Preimplantation Genetic Testing for Monogenic Disorders


{kind=link}
Abstract
1. Introduction
2. Indications for PGT-M
3. Genetic and Reproductive Counseling—Preclinical Workup
4. IVF, Embryo Biopsy, Transfer, and Cryopreservation
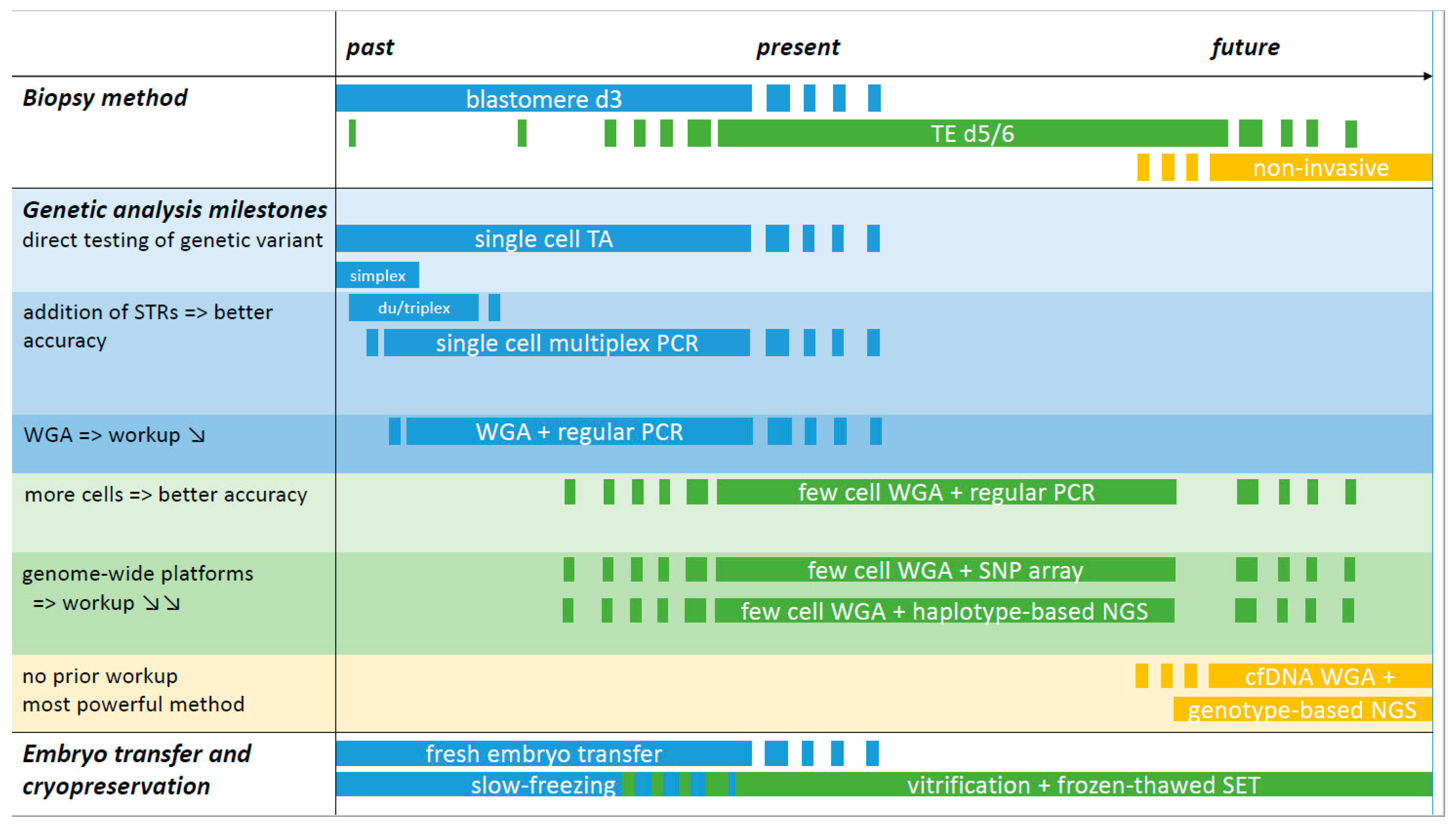
4.1. IVF and Current Embryo Biopsy Methods
4.2. Current Developments and Future Sampling Methods
4.3. Embryo Transfer and Cryopreservation
5. Diagnostic Methods
5.1. Early Methods of PGT-M
5.2. Whole Genome Amplification Approaches
5.3. SNP Array for PGT-M
5.4. PGT-M for De Novo Pathogenic Variants
5.5. SNP Array for Concurrent PGT-M and PGT-A
5.6. NGS for Concurrent PGT-M and PGT-A
6. Clinical Outcome
7. Children Follow-Up
8. Conclusions and Future Perspectives
Author Contributions
Funding
Conflicts of Interest
References
- Zegers-Hochschild, F.; Adamson, G.D.; Dyer, S.; Racowsky, C.; de Mouzon, J.; Sokol, R.; Rienzi, L.; Sunde, A.; Schmidt, L.; Cooke, I.D.; et al. The International Glossary on Infertility and Fertility Care, 2017. Hum. Reprod. 2017, 32, 1786–1801. [Google Scholar] [CrossRef] [PubMed]
- Handyside, A.H.; Kontogianni, E.H.; Hardy, K.; Winston, R.M. Pregnancies from biopsied human preimplantation embryos sexed by Y-specific DNA amplification. Nature 1990, 344, 768–770. [Google Scholar] [CrossRef] [PubMed]
- Laurie, A.D.; Hill, A.M.; Harraway, J.R.; Fellowes, A.P.; Phillipson, G.T.; Benny, P.S.; Smith, M.P.; George, P.M. Preimplantation genetic diagnosis for hemophilia A using indirect linkage analysis and direct genotyping approaches. J. Thromb. Haemost. 2010, 8, 783–789. [Google Scholar] [CrossRef] [PubMed]
- Treff, N.R.; Fedick, A.M.; Tao, X.; Devkota, B.; Taylor, D.; Scott, R.T., Jr. Evaluation of targeted next-generation sequencing–based preimplantation genetic diagnosis of monogenic disease. Fertil. Steril. 2013, 99, 1377–1384. [Google Scholar] [CrossRef] [PubMed]
- Natesan, S.A.; Bladon, A.J.; Coskun, S.; Qubbaj, W.; Prates, R.; Munné, S.; Coonen, E.; Dreesen, J.C.; Stevens, S.J.; Paulussen, A.D.; et al. Genome-wide karyomapping accurately identifies the inheritance of single-gene defects in human preimplantation embryos in vitro. Genet. Med. 2014, 16, 838–845. [Google Scholar] [CrossRef]
- Esteki, M.Z.; Dimitriadou, E.; Mateiu, L.; Melotte, C.; Van Der Aa, N.; Kumar, P.; Das, R.; Theunis, K.; Cheng, J.; Legius, E.; et al. Concurrent Whole-Genome Haplotyping and Copy-Number Profiling of Single Cells. Am. J. Hum. Genet. 2015, 96, 894–912. [Google Scholar] [CrossRef]
- Backenroth, D.; Zahdeh, F.; Kling, Y.; Peretz, A.; Rosen, T.; Kort, D.; Zeligson, S.; Dror, T.; Kirshberg, S.; Burak, E.; et al. Haploseek: A 24-hour all-in-one method for preimplantation genetic diagnosis (PGD) of monogenic disease and aneuploidy. Genet. Med. 2018, 21, 1390–1399. [Google Scholar] [CrossRef]
