Optimized NGS Approach for Detection of Aneuploidies and Mosaicism in PGT-A and Imbalances in PGT-SR

 ,
, 
Abstract
1. Introduction
2. Materials and Methods
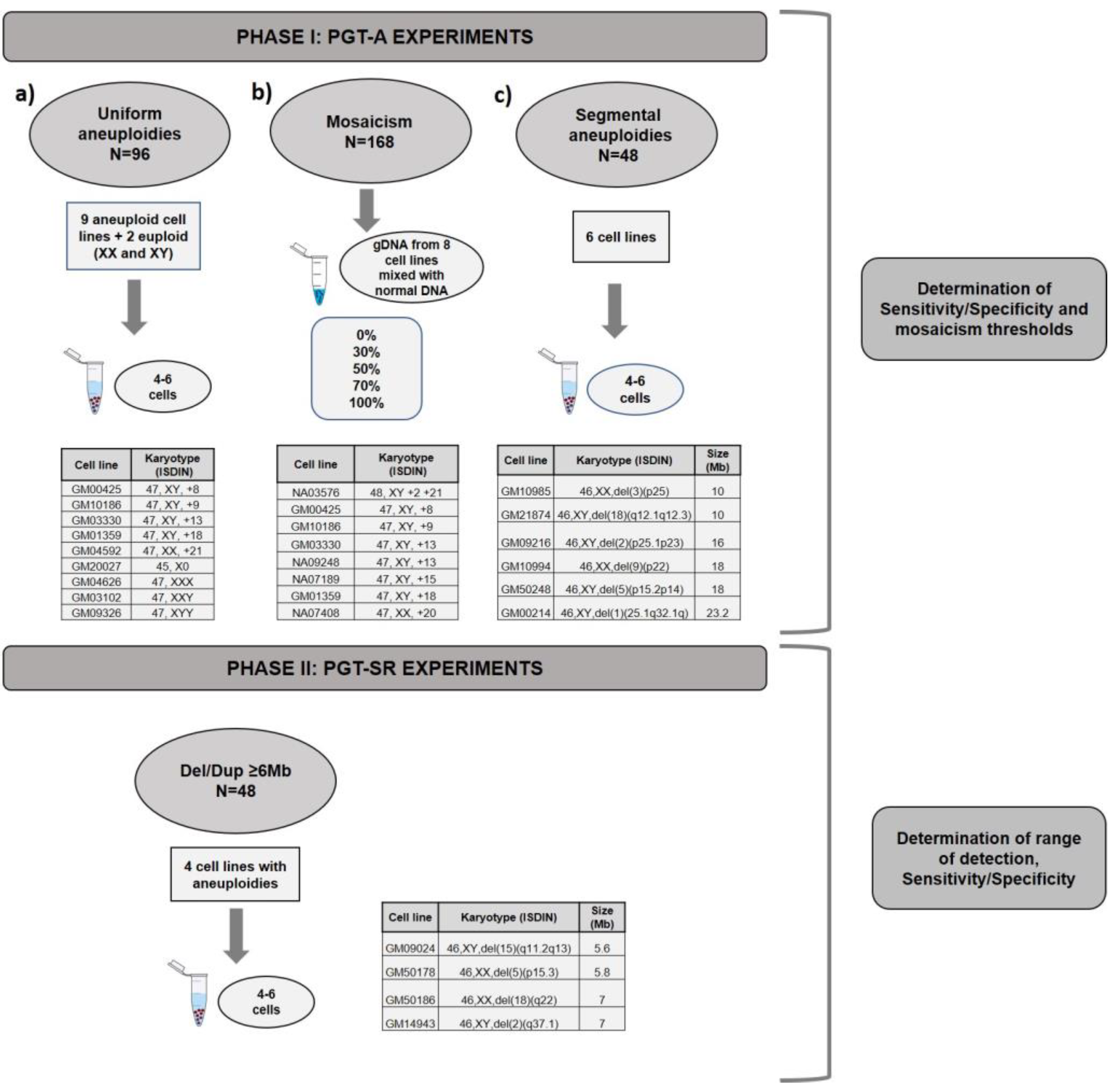
2.1. Experimental Design
2.2. NGS Protocol for PGT-A and PGT-SR
2.3. Bioinformatics Analysis and Interpretation of Results
- -
- Sample Normalized Mean Coverage (SMNC) is the observed ratio of reads in the sample;
- -
- Control Normalized Mean Coverage for 1 copy (CNMP1) is the expected ratio of reads for one copy if the sample is normal;
- -
- Expected Ploidy (EP) is the expected number of copies.
