PTEN Alterations and Their Role in Cancer Management: Are We Making Headway on Precision Medicine?

 ,
,  ,
, 
 , ,
, , 

Abstract
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Structure and Cellular Roles of PTEN Tumor Suppressor
2.1. PTEN as a Downregulator of the PI3K/Akt/mTOR Pathway
2.2. Nuclear PTEN and Modulation of the DNA Damage Response
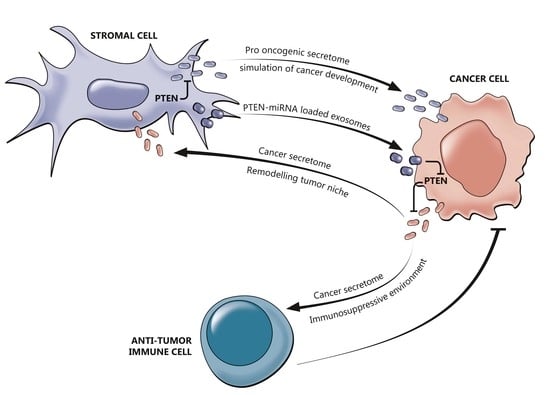
2.3. The Interplay between PTEN and the Tumor Immune Microenvironment
3. PTEN Molecular Aberrations Clinical Roles
3.1. Biomarker in Molecular Epidemiology
3.2. Phenotypic Variability in PTEN Germline Mutation Carriers and Genotype-Phenotype Relationship
3.3. Inactivating Mutations of PTEN in Non-Familial Solid Tumors
4. Clinical Actionability of PTEN Alterations
5. Concluding Remarks
6. Patents
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Masson, G.R.; Williams, R.L. Structural Mechanisms of PTEN Regulation. Cold Spring Harb. Perspect. Med. 2020, 10. [Google Scholar] [CrossRef] [PubMed]
- Steck, P.A.; Pershouse, M.A.; Jasser, S.A.; Yung, W.K.; Lin, H.; Ligon, A.H.; Langford, L.A.; Baumgard, M.L.; Hattier, T.; Davis, T.; et al. Identification of a candidate tumour suppressor gene, MMAC1, at chromosome 10q23.3 that is mutated in multiple advanced cancers. Nat. Genet. 1997, 15, 356–362. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Yen, C.; Liaw, D.; Podsypanina, K.; Bose, S.; Wang, S.I.; Puc, J.; Miliaresis, C.; Rodgers, L.; McCombie, R.; et al. PTEN, a putative protein tyrosine phosphatase gene mutated in human brain, breast, and prostate cancer. Science 1997, 275, 1943–1947. [Google Scholar] [CrossRef]
- Vidwans, S.J.; Turski, M.L.; Janku, F.; Garrido-Laguna, I.; Munoz, J.; Schwab, R.; Subbiah, V.; Rodon, J.; Kurzrock, R. A framework for genomic biomarker actionability and its use in clinical decision making. Oncoscience 2014, 1, 614–623. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.R.; Chen, M.; Pandolfi, P.P. The functions and regulation of the PTEN tumour suppressor: New modes and prospects. Nat. Rev. Mol. Cell Biol. 2018, 19, 547–562. [Google Scholar] [CrossRef]
- Kobayashi, Y.; Lim, S.O.; Yamaguchi, H. Oncogenic signaling pathways associated with immune evasion and resistance to immune checkpoint inhibitors in cancer. Semin. Cancer Biol. 2019. [Google Scholar] [CrossRef]
- Ma, J.; Benitez, J.A.; Li, J.; Miki, S.; Ponte de Albuquerque, C.; Galatro, T.; Orellana, L.; Zanca, C.; Reed, R.; Boyer, A.; et al. Inhibition of Nuclear PTEN Tyrosine Phosphorylation Enhances Glioma Radiation Sensitivity through Attenuated DNA Repair. Cancer Cell 2019, 35, 816. [Google Scholar] [CrossRef]