- Masset, H.; Esteki, M.Z.; Dimitriadou, E.; Dreesen, J.; Debrock, S.; Derhaag, J.; Derks, K.; Destouni, A.; Drüsedau, M.; Meekels, J.; et al. Multi-centre evaluation of a comprehensive preimplantation genetic test through haplotyping-by-sequencing. Hum. Reprod. 2019, 34, 1608–1619. [Google Scholar] [CrossRef]
- Carvalho, F.; Coonen, E.; Goossens, V.; Kokkali, G.; Rubio, C.; Meijer-Hoogeveen, M.; Moutou, C.; Vermeulen, N.; De Rycke, M. ESHRE PGT Consortium good practice recommendations for the organisation of PGT. Hum. Reprod. Open 2020, 2020. [Google Scholar] [CrossRef]
- Kokkali, G.; Coticchio, G.; Bronet, F.; Celebi, C.; Cimadomo, D.; Goossens, V.; Liss, J.; Nunes, S.; Sfontouris, I.; Vermeulen, N.; et al. ESHRE PGT Consortium and SIG Embryology good practice recommendations for polar body and embryo biopsy for PGT. Hum. Reprod. Open 2020, 2020. [Google Scholar] [CrossRef]
- Carvalho, F.; Moutou, C.; Dimitriadou, E.; Dreesen, J.; Giménez, C.; Goossens, V.; Kakourou, G.; Vermeulen, N.; Zuccarello, D.; De Rycke, M. ESHRE PGT Consortium good practice recommendations for the detection of monogenic disorders. Hum. Reprod. Open 2020, 2020. [Google Scholar] [CrossRef] [PubMed]
- Coonen, E.; Rubio, C.; Christopikou, D.; Dimitriadou, E.; Gontar, J.; Goossens, V.; Maurer, M.; Spinella, F.; Vermeulen, N.; De Rycke, M. ESHRE PGT Consortium good practice recommendations for the detection of structural and numerical chromosomal aberrations. Hum. Reprod. Open 2020, 2020. [Google Scholar] [CrossRef] [PubMed]
- Ginoza, M.E.C.; Isasi, R. Regulating Preimplantation Genetic Testing across the World: A Comparison of International Policy and Ethical Perspectives. Cold Spring Harb. Perspect. Med. 2019, 10. [Google Scholar] [CrossRef] [PubMed]
- Bayefsky, M.J. Comparative preimplantation genetic diagnosis policy in Europe and the USA and its implications for reproductive tourism. Reprod. Biomed. Soc. Online 2016, 3, 41–47. [Google Scholar] [CrossRef]
- Calhaz-Jorge, C.; De Geyter, C.H.; Kupka, M.S.; Wyns, C.; Mocanu, E.; Motrenko, T.; Scaravelli, G.; Smeenk, J.; Vidakovic, S.; Goossens, V. Survey on ART and IUI: Legislation, regulation, funding and registries in European countries: The European IVF-monitoring Consortium (EIM) for the European Society of Human Reproduction and Embryology (ESHRE). Hum. Reprod. Open 2020, 2020. [Google Scholar] [CrossRef]
