.; Manso-Silvàn, L.; Thiaucourt, F.; Thébault, P.; Breton, M.; Sirand-Pugnet, P.; Blanchard, A.; et al. Emergence of atypical Mycoplasma agalactiae strains harboring a new prophage and associated with an alpine wild ungulate mortality episode. Appl. Environ. Microbiol. 2012, 78, 4659–4668. [Google Scholar] [CrossRef]
- Voelker, L.L.; Weaver, K.E.; Ehle, L.J.; Washburn, L.R. Association of lysogenic bacteriophage MAV1 with virulence of Mycoplasma arthritidis. Infect. Immun. 1995, 63, 4016–4023. [Google Scholar] [CrossRef]
- Voelker, L.L.; Dybvig, K. Characterization of the lysogenic bacteriophage MAV1 from Mycoplasma arthritidis. J. Bacteriol. 1998, 180, 5928–5931. [Google Scholar] [CrossRef]
- Voelker, L.L.; Dybvig, K. Sequence analysis of the Mycoplasma arthritidis bacteriophage MAV1 genome identifies the putative virulence factor. Gene 1999, 233, 101–107. [Google Scholar] [CrossRef]
- Hata, E.; Nagai, K.; Murakami, K. Complete genome sequence of Mycoplasma bovirhinis strain HAZ141_2 from bovine nasal discharge in Japan. Genome Announc. 2017, 5. [Google Scholar] [CrossRef]
- Chen, S.; Hao, H.; Zhao, P.; Liu, Y.; Chu, Y. Genome-wide analysis of Mycoplasma bovirhinis GS01 reveals potential virulence factors and Phylogenetic relationships. G3 Genes Genomes Genet. 2018, 8, 1417–1424. [Google Scholar] [CrossRef] [PubMed]