- Pagni, F.; Guerini-Rocco, E.; Schultheis, A.M.; Grazia, G.; Rijavec, E.; Ghidini, M.; Lopez, G.; Venetis, K.; Croci, G.A.; Malapelle, U.; et al. Targeting Immune-Related Biological Processes in Solid Tumors: We do Need Biomarkers. Int. J. Mol. Sci. 2019, 20, 5452. [Google Scholar] [CrossRef]
- Taylor, H.; Laurence, A.D.J.; Uhlig, H.H. The Role of PTEN in Innate and Adaptive Immunity. Cold Spring Harb. Perspect. Med. 2019, 9. [Google Scholar] [CrossRef] [PubMed]
- Pezzolesi, M.G.; Zbuk, K.M.; Waite, K.A.; Eng, C. Comparative genomic and functional analyses reveal a novel cis-acting PTEN regulatory element as a highly conserved functional E-box motif deleted in Cowden syndrome. Hum. Mol. Genet. 2007, 16, 1058–1071. [Google Scholar] [CrossRef]
- Iijima, M.; Huang, Y.E.; Luo, H.R.; Vazquez, F.; Devreotes, P.N. Novel mechanism of PTEN regulation by its phosphatidylinositol 4,5-bisphosphate binding motif is critical for chemotaxis. J. Biol. Chem. 2004, 279, 16606–16613. [Google Scholar] [CrossRef] [PubMed]
- Haynie, D.T.; Xue, B. Superdomains in the protein structure hierarchy: The case of PTP-C2. Protein Sci. 2015, 24, 874–882. [Google Scholar] [CrossRef]
- Chen, Z.; Dempsey, D.R.; Thomas, S.N.; Hayward, D.; Bolduc, D.M.; Cole, P.A. Molecular Features of Phosphatase and Tensin Homolog (PTEN) Regulation by C-terminal Phosphorylation. J. Biol. Chem. 2016, 291, 14160–14169. [Google Scholar] [CrossRef]
- Chen, C.Y.; Chen, J.; He, L.; Stiles, B.L. PTEN: Tumor Suppressor and Metabolic Regulator. Front. Endocrinol. 2018, 9, 338. [Google Scholar] [CrossRef]
- Lawrence, M.S.; Stojanov, P.; Mermel, C.H.; Robinson, J.T.; Garraway, L.A.; Golub, T.R.; Meyerson, M.; Gabriel, S.B.; Lander, E.S.; Getz, G. Discovery and saturation analysis of cancer genes across 21 tumour types. Nature 2014, 505, 495–501. [Google Scholar] [CrossRef] [PubMed]
- Hoxhaj, G.; Manning, B.D. The PI3K-AKT network at the interface of oncogenic signalling and cancer metabolism. Nat. Rev. Cancer 2020, 20, 74–88. [Google Scholar] [CrossRef] [PubMed]
- Porta, C.; Paglino, C.; Mosca, A. Targeting PI3K/Akt/mTOR Signaling in Cancer. Front. Oncol. 2014, 4, 64. [Google Scholar] [CrossRef] [PubMed]
- Grizzi, G.; Ghidini, M.; Botticelli, A.; Tomasello, G.; Ghidini, A.; Grossi, F.; Fusco, N.; Cabiddu, M.; Savio, T.; Petrelli, F. Strategies for Increasing the Effectiveness of Aromatase Inhibitors in Locally Advanced Breast Cancer: An Evidence-Based Review on Current Options. Cancer Manag. Res. 2020, 12, 675–686. [Google Scholar] [CrossRef]
- Ramapriyan, R.; Caetano, M.S.; Barsoumian, H.B.; Mafra, A.C.P.; Zambalde, E.P.; Menon, H.; Tsouko, E.; Welsh, J.W.; Cortez, M.A. Altered cancer metabolism in mechanisms of immunotherapy resistance. Pharmacol. Ther. 2019, 195, 162–171. [Google Scholar] [CrossRef]