- Dondorp, W.; De Wert, G. Refining the ethics of preimplantation genetic diagnosis: A plea for contextualized proportionality. Bioethics 2019, 33, 294–301. [Google Scholar] [CrossRef]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. Standards and Guidelines for the Interpretation of Sequence Variants: A Joint Consensus Recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 2015, 17, 405–424. [Google Scholar] [CrossRef]
- Kakourou, G.; Kahraman, S.; Ekmekci, G.C.; Tac, H.A.; Kourlaba, G.; Kourkouni, E.; Sanz, A.C.; Martin, J.; Malmgren, H.; Giménez, C.; et al. The clinical utility of PGD with HLA matching: A collaborative multi-centre ESHRE study. Hum. Reprod. 2018, 33, 520–530. [Google Scholar] [CrossRef]
- Van Rij, M.C.; De Rademaeker, M.; Moutou, C.; Dreesen, J.C.; De Rycke, M.; Liebaers, I.; Geraedts, J.P.; De Die-Smulders, C.E.; Viville, S. Preimplantation genetic diagnosis (PGD) for Huntington’s disease: The experience of three European centres. Eur. J. Hum. Genet. 2012, 20, 368–375. [Google Scholar] [CrossRef]
- Shenfield, F.; Pennings, G.; Devroey, P.; Sureau, C.; Tarlatzis, B.; Cohen, J. Taskforce 5: Preimplantation genetic diagnosis. Hum. Reprod. 2003, 18, 649–651. [Google Scholar] [CrossRef]
- Smeets, B.; Sallevelt, S.C.E.H.; Dreesen, J.C.; De Die-Smulders, C.E.; De Coo, I.; Die-Smulders, C.E. Preventing the transmission of mitochondrial DNA disorders using prenatal or preimplantation genetic diagnosis. Ann. N. Y. Acad. Sci. 2015, 1350, 29–36. [Google Scholar] [CrossRef]
- Berckmoes, V.; Verdyck, P.; De Becker, P.; De Vos, A.; Verheyen, G.; Van Der Niepen, P.; Verpoest, W.; Liebaers, I.; Bonduelle, M.; Keymolen, K.; et al. Factors influencing the clinical outcome of preimplantation genetic testing for polycystic kidney disease. Hum. Reprod. 2019, 34, 949–958. [Google Scholar] [CrossRef] [PubMed]
- Ubaldi, F.M.; Capalbo, A.; Vaiarelli, A.; Cimadomo, D.; Colamaria, S.; Alviggi, C.; Trabucco, E.; Venturella, R.; Vajta, G.; Rienzi, L. Follicular versus luteal phase ovarian stimulation during the same menstrual cycle (DuoStim) in a reduced ovarian reserve population results in a similar euploid blastocyst formation rate: New insight in ovarian reserve exploitation. Fertil. Steril. 2016, 105, 1488–1495. [Google Scholar] [CrossRef] [PubMed]
- Chamayou, S.; Sicali, M.; Alecci, C.; Ragolia, C.; Liprino, A.; Nibali, D.; Storaci, G.; Cardea, A.; Guglielmino, A. The accumulation of vitrified oocytes is a strategy to increase the number of euploid available blastocysts for transfer after preimplantation genetic testing. J. Assist. Reprod. Genet. 2017, 34, 479–486. [Google Scholar] [CrossRef] [PubMed]
- Hu, X.; Ding, C.; Zhang, D.; Zhou, W.; Wang, J.; Zeng, Y.; Lv, J.; Xu, Y.; Zhou, C.-Q. Embryo pooling: A promising strategy for managing insufficient number of embryos in preimplantation genetic diagnosis. Gynecol. Endocrinol. 2017, 33, 867–871. [Google Scholar] [CrossRef]