2.4. Evaluation of Efficiency, Concordance, Sensitivity and Specificity
3. Results
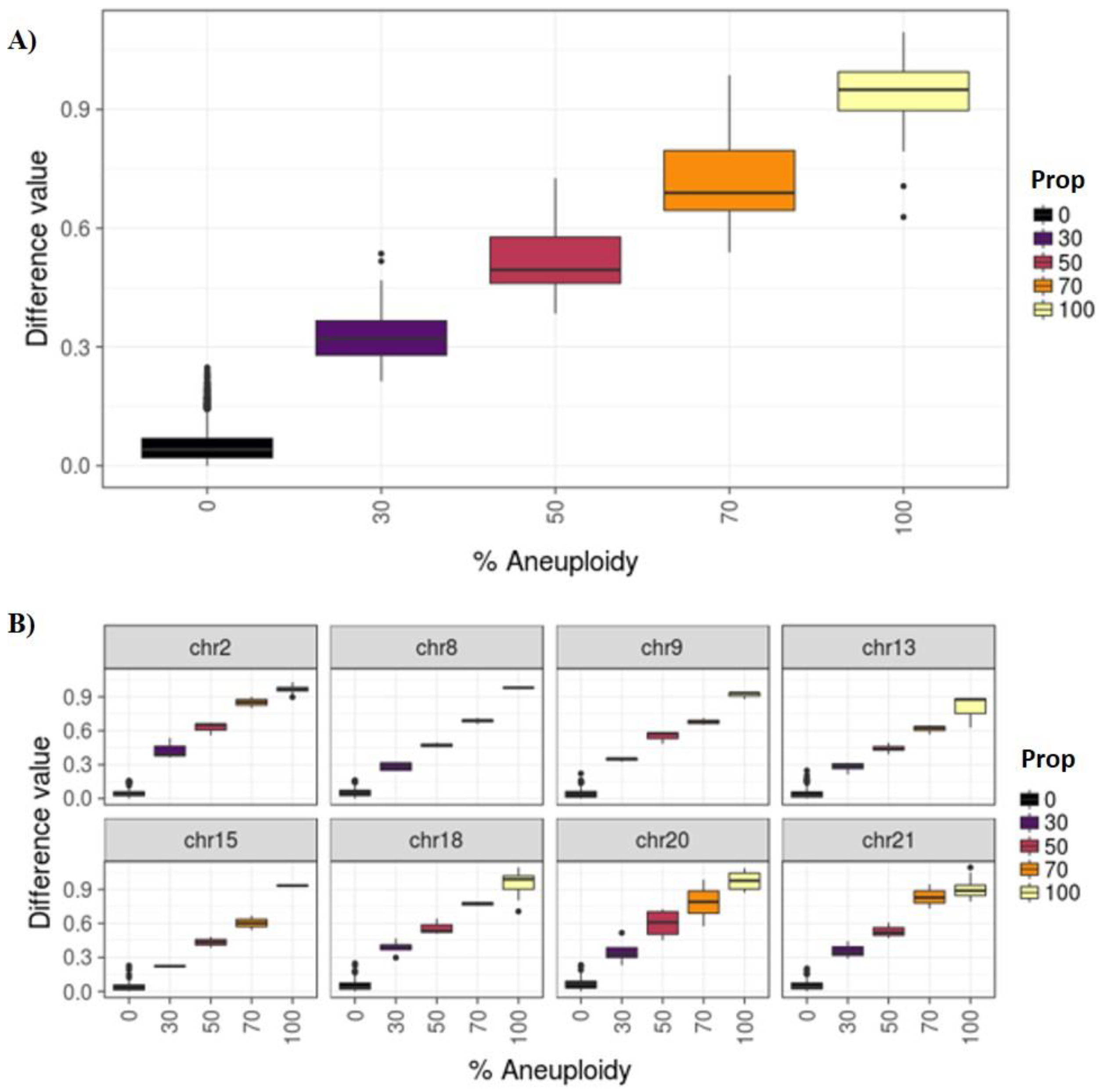
3.1. Phase I: PGT-A for Uniform Whole Aneuploidies, Mosaicism, and Segmental Aneuploidies (≥10 Mb)
3.2. Phase II: PGT-SR
4. Discussion
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Kung, A.; Munné, S.; Bankowski, B.; Coates, A.; Wells, D. Validation of next-generation sequencing for comprehensive chromosome screening of embryos. Reprod. Biomed. Online 2015, 31, 760–769. [Google Scholar] [CrossRef] [PubMed]
- Franasiak, J.M.; Forman, E.J.; Hong, K.H.; Werner, M.D.; Upham, K.M.; Treff, N.R.; Scott, R.T., Jr. The nature of aneuploidy with increasing age of the female partner: A review of 15,169 consecutive trophectoderm biopsies evaluated with comprehensive chromosomal screening. Fertil. Steril. 2014, 101, 656–663.e1. [Google Scholar] [CrossRef] [PubMed]
- Rubio, C.; Rodrigo, L.; Garcia-Pascual, C.; Peinado, V.; Campos-Galindo, I.; Garcia-Herrero, S.; Simón, C. Clinical application of embryo aneuploidy testing by NGS. Biol. Reprod. 2019. [Google Scholar] [CrossRef] [PubMed]
- Scott, R.T., Jr.; Ferry, K.; Su, J.; Tao, X.; Scott, K.; Treff, N.R. Comprehensive chromosome screening is highly predictive of the reproductive potential of human embryos: A prospective, blinded, nonselection study. Fertil. Steril. 2012, 97, 870–875. [Google Scholar] [CrossRef]
- Rubio, C.; Bellver, J.; Rodrigo, L.; Castillón, G.; Guillén, A.; Vidal, C.; Giles, J.; Ferrando, M.; Cabanillas, S.; Remohí, J.; et al. In vitro fertilization with preimplantation genetic diagnosis for aneuploidies in advanced maternal age: A randomized, controlled study. Fertil. Steril. 2017, 107, 1122–1129. [Google Scholar] [CrossRef]
- Lean, S.C.; Derricott, H.; Jones, R.L.; Heazell, A.E.P. Advanced maternal age and adverse pregnancy outcomes: A systematic review and meta-analysis. PLoS ONE 2017, 12, e0186287. [Google Scholar] [CrossRef]
- Pinheiro, R.L.; Areia, A.L.; Mota Pinto, A.; Donato, H. Advanced maternal age: Adverse outcomes of pregnancy, a meta-analysis. Acta Med. Port. 2019, 32, 219–226. [Google Scholar] [CrossRef]
- Neal, S.A.; Morin, S.J.; Franasiak, J.M.; Goodman, L.R.; Juneau, C.R.; Forman, E.J.; Werner, M.D.; Scott, R.T., Jr. Preimplantation genetic testing for aneuploidy is cost-effective, shortens treatment time, and reduces the risk of failed embryo transfer and clinical miscarriage. Fertil. Steril. 2018, 110, 896–904. [Google Scholar] [CrossRef]
- Somigliana, E.; Busnelli, A.; Paffoni, A.; Vigano, P.; Riccaboni, A.; Rubio, C.; Capalbo, A. Cost-effectiveness of preimplantation genetic testing for aneuploidies. Fertil. Steril. 2019, 111, 1169–1176. [Google Scholar] [CrossRef]