- Gozzelino, L.; De Santis, M.C.; Gulluni, F.; Hirsch, E.; Martini, M. PI(3,4)P2 Signaling in Cancer and Metabolism. Front. Oncol. 2020, 10, 360. [Google Scholar] [CrossRef]
- Bilanges, B.; Posor, Y.; Vanhaesebroeck, B. PI3K isoforms in cell signalling and vesicle trafficking. Nat. Rev. Mol. Cell Biol. 2019, 20, 515–534. [Google Scholar] [CrossRef] [PubMed]
- Fruman, D.A.; Chiu, H.; Hopkins, B.D.; Bagrodia, S.; Cantley, L.C.; Abraham, R.T. The PI3K Pathway in Human Disease. Cell 2017, 170, 605–635. [Google Scholar] [CrossRef] [PubMed]
- Thorpe, L.M.; Yuzugullu, H.; Zhao, J.J. PI3K in cancer: Divergent roles of isoforms, modes of activation and therapeutic targeting. Nat. Rev. Cancer 2015, 15, 7–24. [Google Scholar] [CrossRef] [PubMed]
- Zbuk, K.M.; Eng, C. Cancer phenomics: RET and PTEN as illustrative models. Nat. Rev. Cancer 2007, 7, 35–45. [Google Scholar] [CrossRef]
- Papa, A.; Pandolfi, P.P. The PTEN(-)PI3K Axis in Cancer. Biomolecules 2019, 9, 153. [Google Scholar] [CrossRef]
- Song, M.S.; Salmena, L.; Pandolfi, P.P. The functions and regulation of the PTEN tumour suppressor. Nat. Rev. Mol. Cell Biol. 2012, 13, 283–296. [Google Scholar] [CrossRef]
- Ho, J.; Cruise, E.S.; Dowling, R.J.O.; Stambolic, V. PTEN Nuclear Functions. Cold Spring Harb. Perspect. Med. 2020, 10. [Google Scholar] [CrossRef]
- Naderali, E.; Khaki, A.A.; Rad, J.S.; Ali-Hemmati, A.; Rahmati, M.; Charoudeh, H.N. Regulation and modulation of PTEN activity. Mol. Biol. Rep. 2018, 45, 2869–2881. [Google Scholar] [CrossRef]
- Milella, M.; Falcone, I.; Conciatori, F.; Cesta Incani, U.; Del Curatolo, A.; Inzerilli, N.; Nuzzo, C.M.; Vaccaro, V.; Vari, S.; Cognetti, F.; et al. PTEN: Multiple Functions in Human Malignant Tumors. Front. Oncol. 2015, 5, 24. [Google Scholar] [CrossRef]
- Song, M.S.; Carracedo, A.; Salmena, L.; Song, S.J.; Egia, A.; Malumbres, M.; Pandolfi, P.P. Nuclear PTEN regulates the APC-CDH1 tumor-suppressive complex in a phosphatase-independent manner. Cell 2011, 144, 187–199. [Google Scholar] [CrossRef]
- Malaney, P.; Palumbo, E.; Semidey-Hurtado, J.; Hardee, J.; Stanford, K.; Kathiriya, J.J.; Patel, D.; Tian, Z.; Allen-Gipson, D.; Dave, V. PTEN Physically Interacts with and Regulates E2F1-mediated Transcription in Lung Cancer. Cell Cycle 2018, 17, 947–962. [Google Scholar] [CrossRef] [PubMed]
- Ge, M.K.; Zhang, N.; Xia, L.; Zhang, C.; Dong, S.S.; Li, Z.M.; Ji, Y.; Zheng, M.H.; Sun, J.; Chen, G.Q.; et al. FBXO22 degrades nuclear PTEN to promote tumorigenesis. Nat. Commun. 2020, 11, 1720. [Google Scholar] [CrossRef] [PubMed]
- Ming, M.; He, Y.Y. PTEN in DNA damage repair. Cancer Lett. 2012, 319, 125–129. [Google Scholar] [CrossRef]