- De Vos, A.; Staessen, C.; De Rycke, M.; Verpoest, W.; Haentjens, P.; Devroey, P.; Liebaers, I.; Van de Velde, H. Impact of cleavage-stage embryo biopsy in view of PGD on human blastocyst implantation: A prospective cohort of single embryo transfers. Hum. Reprod. 2009, 24, 2988–2996. [Google Scholar] [CrossRef] [PubMed]
- De Rycke, M.; Goossens, V.; Kokkali, G.; Meijer-Hoogeveen, M.; Coonen, E.; Moutou, C. ESHRE PGD Consortium data collection XIV-XV: Cycles from January 2011 to December 2012 with pregnancy follow-up to October 2013. Hum. Reprod. 2017, 32, 1974–1994. [Google Scholar] [CrossRef] [PubMed]
- Cimadomo, D.; Rienzi, L.; Capalbo, A.; Rubio, C.; Innocenti, F.; García-Pascual, C.M.; Ubaldi, F.M.; Handyside, A. The dawn of the future: 30 years from the first biopsy of a human embryo. The detailed history of an ongoing revolution. Hum. Reprod. Update 2020, 26, 453–473. [Google Scholar] [CrossRef] [PubMed]
- Scott, R.T.; Upham, K.M.; Forman, E.J.; Zhao, T.; Treff, N.R. Cleavage-stage biopsy significantly impairs human embryonic implantation potential while blastocyst biopsy does not: A randomized and paired clinical trial. Fertil. Steril. 2013, 100, 624–630. [Google Scholar] [CrossRef]
- Zakharova, E.E.; Zaletova, V.V.; Krivokharchenko, A.S. Biopsy of Human Morula-Stage Embryos: Outcome of 215 IVF/ICSI Cycles with PGS. PLoS ONE 2014, 9. [Google Scholar] [CrossRef]
- Irani, M.; Zaninovic, N.; Canon, C.; O’Neill, C.; Gunnala, V.; Zhan, Q.; Palermo, G.; Reichman, D.; Rosenwaks, Z. A rationale for biopsying embryos reaching the morula stage on Day 6 in women undergoing preimplantation genetic testing for aneuploidy. Hum. Reprod. 2018, 33, 935–941. [Google Scholar] [CrossRef] [PubMed]
- Magli, M.C.; Albanese, C.; Crippa, A.; Tabanelli, C.; Ferraretti, A.P.; Gianaroli, L. Deoxyribonucleic acid detection in blastocoelic fluid: A new predictor of embryo ploidy and viable pregnancy. Fertil. Steril. 2019, 111, 77–85. [Google Scholar] [CrossRef] [PubMed]
- Van Landuyt, L.; Polyzos, N.P.; De Munck, N.; Blockeel, C.; Van de Velde, H.; Verheyen, G. A prospective randomized controlled trial investigating the effect of artificial shrinkage (collapse) on the implantation potential of vitrified blastocysts. Hum. Reprod. 2015, 30, 2509–2518. [Google Scholar] [CrossRef] [PubMed]
- Brouillet, S.; Martinez, G.; Coutton, C.; Hamamah, S.; Sophie, B.; Guillaume, M.; Charles, C.; Samir, H. Is cell-free DNA in spent embryo culture medium an alternative to embryo biopsy for preimplantation genetic testing? A systematic review. Reprod. Biomed. Online 2020, 40, 779–796. [Google Scholar] [CrossRef]