- Harper, J.C.; Harton, G. The use of arrays in preimplantation genetic diagnosis and screening. Fertil. Steril. 2010, 94, 1173–1177. [Google Scholar] [CrossRef]
- Fiorentino, F.; Caiazzo, F.; Napolitano, S.; Spizzichino, L.; Bono, S.; Sessa, M.; Nuccitelli, A.; Biricik, A.; Gordon, A.; Rizzo, G.; et al. Introducing array comparative genomic hybridization into routine prenatal diagnosis practice: A prospective study on over 1000 consecutive clinical cases. Prenat. Diagn. 2011, 31, 1270–1282. [Google Scholar] [CrossRef] [PubMed]
- Treff, N.R.; Tao, X.; Ferry, K.M.; Su, J.; Taylor, D.; Scott, R.T., Jr. Development and validation of an accurate quantitative real-time polymerase chain reaction-based assay for human blastocyst comprehensive chromosomal aneuploidy screening. Fertil. Steril. 2012, 97, 819–824. [Google Scholar] [CrossRef] [PubMed]
- Rodrigo, L.; Mateu, E.; Mercader, A.; Cobo, A.C.; Peinado, V.; Milán, M.; Al-Asmar, N.; Campos-Galindo, I.; García-Herrero, S.; Mir, P.; et al. New tools for embryo selection: Comprehensive chromosome screening by array comparative genomic hybridization. Biomed. Res. Int. 2014, 2014, 517125. [Google Scholar] [CrossRef] [PubMed]
- Vera-Rodríguez, M.; Michel, C.E.; Mercader, A.; Bladon, A.J.; Rodrigo, L.; Kokocinski, F.; Mateu, E.; Al-Asmar, N.; Blesa, D.; Simón, C.; et al. Distribution patterns of segmental aneuploidies in human blastocysts identified by next-generation sequencing. Fertil. Steril. 2016, 105, 1047–1055. [Google Scholar] [CrossRef] [PubMed]
- Lai, H.H.; Chuang, T.H.; Wong, L.K.; Lee, M.J.; Hsieh, C.L.; Wang, H.L.; Chen, S.U. Identification of mosaic and segmental aneuploidies by next-generation sequencing in preimplantation genetic screening can improve clinical outcomes compared to array-comparative genomic hybridization. Mol. Cytogenet. 2017, 10, 14. [Google Scholar] [CrossRef] [PubMed]
- Wells, D.; Kaur, K.; Grifo, J.; Glassner, M.; Taylor, J.C.; Fragouli, E.; Munne, S. Clinical utilisation of a rapid low-pass whole genome sequencing technique for the diagnosis of aneuploidy in human embryos prior to implantation. J. Med. Genet. 2014, 51, 553–562. [Google Scholar] [CrossRef]
- Munné, S.; Blazek, J.; Large, M.; Martinez-Ortiz, P.A.; Nisson, H.; Liu, E.; Tarozzi, N.; Borini, A.; Becker, A.; Zhang, J.; et al. Detailed investigation into the cytogenetic constitution and pregnancy outcome of replacing mosaic blastocysts detected with the use of high-resolution next-generation sequencing. Fertil. Steril. 2017, 108, 62–71. [Google Scholar] [CrossRef]
- Miroslav, H.; Jakub, H.; David, K.; Rostislav, N.; Gabriela, T.; Pavel, T.; Katerina, V. The incidence and origin of segmental chromosome abnormalities in human IVF embryos detected during PGD and PGS. Rep. Biomed. Online 2018, 36, e13–e14. [Google Scholar]