- Vidotto, T.; Melo, C.M.; Castelli, E.; Koti, M.; Dos Reis, R.B.; Squire, J.A. Emerging role of PTEN loss in evasion of the immune response to tumours. Br. J. Cancer 2020, 122, 1732–1743. [Google Scholar] [CrossRef]
- Hou, S.Q.; Ouyang, M.; Brandmaier, A.; Hao, H.; Shen, W.H. PTEN in the maintenance of genome integrity: From DNA replication to chromosome segregation. BioEssays News Rev. Mol. Cell. Dev. Biol. 2017, 39. [Google Scholar] [CrossRef] [PubMed]
- Brandmaier, A.; Hou, S.Q.; Shen, W.H. Cell Cycle Control by PTEN. J. Mol. Biol. 2017, 429, 2265–2277. [Google Scholar] [CrossRef]
- Iyer, D.R.; Rhind, N. The Intra-S Checkpoint Responses to DNA Damage. Genes 2017, 8, 74. [Google Scholar] [CrossRef]
- Liu, T.; Wang, Y.; Wang, Y.; Chan, A.M. Multifaceted Regulation of PTEN Subcellular Distributions and Biological Functions. Cancers 2019, 11, 1247. [Google Scholar] [CrossRef]
- Chen, Z.H.; Zhu, M.; Yang, J.; Liang, H.; He, J.; He, S.; Wang, P.; Kang, X.; McNutt, M.A.; Yin, Y.; et al. PTEN interacts with histone H1 and controls chromatin condensation. Cell Rep. 2014, 8, 2003–2014. [Google Scholar] [CrossRef]
- Yang, J.; Yin, Y. PTEN in Chromatin Remodeling. Cold Spring Harb. Perspect. Med. 2020, 10. [Google Scholar] [CrossRef]
- Fan, X.; Kraynak, J.; Knisely, J.P.S.; Formenti, S.C.; Shen, W.H. PTEN as a Guardian of the Genome: Pathways and Targets. Cold Spring Harb. Perspect. Med. 2020. [Google Scholar] [CrossRef] [PubMed]
- Alvarez-Garcia, V.; Tawil, Y.; Wise, H.M.; Leslie, N.R. Mechanisms of PTEN loss in cancer: It’s all about diversity. Semin. Cancer Biol. 2019, 59, 66–79. [Google Scholar] [CrossRef] [PubMed]
- The Cancer Genome Atlas Research Network; Kandoth, C.; Schultz, N.; Cherniack, A.D.; Akbani, R.; Liu, Y.; Shen, H.; Robertson, A.G.; Pashtan, I.; Shen, R.; et al. Integrated genomic characterization of endometrial carcinoma. Nature 2013, 497, 67–73. [Google Scholar] [CrossRef]
- Huang, H.N.; Kuo, C.W.; Lin, M.C.; Mao, T.L.; Kuo, K.T. Frequent CTNNB1 or PIK3CA Mutations Occurred in Endometrial Endometrioid Adenocarcinoma With High Levels of Microsatellite Instability and Loss of MSH2/MSH6 Expression. Appl. Immunohistochem. Mol. Morphol. 2020, 28, 284–289. [Google Scholar] [CrossRef] [PubMed]
- Chang, H.; Cai, Z.; Roberts, T.M. The Mechanisms Underlying PTEN Loss in Human Tumors Suggest Potential Therapeutic Opportunities. Biomolecules 2019, 9, 713. [Google Scholar] [CrossRef]
- Djordjevic, B.; Barkoh, B.A.; Luthra, R.; Broaddus, R.R. Relationship between PTEN, DNA mismatch repair, and tumor histotype in endometrial carcinoma: Retained positive expression of PTEN preferentially identifies sporadic non-endometrioid carcinomas. Modern Pathol. 2013, 26, 1401–1412. [Google Scholar] [CrossRef]