- Capalbo, A.; Romanelli, V.; Patassini, C.; Poli, M.; Girardi, L.; Giancani, A.; Stoppa, M.; Cimadomo, D.; Ubaldi, F.M.; Rienzi, L. Diagnostic efficacy of blastocoel fluid and spent media as sources of DNA for preimplantation genetic testing in standard clinical conditions. Fertil. Steril. 2018, 110, 870–879. [Google Scholar] [CrossRef]
- Leaver, M.; Wells, D. Non-invasive preimplantation genetic testing (niPGT): The next revolution in reproductive genetics? Hum. Reprod. Update 2019, 26, 16–42. [Google Scholar] [CrossRef]
- Loutradi, K.E.; Kolibianakis, E.; Venetis, C.A.; Papanikolaou, E.G.; Pados, G.; Bontis, I.; Tarlatzis, B.C. Cryopreservation of human embryos by vitrification or slow freezing: A systematic review and meta-analysis. Fertil. Steril. 2008, 90, 186–193. [Google Scholar] [CrossRef]
- Bosch, E.; De Vos, M.; Humaidan, P. The Future of Cryopreservation in Assisted Reproductive Technologies. Front. Endocrinol. 2020, 11. [Google Scholar] [CrossRef]
- Rechitsky, S.; Ström, C.; Verlinsky, O.; Amet, T.; Ivakhnenko, V.; Kukharenko, V.; Kuliev, A.; Verlinsky, Y. Accuracy of Preimplantation Diagnosis of Single-Gene Disorders by Polar Body Analysis of Oocytes. J. Assist. Reprod. Genet. 1999, 16, 192–198. [Google Scholar] [CrossRef]
- Spits, C.; De Rycke, M.; Verpoest, W.; Lissens, W.; Van Steirteghem, A.; Liebaers, I.; Sermon, K. Preimplantation genetic diagnosis for Marfan syndrome. Fertil. Steril. 2006, 86, 310–320. [Google Scholar] [CrossRef]
- Coskun, S.; Alsmadi, O. Whole genome amplification from a single cell: A new era for preimplantation genetic diagnosis. Prenat. Diagn. 2007, 27, 297–302. [Google Scholar] [CrossRef] [PubMed]
- Spits, C.; Le Caignec, C.; De Rycke, M.; Van Haute, L.; Van Steirteghem, A.; Liebaers, I.; Sermon, K. Whole-genome multiple displacement amplification from single cells. Nat. Protoc. 2006, 1, 1965–1970. [Google Scholar] [CrossRef] [PubMed]
- Langmore, J.P. Rubicon Genomics, Inc. Pharmacogenomics 2002, 3, 557–560. [Google Scholar] [CrossRef] [PubMed]
- Zong, C.; Lu, S.; Chapman, A.R.; Xie, X.S. Genome-Wide Detection of Single-Nucleotide and Copy-Number Variations of a Single Human Cell. Science 2012, 338, 1622–1626. [Google Scholar] [CrossRef] [PubMed]
- Deleye, L.; Coninck, D.D.; Christodoulou, C.; Sante, T.; Dheedene, A.; Heindryckx, B.; Abbeel, E.V.D.; Sutter, P.D.; Menten, B.; Deforce, D.; et al. Whole genome amplification with SurePlex results in better copy number alteration detection using sequencing data compared to the MALBAC method. Sci. Rep. 2015, 5, 11711. [Google Scholar] [CrossRef]
- De Bourcy, C.F.A.; Vlaminck, I.D.; Kanbar, J.N.; Wang, J.; Gawad, C.; Quake, S.R. A Quantitative Comparison of Single-Cell Whole Genome Amplification Methods. PLoS ONE 2014, 9, e105585. [Google Scholar] [CrossRef]