- Zhu, X.; Dordet-Frisoni, E.; Gillard, L.; Ba, A.; Hygonenq, M.-C.; Sagné, E.; Nouvel, L.X.; Maillard, R.; Assié, S.; Guo, A.; et al. Extracellular DNA: A nutritional trigger of Mycoplasma bovis Cytotoxicity. Front. Microbiol. 2019, 10, 2753. [Google Scholar] [CrossRef]
- Röske, K.; Calcutt, M.J.; Wise, K.S. The Mycoplasma fermentans prophage phiMFV1: Genome organization, mobility and variable expression of an encoded surface protein. Mol. Microbiol. 2004, 52, 1703–1720. [Google Scholar] [CrossRef]
- Calcutt, M.J.; Foecking, M.F. Analysis of the complete Mycoplasma hominis LBD-4 genome sequence reveals strain-variable Prophage insertion and distinctive repeat-containing surface protein arrangements. Genome Announc. 2015, 3. [Google Scholar] [CrossRef] [PubMed]
- Gourlay, R.N.; Wyld, S.G.; Poulton, M.E. Some characteristics of mycoplasma virus Hr 1, isolated from and infecting Mycoplasma hyorhinis. Brief report. Arch. Virol. 1983, 77, 81–85. [Google Scholar] [CrossRef] [PubMed]
- Bumgardner, E.A.; Kittichotirat, W.; Bumgarner, R.E.; Lawrence, P.K. Comparative genomic analysis of seven Mycoplasma hyosynoviae strains. Microbiologyopen 2015, 4, 343–359. [Google Scholar] [CrossRef]
- Dybvig, K.; Liss, A.; Alderete, J.; Cole, R.M.; Cassell, G.H. Isolation of a virus from Mycoplasma pulmonis. ISR J. Med. Sci. 1987, 23, 418–422. [Google Scholar] [PubMed]