- Popovic, M.; Dheedene, A.; Christodoulou, C.; Taelman, J.; Dhaenens, L.; Van Nieuwerburgh, F.; Deforce, D.; Van den Abbeel, E.; De Sutter, P.; Menten, B.; et al. Chromosomal mosaicism in human blastocysts: The ultimate challenge of preimplantation genetic testing? Hum. Reprod. 2018, 33, 1342–1354. [Google Scholar] [CrossRef]
- Victor, A.R.; Tyndall, J.C.; Brake, A.J.; Lepkowsky, L.T.; Murphy, A.E.; Griffin, D.K.; McCoy, R.C.; Barnes, F.L.; Zouves, C.G.; Viotti, M. One hundred mosaic embryos transferred prospectively in a single clinic: Exploring when and why they result in healthy pregnancies. Fertil. Steril. 2019, 111, 280–293. [Google Scholar] [CrossRef]
- Greco, E.; Minasi, M.G.; Fiorentino, F. Healthy babies after intrauterine transfer of mosaic Aneuploid blastocysts. N. Engl. J. Med. 2015, 373, 2089–2090. [Google Scholar] [CrossRef] [PubMed]
- Fragouli, E.; Alfarawati, S.; Spath, K.; Babariya, D.; Tarozzi, N.; Borini, A.; Wells, D. Analysis of implantation and ongoing pregnancy rates following the transfer of mosaic diploid-aneuploid blastocysts. Hum. Genet. 2017, 136, 805–819. [Google Scholar] [CrossRef]
- Chuang, T.-H.; Hsieh, J.-Y.; Lee, M.-J.; Lai, H.-L.; Hsieh, C.-L.; Wang, H.-L.; Chang, Y.-J.; Chen, S.-U. Concordance between different trophectoderm biopsy sites and the inner cell mass of chromosomal composition measured with a next-generation sequencing platform. Mol. Hum. Rep. 2018, 21, 593–601. [Google Scholar] [CrossRef] [PubMed]
- Spinella, F.; Fiorentino, F.; Biricik, A.; Bono, S.; Ruberti, A.; Cotroneo, E.; Baldi, M.; Cursio, E.; Minasi, M.G.; Greco, E. Extent of chromosomal mosaicism influences the clinical outcome of in vitro fertilization treatments. Fertil. Steril. 2018, 109, 77–83. [Google Scholar] [CrossRef] [PubMed]
- Grati, F.R.; Gallazzi, G.; Branca, L.; Maggi, F.; Simoni, G.; Yaron, Y. An evidence-based scoring system for prioritizing mosaic aneuploid embryos following preimplantation genetic screening. Reprod. Biomed. Online 2018, 36, 442–449. [Google Scholar] [CrossRef]
- Maxwell, S.M.; Colls, P.; Hodes-Wertz, B.; McCulloh, D.H.; McCaffrey, C.; Wells, D.; Munné, S.; Grifo, J.A. Why do euploid embryos miscarry? A case-control study comparing the rate of aneuploidy within presumed euploid embryos that resulted in miscarriage or live birth using next-generation sequencing. Fertil. Steril. 2016, 106, 1414–1419.e5. [Google Scholar] [CrossRef]
- Tsuji, K.; Ojima, M.; Otabe, K.; Horie, M.; Koga, H.; Sekiya, I.; Muneta, T. Effects of different cell-detaching methods on the viability and cell surface antigen expression of synovial mesenchymal stem cells. Cell Transpl. 2017, 26, 1089–1102. [Google Scholar] [CrossRef]