- Deleye, L.; Gansemans, Y.; De Coninck, D.; Van Nieuwerburgh, F.; Deforce, D. Massively parallel sequencing of micro-manipulated cells targeting a comprehensive panel of disease-causing genes: A comparative evaluation of upstream whole-genome amplification methods. PLoS ONE 2018, 13, e0196334. [Google Scholar] [CrossRef]
- Renwick, P.; Trussler, J.; Lashwood, A.; Braude, P.; Ogilvie, C.M. Preimplantation genetic haplotyping: 127 diagnostic cycles demonstrating a robust, efficient alternative to direct mutation testing on single cells. Reprod. Biomed. Online 2010, 20, 470–476. [Google Scholar] [CrossRef]
- LaFramboise, T. Single nucleotide polymorphism arrays: A decade of biological, computational and technological advances. Nucleic Acids Res. 2009, 37, 4181–4193. [Google Scholar] [CrossRef]
- Handyside, A.H.; Harton, G.L.; Mariani, B.; Thornhill, A.R.; Affara, N.; Shaw, M.-A.; Griffin, D.K. Karyomapping: A universal method for genome wide analysis of genetic disease based on mapping crossovers between parental haplotypes. J. Med. Genet. 2009, 47, 651–658. [Google Scholar] [CrossRef]
- García-Bermúdez, M.; Piyamongkol, W.; Tomaz, S.; Dudman, E.; Sherlock, J.K.; Wells, D. Single-cell sequencing and mini-sequencing for preimplantation genetic diagnosis. Prenat. Diagn. 2003, 23, 669–677. [Google Scholar] [CrossRef] [PubMed]
- Chow, J.F.C.; Cheng, H.H.Y.; Lau, E.Y.L.; Yeung, W.S.B.; Ng, E.H.Y. Distinguishing between carrier and noncarrier embryos with the use of long-read sequencing in preimplantation genetic testing for reciprocal translocations. Genomics 2020, 112, 494–500. [Google Scholar] [CrossRef] [PubMed]
- Kubicek, D.; Hornak, M.; Horak, J.; Navratil, R.; Tauwinklova, G.; Rubes, J.; Vesela, K. Incidence and origin of meiotic whole and segmental chromosomal aneuploidies detected by karyomapping. Reprod. Biomed. Online 2019, 38, 330–339. [Google Scholar] [CrossRef] [PubMed]
- Yin, X.; Tan, K.; Vajta, G.; Jiang, H.; Tan, Y.; Zhang, C.; Chen, F.; Chen, S.; Zhang, C.; Pan, X.; et al. Massively Parallel Sequencing for Chromosomal Abnormality Testing in Trophectoderm Cells of Human Blastocysts1. Boil. Reprod. 2013, 88, 69. [Google Scholar] [CrossRef] [PubMed]
- Yan, L.; Huang, L.; Xu, L.; Huang, J.; Ma, F.; Zhu, X.; Tang, Y.; Liu, M.; Lian, Y.; Liu, P.; et al. Live births after simultaneous avoidance of monogenic diseases and chromosome abnormality by next-generation sequencing with linkage analyses. Proc. Natl. Acad. Sci. USA 2015, 112, 15964–15969. [Google Scholar] [CrossRef] [PubMed]