- Lopez, G.; Noale, M.; Corti, C.; Gaudioso, G.; Sajjadi, E.; Venetis, K.; Gambini, D.; Runza, L.; Costanza, J.; Pesenti, C.; et al. PTEN Expression as a Complementary Biomarker for Mismatch Repair Testing in Breast Cancer. Int. J. Mol. Sci. 2020, 21, 1461. [Google Scholar] [CrossRef]
- Lemery, S.; Keegan, P.; Pazdur, R. First FDA Approval Agnostic of Cancer Site - When a Biomarker Defines the Indication. N. Engl. J. Med. 2017, 377, 1409–1412. [Google Scholar] [CrossRef]
- Davies, H.; Morganella, S.; Purdie, C.A.; Jang, S.J.; Borgen, E.; Russnes, H.; Glodzik, D.; Zou, X.; Viari, A.; Richardson, A.L.; et al. Whole-Genome Sequencing Reveals Breast Cancers with Mismatch Repair Deficiency. Cancer Res. 2017, 77, 4755–4762. [Google Scholar] [CrossRef]
- Cheng, A.S.; Leung, S.C.Y.; Gao, D.; Burugu, S.; Anurag, M.; Ellis, M.J.; Nielsen, T.O. Mismatch repair protein loss in breast cancer: Clinicopathological associations in a large British Columbia cohort. Breast Cancer Res. Treat. 2020, 179, 3–10. [Google Scholar] [CrossRef]
- Lee, S.E.; Lee, H.S.; Kim, K.Y.; Park, J.H.; Roh, H.; Park, H.Y.; Kim, W.S. High prevalence of the MLH1 V384D germline mutation in patients with HER2-positive luminal B breast cancer. Sci. Rep. 2019, 9, 10966. [Google Scholar] [CrossRef] [PubMed]
- Le, D.T.; Durham, J.N.; Smith, K.N.; Wang, H.; Bartlett, B.R.; Aulakh, L.K.; Lu, S.; Kemberling, H.; Wilt, C.; Luber, B.S.; et al. Mismatch-repair deficiency predicts response of solid tumors to PD-1 blockade. Science 2017, 357, 409–413. [Google Scholar] [CrossRef] [PubMed]
- Horimoto, Y.; Thinzar Hlaing, M.; Saeki, H.; Kitano, S.; Nakai, K.; Sasaki, R.; Kurisaki-Arakawa, A.; Arakawa, A.; Otsuji, N.; Matsuoka, S.; et al. Microsatellite instability and mismatch repair protein expressions in lymphocyte-predominant breast cancer. Cancer Sci. 2020. [Google Scholar] [CrossRef] [PubMed]
- Fusco, N.; Lopez, G.; Corti, C.; Pesenti, C.; Colapietro, P.; Ercoli, G.; Gaudioso, G.; Faversani, A.; Gambini, D.; Michelotti, A.; et al. Mismatch Repair Protein Loss as a Prognostic and Predictive Biomarker in Breast Cancers Regardless of Microsatellite Instability. JNCI Cancer Spectr. 2018, 2, pky056. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Guo, D. The functions of tumor suppressor PTEN in innate and adaptive immunity. Cell. Mol. Immunol. 2017, 14, 581–589. [Google Scholar] [CrossRef]
- Bensinger, S.J.; Walsh, P.T.; Zhang, J.; Carroll, M.; Parsons, R.; Rathmell, J.C.; Thompson, C.B.; Burchill, M.A.; Farrar, M.A.; Turka, L.A. Distinct IL-2 receptor signaling pattern in CD4+CD25+ regulatory T cells. J. Immunol. 2004, 172, 5287–5296. [Google Scholar] [CrossRef]
- Dong, Y.; Richards, J.; Gupta, R.; Aung, P.; Emley, A.; Kluger, Y.; Dogra, S.; Mahalingam, M.; Wajapeyee, N. PTEN Functions as a Melanoma Tumor Suppressor by Promoting Host Immune Response. Oncogene 2014, 33. [Google Scholar] [CrossRef]