- Zou, N.; Park, K.; Dybvig, K. Mycoplasma virus P1 has a linear, double-stranded DNA genome with inverted terminal repeats. Plasmid 1995, 33, 41–49. [Google Scholar] [CrossRef]
- Tu, A.H.; Voelker, L.L.; Shen, X.; Dybvig, K. Complete nucleotide sequence of the mycoplasma virus P1 genome. Plasmid 2001, 45, 122–126. [Google Scholar] [CrossRef]
- Roachford, O.S.E.; Nelson, K.E.; Mohapatra, B.R. Comparative genomics of four Mycoplasma species of the human urogenital tract: Analysis of their core genomes and virulence genes. Int. J. Med. Microbiol. 2017, 307, 508–520. [Google Scholar] [CrossRef]
- Hurwitz, B.L.; Ponsero, A.; Thornton, J.; U’Ren, J.M. Phage hunters: Computational strategies for finding phages in large-scale ‘omics datasets. Virus Res. 2018, 244, 110–115. [Google Scholar] [CrossRef] [PubMed]
- Canchaya, C.; Proux, C.; Fournous, G.; Bruttin, A.; Brüssow, H. Prophage genomics. Microbiol. Mol. Biol. Rev. 2003, 67, 238–276. [Google Scholar] [CrossRef] [PubMed]
- Clapper, B.; Tu, A.-H.T.; Elgavish, A.; Dybvig, K. The vir gene of bacteriophage MAV1 confers resistance to phage infection on Mycoplasma arthritidis. J. Bacteriol. 2004, 186, 5715–5720. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Tu, A.-H.T.; Lindsey, J.R.; Schoeb, T.R.; Elgavish, A.; Yu, H.; Dybvig, K. Role of bacteriophage MAV1 as a mycoplasmal virulence factor for the development of arthritis in mice and rats. J. Infect. Dis. 2002, 186, 432–435. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Clapper, B.; Tu, A.-H.T.; Simmons, W.L.; Dybvig, K. Bacteriophage MAV1 is not associated with virulence of Mycoplasma arthritidis. Infect. Immun. 2004, 72, 7322–7325. [Google Scholar] [CrossRef]
- Arndt, D.; Grant, J.R.; Marcu, A.; Sajed, T.; Pon, A.; Liang, Y.; Wishart, D.S. PHASTER: A better, faster version of the PHAST phage search tool. Nucleic Acids Res. 2016, 44, W16–W21. [Google Scholar] [CrossRef]
- Thomas, C.M.; Nielsen, K.M. Mechanisms of, and barriers to, horizontal gene transfer between bacteria. Nat. Rev. Microbiol. 2005, 3, 711–721. [Google Scholar] [CrossRef]
- Ipoutcha, T.; Tsarmpopoulos, I.; Talenton, V.; Gaspin, C.; Moisan, A.; Walker, C.A.; Brownlie, J.; Blanchard, A.; Thebault, P.; Sirand-Pugnet, P. Multiple origins and specific evolution of CRISPR/Cas9 systems in minimal bacteria (Mollicutes). Front. Microbiol. 2019, 10, 2701. [Google Scholar] [CrossRef]
- Popa, O.; Dagan, T. Trends and barriers to lateral gene transfer in prokaryotes. Curr. Opin. Microbiol. 2011, 14, 615–623. [Google Scholar] [CrossRef]







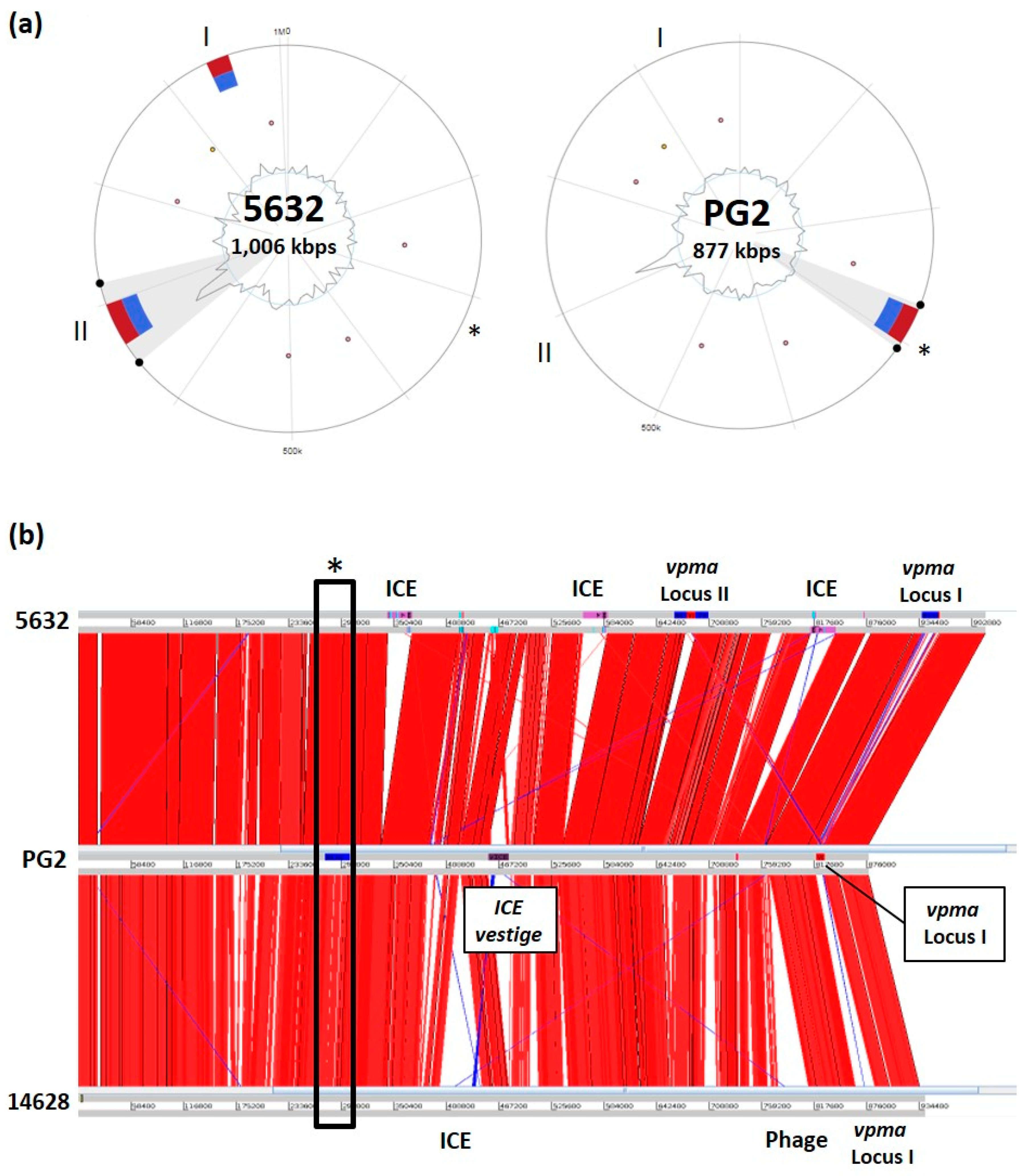{kind=link}
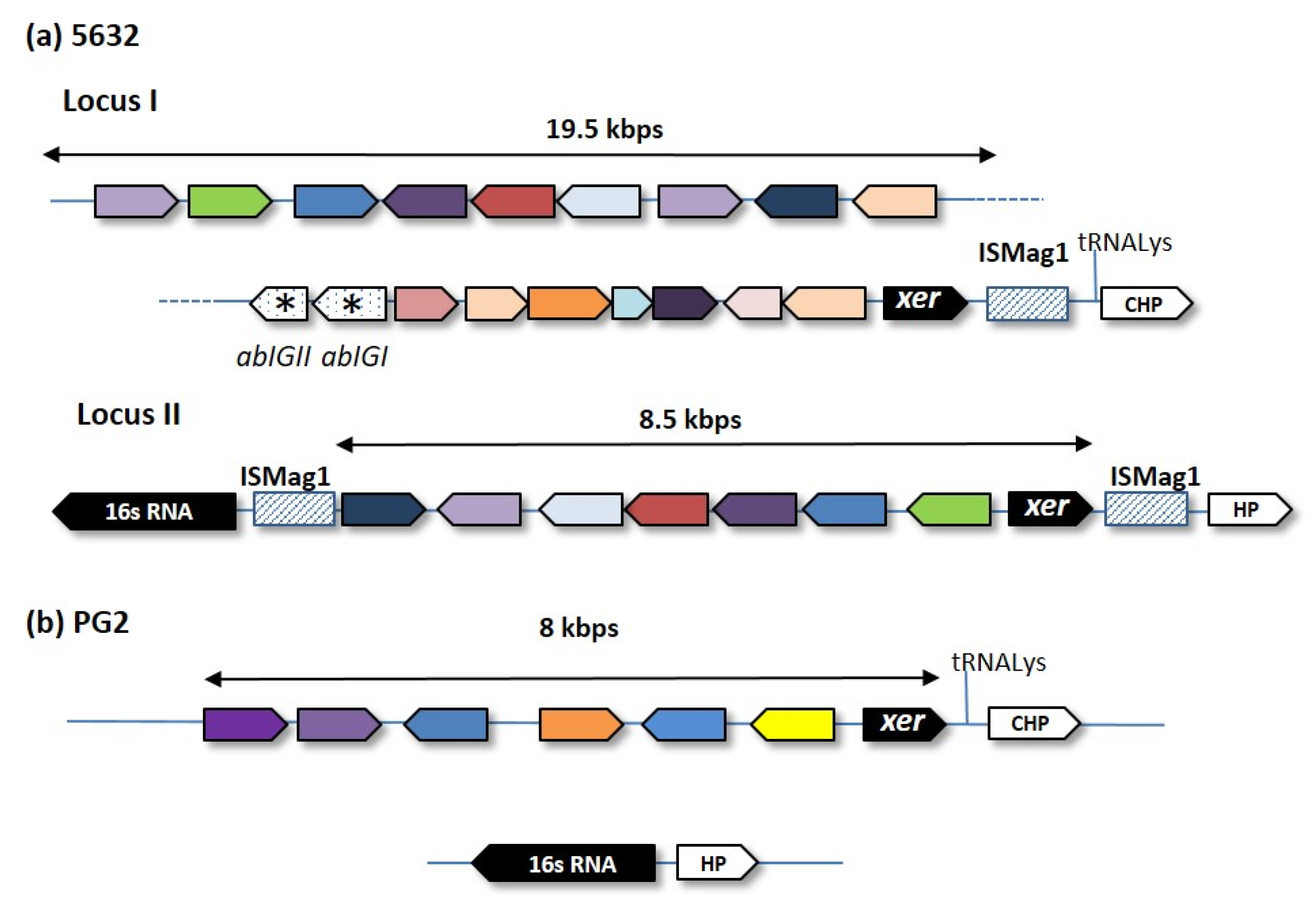{kind=link}
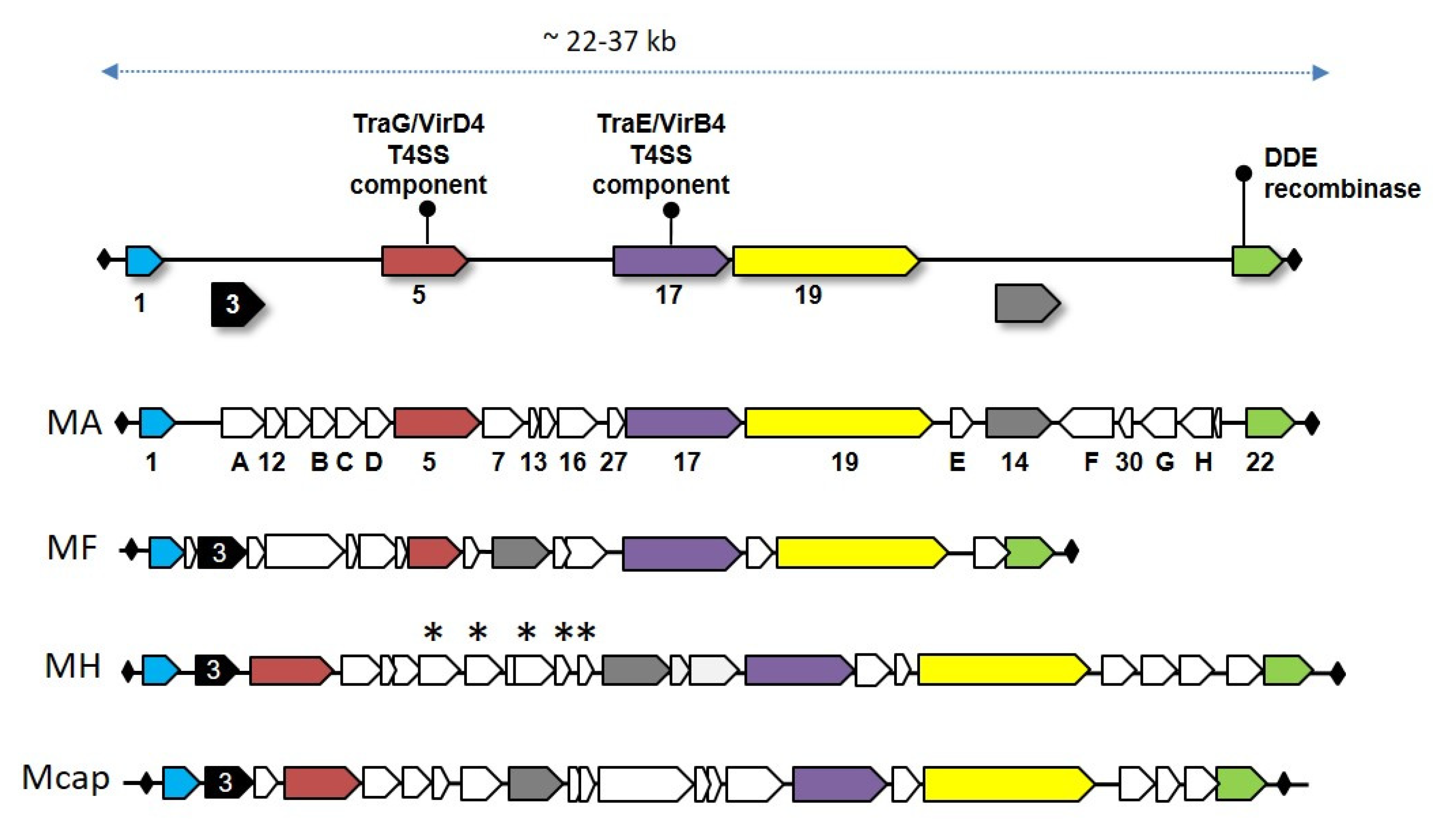{kind=link}
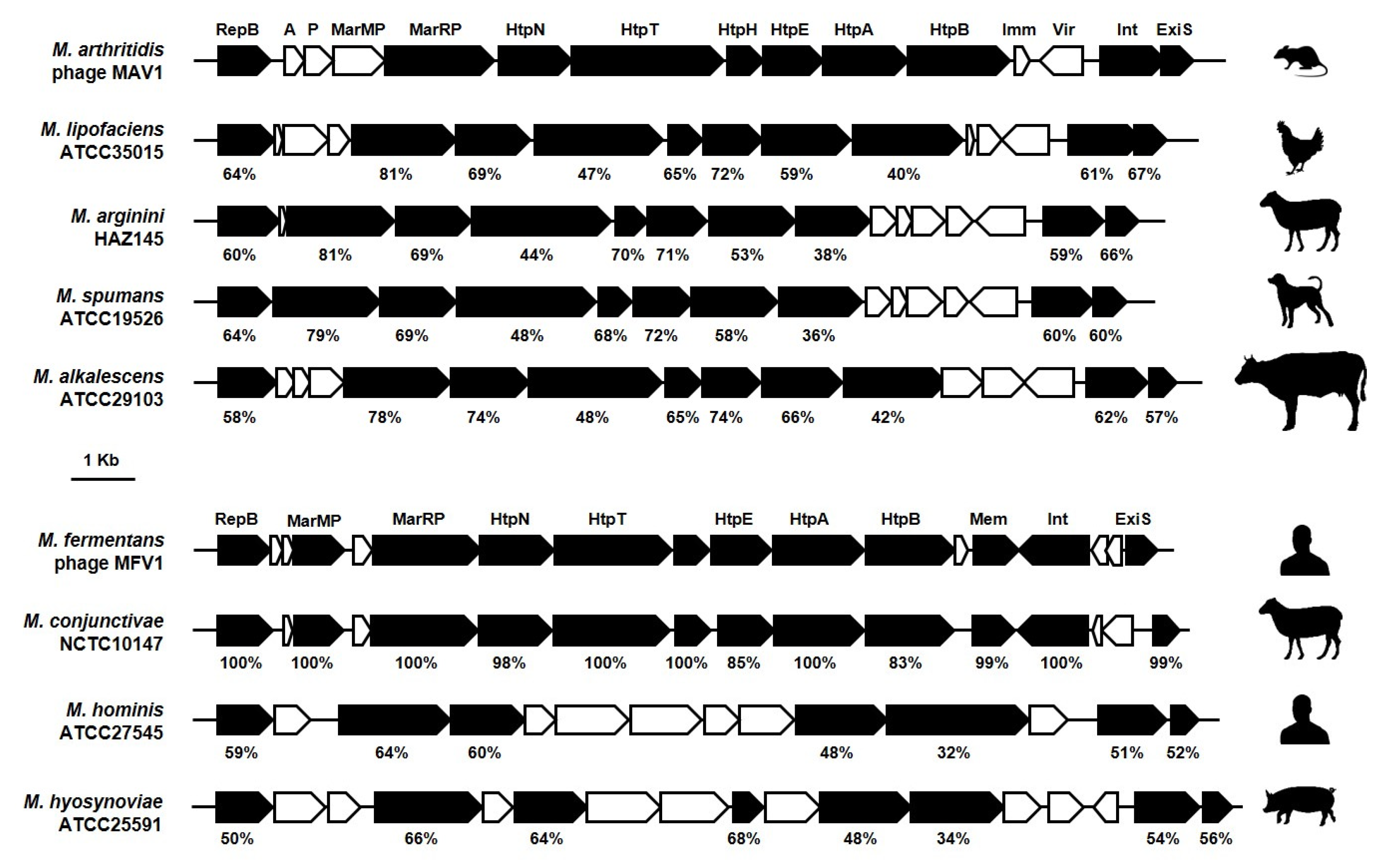{kind=link}
{kind=link}
{kind=link}
| Phylogenic Group | Species | Hosts 1 | Strain | ICE Occurrence | ICE Designation | Copy Number | Size (Kbp) | CDS Number | Reference |