- Chamayou, S.; Sicali, M.; Lombardo, D.; Alecci, C.; Ragolia, C.; Maglia, E.; Liprino, A.; Cardea, C.; Storaci, G.; Romano, S.; et al. Universal strategy for preimplantation genetic testing for cystic fibrosis based on next generation sequencing. J. Assist. Reprod. Genet. 2019, 37, 213–222. [Google Scholar] [CrossRef]
- Del Rey, J.; Vidal, F.; Ramírez, L.; Borràs, N.; Corrales, I.; Garcia, I.; Garcia-Martínez, I.; Fernandez, S.F.; Garcia-Cruz, R.; Pujol, A.; et al. Novel Double Factor PGT strategy analyzing blastocyst stage embryos in a single NGS procedure. PLoS ONE 2018, 13, e0205692. [Google Scholar] [CrossRef]
- De Geyter, C.; Calhaz-Jorge, C.; Kupka, M.S.; Wyns, C.; Mocanu, E.; Motrenko, T.; Scaravelli, G.; Smeenk, J.; Vidakovic, S.; Goossens, V.; et al. ART in Europe, 2015: Results generated from European registries by ESHRE. Hum. Reprod. Open 2020, 2020. [Google Scholar] [CrossRef]
- De Rycke, M.; De Vos, A.; Belva, F.; Berckmoes, V.; Bonduelle, M.; Buysse, A.; Keymolen, K.; Liebaers, I.; Nekkebroeck, J.; Verdyck, P.; et al. Preimplantation genetic testing with HLA matching: From counseling to birth and beyond. J. Hum. Genet. 2020, 65, 445–454. [Google Scholar] [CrossRef]
- Davies, M.J.; Moore, V.M.; Willson, K.J.; Van Essen, P.; Priest, K.; Scott, H.; Haan, E.A.; Chan, A. Reproductive Technologies and the Risk of Birth Defects. Obstet. Gynecol. Surv. 2012, 67, 527–528. [Google Scholar] [CrossRef]
- Pandey, S.; Shetty, A.; Hamilton, M.; Bhattacharya, S.; Maheshwari, A. Obstetric and perinatal outcomes in singleton pregnancies resulting from IVF/ICSI: A systematic review and meta-analysis. Hum. Reprod. Update 2012, 18, 485–503. [Google Scholar] [CrossRef] [PubMed]
- Zhu, J.; Zhu, Q.; Wang, Y.; Wang, B.; Lyu, Q.; Kuang, Y. Comparative study on risk for birth defects among infants after in vitro fertilization and intracytoplasmic sperm injection. Syst. Boil. Reprod. Med. 2018, 65, 54–60. [Google Scholar] [CrossRef] [PubMed]
- Desmyttere, S.; De Rycke, M.; De Schrijver, F.; Verpoest, W.; Haentjens, P.; Staessen, C.; Liebaers, I.; Bonduelle, M. Neonatal follow-up of 995 consecutively born children after embryo biopsy for PGD. Hum. Reprod. 2011, 27, 288–293. [Google Scholar] [CrossRef] [PubMed]
- Bay, B.; Ingerslev, H.J.; Lemmen, J.G.; Degn, B.; Rasmussen, I.A.; Kesmodel, U.S. Preimplantation genetic diagnosis: A national multicenter obstetric and neonatal follow-up study. Fertil. Steril. 2016, 106, 1363–1369. [Google Scholar] [CrossRef] [PubMed]
- Heijligers, M.; Van Montfoort, A.; Meijer-Hoogeveen, M.; Broekmans, F.; Bouman, K.; Homminga, I.; Dreesen, J.; Paulussen, A.; Engelen, J.; Coonen, E.; et al. Perinatal follow-up of children born after preimplantation genetic diagnosis between 1995 and 2014. J. Assist. Reprod. Genet. 2018, 35, 1995–2002. [Google Scholar] [CrossRef]