- Goodrich, D.; Xing, T.; Tao, X.; Lonczak, A.; Zhan, Y.; Landis, J.; Zimmerman, R.; Scott, R.T., Jr.; Treff, N.R. Evaluation of comprehensive chromosome screening platforms for the detection of mosaic segmental aneuploidy. J. Assist. Reprod. Genet. 2017, 34, 975–981. [Google Scholar] [CrossRef] [PubMed]
- Fiorentino, F.; Biricik, A.; Bono, S.; Spizzichino, L.; Cotroneo, E.; Cottone, G.; Kokocinski, F.; Michel, C.E. Development and validation of a next-generation sequencing-based protocol for 24-chromosome aneuploidy screening of embryos. Fertil. Steril. 2014, 101, 1375–1382. [Google Scholar] [CrossRef] [PubMed]
- Goodrich, D.; Tao, X.; Bohrer, C.; Lonczak, A.; Xing, T.; Zimmerman, R.; Zhan, Y.; Scott, R.T., Jr.; Treff, N.R. A randomized and blinded comparison of qPCR and NGS based detection of aneuploidy in a cell line mixture model of blastocyst biopsy mosaicism. J. Assist. Reprod. Genet. 2016, 33, 1473–1480. [Google Scholar] [CrossRef] [PubMed]
- Kebschull, J.M.; Zador, A.M. Sources of PCR-induced distortions in high-throughput sequencing data sets. Nucleic Acids Res. 2012, 43, e143. [Google Scholar] [CrossRef] [PubMed]
- Munné, S. Origins of mosaicism and criteria for the transfer of mosaic embryos. Reprod. Biomed. Online 2018, 36, 369–370. [Google Scholar] [CrossRef] [PubMed]
- Cram, D.S.; Leigh, D.; Handyside, A.; Rechitsky, L.; Xu, K.; Harton, G.; Grifo, J.; Rubio, C.; Fragouli, E.; Kahraman, S.; et al. PGDIS newsletter, 27 May 2019. In Proceedings of the Pgdis Position Statement On The Transfer Of Mosaic Embryos In Preimplantation Ganetic Testing For Aneuploidy (PGT-A) * Based On Materials Of 18th International Conference On Preimplantation Genetics, Geneva, Switzerland, 15–18 April 2019. [Google Scholar]
- Kushnir, V.A.; Darmon, S.K.; Barad, D.H.; Gleicher, N. Degree of mosaicism in trophectoderm does not predict pregnancy potential: A corrected analysis of pregnancy outcomes following transfer of mosaic embryos. Reprod. Biol. Endocrinol. 2018, 16, 6. [Google Scholar] [CrossRef]
- Girardi, L.; Romanelli, V.; Fabiani, M.; Cimadomo, D.; Rienzi, L.; Ubaldi, F.M.; Serdarogulları, M.; Coban, O.; Findikli, N.; Boynukalin, K.; et al. Segmental aneuploidies show mosaic pattern reducing predictive value compared to high whole chromosome aneuploidies representativeness. Reprod. Biomed. Online 2019, 39, 18–19. [Google Scholar] [CrossRef]
- Zhou, S.; Cheng, D.; Ouyang, Q.; Xie, P.; Lu, C.; Gong, F.; Hu, L.; Tan, Y.; Lu, G.; Lin, G. Prevalence and authenticity of de-novo segmental aneuploidy (>16 Mb) in human blastocysts as detected by next-generation sequencing. Reprod. Biomed. Online 2018, 37, 511–520. [Google Scholar] [CrossRef] [PubMed]
- Capalbo, A.; Ubaldi, F.; Rienzi, L.; Scott, R.; Treff, N. Detecting mosaicism in trophectoderm biopsies: Current challenges and future possibilities. Hum. Reprod. 2017, 32, 492–498. [Google Scholar] [CrossRef]