|---|---|---|---|---|---|---|---|---|---|
| Hominis | M. conjunctivae | Small R | HRC/581 T | + | ICECJ | 2 | 28.5 | 23 | [32] |
| M. hyopneumoniae | Swine | 7448 | + | ICEH | 1 | 22.3 | 14 | [33] | |
| 168 | + | ICEH | 1 | 23.4 | 19 | [34] | |||
| 232 | + | ICEH | 1 | 22.5 | 19 | [35] | |||
| M. fermentans | Human | PG18 T | + | ICEF | 4 | 23.2 | 22 | [26] | |
| JER | + | ICEF | 2 | 24.4 | 23 | [36] | |||
| M64 | + | ICEF | 7 | 22.7–26.0 | 22–26 | [37] | |||
| M. agalactiae | Small R | PG2 T | vestige | vestige | 1 | nd | nd | [9] | |
| 5632 | + | ICEA | 3 | 27.2 | 23 | [25] | |||
| Wild R | 14628 | + | ICEA | 1 | 27.2 | 23 | [30] | ||
| M. bovis | Cattle | PG45 T | + | ICEB | 1 | 37.1 | 22 | [38] | |
| Hubei | vestige | vestige | un 4 | nd | nd | [39] | |||
| HB0801 | vestige | vestige | un | nd | nd | [40] | |||
| M. auris | Small R | 15026 | D 2 | nd 3 | un | nd | nd | [31] | |
| M. bovigenitalium | Cattle | 51080 | D | nd | un | nd | nd | [41] | |
| M. alkalescens | Cattle | 14918 | D | nd | un | nd | nd | [41] | |
| M. hominis | Human | 4788 | + | ICEHo | 1 | 29.1 | 25 | [42] | |
| 4235 | + | ICEHo | 2 | 30.5 | 25 | [42] | |||
| 35 | + | ICEHo | 2 | 29.1; 30.3 | 25 | [42] | |||
| Sprott | + | TetM mosaic transposon | 1 | 25.2 | 11 | [43] | |||
| Spiroplasma | M. mycoides subsp. capri | Small R | GM12 T | + | ICEM | 1 | 29.6 | 21 | [30] |