- He, H.; Jing, S.; Lu, C.F.; Tan, Y.Q.; Luo, K.L.; Zhang, S.P.; Gong, F.; Lu, G.X.; Lin, G. Neonatal outcomes of live births after blastocyst biopsy in preimplantation genetic testing cycles: A follow-up of 1721 children. Fertil. Steril. 2019, 112, 82–88. [Google Scholar] [CrossRef]
- Winter, C.; Van Acker, F.; Bonduelle, M.; Desmyttere, S.; De Schrijver, F.; Nekkebroeck, J. Cognitive and psychomotor development of 5- to 6-year-old singletons born after PGD: A prospective case-controlled matched study. Hum. Reprod. 2014, 29, 1968–1977. [Google Scholar] [CrossRef]
- Winter, C.; Van Acker, F.; Bonduelle, M.; Desmyttere, S.; Nekkebroeck, J. Psychosocial development of full term singletons, born after preimplantation genetic diagnosis (PGD) at preschool age and family functioning: A prospective case-controlled study and multi-informant approach. Hum. Reprod. 2015, 30, 1122–1136. [Google Scholar] [CrossRef]
- Sacks, G.C.; Altarescu, G.; Guedalia, J.; Varshaver, I.; Gilboa, T.; Levy-Lahad, E.; Eldar-Geva, T. Developmental neuropsychological assessment of 4- to 5-year-old children born following Preimplantation Genetic Diagnosis (PGD): A pilot study. Child Neuropsychol. 2016, 22, 458–471. [Google Scholar] [CrossRef]
- Heijligers, M.; Peeters, A.; Van Montfoort, A.; Nijsten, J.; Janssen, E.; Gunnewiek, F.K.; De Rooy, R.; Van Golde, R.; Coonen, E.; Meijer-Hoogeveen, M.; et al. Growth, health, and motor development of 5-year-old children born after preimplantation genetic diagnosis. Fertil. Steril. 2019, 111, 1151–1158. [Google Scholar] [CrossRef]
- Belva, F.; Roelants, M.; Kluijfhout, S.; Winter, C.; De Schrijver, F.; Desmyttere, S.; De Rycke, M.; Tournaye, H.; Liebaers, I.; Bonduelle, M. Body composition and blood pressure in 6-year-old singletons born after pre-implantation genetic testing for monogenic and structural chromosomal aberrations: A matched cohort study. Hum. Reprod. Open 2018, 2018. [Google Scholar] [CrossRef] [PubMed]

© 2020 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
De Rycke, M.; Berckmoes, V. Preimplantation Genetic Testing for Monogenic Disorders. Genes 2020, 11, 871. https://doi.org/10.3390/genes11080871
De Rycke M, Berckmoes V. Preimplantation Genetic Testing for Monogenic Disorders. Genes. 2020; 11(8):871. https://doi.org/10.3390/genes11080871
Chicago/Turabian StyleDe Rycke, Martine, and Veerle Berckmoes. 2020. "Preimplantation Genetic Testing for Monogenic Disorders" Genes 11, no. 8: 871. https://doi.org/10.3390/genes11080871
APA StyleDe Rycke, M., & Berckmoes, V. (2020). Preimplantation Genetic Testing for Monogenic Disorders. Genes, 11(8), 871. https://doi.org/10.3390/genes11080871