- Monahan, D.; Harton, G.; Griffin, D.; Angle, D.; Smikle, C. Clinical comparison of two PGT-A platforms utilizing different thresholds to determine ploidy status. Reprod. Biomed. Online 2019, 39, 27–28. [Google Scholar] [CrossRef]
- Bono, S.; Biricik, A.; Spizzichino, L.; Nuccitelli, A.; Minasi, M.; Greco, E.; Spinella, F.; Fiorentino, F. Validation of a semiconductor next-generation sequencing-based protocol for preimplantation genetic diagnosis of reciprocal translocations. Prenat. Diagn. 2015, 35, 938–944. [Google Scholar] [CrossRef]
- Blanca, H.; González-Reig, S.; Penacho, V.; Castejón-Fernández, N.; Amoros, D.; Galán, F.; Alcaraz, L.A. Detection limit of partial insertions and deletions for PGS in terms of NGS by analyzing 242 embryos of couples with balanced translocations. Reprod. Biomed. Online 2018, 36 (Suppl. 1), e17. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
| TEST | Type of Sample | Total Samples | Informativity | Concordance Rates | Reads * | MAPD * | Duplicates |
|---|---|---|---|---|---|---|---|
| PGT-A | Uniform whole aneuploidies | 96 | 100% (96/96) | 100% (96/96) | 173,053 | 0.170 | 10.00% |
| Segmentals (≥ 10 Mb) | 48 | 98% (47/48) | 100% (47/47) | 126,780 | 0.194 | 9.60% | |
| Mosaicism (0%, 40%, 60%, 100%) | 18 | 100% (18/18) | 94.4% (17/18) | 148,874 | 0.174 | 6.63% | |
| PGT-SR | Small rearrangements (≥ 6 Mb) | 48 | 100% (48/48) | 100% (48/48) | 305,287 | 0.145 | 7.00% |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
García-Pascual, C.M.; Navarro-Sánchez, L.; Navarro, R.; Martínez, L.; Jiménez, J.; Rodrigo, L.; Simón, C.; Rubio, C. Optimized NGS Approach for Detection of Aneuploidies and Mosaicism in PGT-A and Imbalances in PGT-SR. Genes 2020, 11, 724. https://doi.org/10.3390/genes11070724
García-Pascual CM, Navarro-Sánchez L, Navarro R, Martínez L, Jiménez J, Rodrigo L, Simón C, Rubio C. Optimized NGS Approach for Detection of Aneuploidies and Mosaicism in PGT-A and Imbalances in PGT-SR. Genes. 2020; 11(7):724. https://doi.org/10.3390/genes11070724
Chicago/Turabian StyleGarcía-Pascual, Carmen M., Luis Navarro-Sánchez, Roser Navarro, Lucía Martínez, Jorge Jiménez, Lorena Rodrigo, Carlos Simón, and Carmen Rubio. 2020. "Optimized NGS Approach for Detection of Aneuploidies and Mosaicism in PGT-A and Imbalances in PGT-SR" Genes 11, no. 7: 724. https://doi.org/10.3390/genes11070724
APA StyleGarcía-Pascual, C. M., Navarro-Sánchez, L., Navarro, R., Martínez, L., Jiménez, J., Rodrigo, L., Simón, C., & Rubio, C. (2020). Optimized NGS Approach for Detection of Aneuploidies and Mosaicism in PGT-A and Imbalances in PGT-SR. Genes, 11(7), 724. https://doi.org/10.3390/genes11070724