| 95010 | + | ICEMx2 | 2 | 30.0 | 21 | [44] | |||
| M. capricolum subsp. capricolum | Small R | CK T | + | ICEC | 1 | 23.8 | 17 | GenBank CP000123.1 | |
| M. putrefaciens | Small R | 9231 | D | nd | un | nd | nd | [45] | |
| M. yeatsii | Small R | 13926 | D | nd | un | nd | nd | [31] | |
| M. feriruminatoris | Wild R | 14/OD_0492 | D | nd | un | nd | TraE detected | GenBank LR739237.1 | |
| 8756-13 | D | nd | un | nd | TraE detected | GenBank LR739235.1 |
| Phylogenetic Group | Species | Host 1 | Strain 2 | Phage Name | Morpho-logy 3 | Genome (Size in Kbp) | Reference |
|---|---|---|---|---|---|---|---|
| Hominis | M. agalactiae | Wild R | 14628 | MAgV1 4 | nk 6 | dsDNA (34) | [56] |
| M. alkalescens | Cattle | ATCC 29103 | MAV1-like | nk | nk | This study | |
| M. arginini | WHR | HAZ145 | MAV1-like | nk | nk | This study | |
| M. arthritidis | Rodent | PG61 * | MAV1 | nk | dsDNA (16) | [57,58,59] | |
| M. bovigenitalium | Cattle | 51080 | MAgV1-like | nk | nk | [41,56] | |
| M. bovirhinis | Cattle | nd | Br1 | PH, LT | nk | [54,55] | |
| HAZ141_2 | nd 5 | nk | dsDNA (54) | [60,61] | |||
| M. bovis | Cattle | RM16 | MAgV1-like | nk | nk | This study, [62] | |
| 3308MB | P1-like | nk | nk | This study | |||
| M. conjunctivae | Small R | HRC/581 T | MAgV1-like | nk | nk | [32,56] | |
| NCTC 10147 | MFV1-like | nk | nk | This study | |||
| M. fermentans | Human | PG18 T* | MFV1 | nk | dsDNA (16) | [63] | |
| M. hominis | Human | LBD4 | MHoV1 | nk | dsDNA (16) | [64] | |
| M. hyorhinis | Swine | GDL-1 | Hr1 | PH, ST | nk | [65] | |
| M. hyosynoviae | Swine | NPL3 * | MFV1-like | nk | nk | [66] | |
| M. lipofaciens | Avian | ATCC 35015 | MAV1-like | nk | nk | This study | |
| M. molare | Canine | ATCC 27746 | MAgV1-like | nk | nk | This study | |
| M. mustelae | Mink | ATCC 35214 | MAgV1-like | nk | nk | This study | |
| M. pulmonis | Rodent | nd | P1 | PH, ST | dsDNA (12) | [67,68,69] | |
| M. spumans | Canine | ATCC 19526 | MAV1-like | nk | nk | This study |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Citti, C.; Baranowski, E.; Dordet-Frisoni, E.; Faucher, M.; Nouvel, L.-X. Genomic Islands in Mycoplasmas. Genes 2020, 11, 836. https://doi.org/10.3390/genes11080836
Citti C, Baranowski E, Dordet-Frisoni E, Faucher M, Nouvel L-X. Genomic Islands in Mycoplasmas. Genes. 2020; 11(8):836. https://doi.org/10.3390/genes11080836
Chicago/Turabian StyleCitti, Christine, Eric Baranowski, Emilie Dordet-Frisoni, Marion Faucher, and Laurent-Xavier Nouvel. 2020. "Genomic Islands in Mycoplasmas" Genes 11, no. 8: 836. https://doi.org/10.3390/genes11080836
APA StyleCitti, C., Baranowski, E., Dordet-Frisoni, E., Faucher, M., & Nouvel, L.-X. (2020). Genomic Islands in Mycoplasmas. Genes, 11(8), 836. https://doi.org/10.3390/genes11080836