- Ying, H.; Elpek, K.; Vinjamoori, A.; Zimmerman, S.; Chu, G.; Yan, H.; Fletcher-Sananikone, E.; Zhang, H.; Liu, Y.; Wang, W.; et al. PTEN Is a Major Tumor Suppressor in Pancreatic Ductal Adenocarcinoma and Regulates an NF-κB-cytokine Network. Cancer Discov. 2011, 1. [Google Scholar] [CrossRef]
- Bezzi, M.; Seitzer, N.; Ishikawa, T.; Reschke, M.; Chen, M.; Wang, G.; Mitchell, C.; Ng, C.; Katon, J.; Lunardi, A.; et al. Diverse Genetic-Driven Immune Landscapes Dictate Tumor Progression Through Distinct Mechanisms. Nat. Med. 2018, 24. [Google Scholar] [CrossRef]
- Bronisz, A.; Godlewski, J.; Wallace, J.; Merchant, A.; Nowicki, M.; Mathsyaraja, H.; Srinivasan, R.; Trimboli, A.; Martin, C.; Li, F.; et al. Reprogramming of the Tumour Microenvironment by Stromal PTEN-regulated miR-320. Nat. Cell Biol. 2011, 14. [Google Scholar] [CrossRef]
- Trimboli, A.J.; Cantemir-Stone, C.Z.; Li, F.; Wallace, J.A.; Merchant, A.; Creasap, N.; Thompson, J.C.; Caserta, E.; Wang, H.; Chong, J.L.; et al. Pten in Stromal Fibroblasts Suppresses Mammary Epithelial Tumors. Nature 2009, 461, 1084–1091. [Google Scholar] [CrossRef] [PubMed]
- Chandler, C.; Liu, T.; Buckanovich, R.; Coffman, L.G. The double edge sword of fibrosis in cancer. Transl. Res. 2019, 209, 55–67. [Google Scholar] [CrossRef] [PubMed]
- Marchiò, C.; De Filippo, M.R.; Ng, C.K.; Piscuoglio, S.; Soslow, R.A.; Reis-Filho, J.S.; Weigelt, B. PIKing the type and pattern of PI3K pathway mutations in endometrioid endometrial carcinomas. Gynecol. Oncol. 2015, 137, 321–328. [Google Scholar] [CrossRef] [PubMed]
- Mecca, C.; Giambanco, I.; Donato, R.; Arcuri, C. Targeting mTOR in Glioblastoma: Rationale and Preclinical/Clinical Evidence. Dis. Markers 2018, 2018, 9230479. [Google Scholar] [CrossRef]
- Phin, S.; Moore, M.W.; Cotter, P.D. Genomic Rearrangements of PTEN in Prostate Cancer. Front. Oncol. 2013, 3, 240. [Google Scholar] [CrossRef]
- Braglia, L.; Zavatti, M.; Vinceti, M.; Martelli, A.M.; Marmiroli, S. Deregulated PTEN/PI3K/AKT/mTOR signaling in prostate cancer: Still a potential druggable target? Biochim. Biophys. Acta Mol. Cell Res. 2020, 1867, 118731. [Google Scholar] [CrossRef]
- Carbognin, L.; Miglietta, F.; Paris, I.; Dieci, M.V. Prognostic and Predictive Implications of PTEN in Breast Cancer: Unfulfilled Promises but Intriguing Perspectives. Cancers 2019, 11, 1401. [Google Scholar] [CrossRef]
- Jamaspishvili, T.; Berman, D.M.; Ross, A.E.; Scher, H.I.; De Marzo, A.M.; Squire, J.A.; Lotan, T.L. Clinical implications of PTEN loss in prostate cancer. Nat. Rev. Urol. 2018, 15, 222–234. [Google Scholar] [CrossRef]
- Gkountakos, A.; Sartori, G.; Falcone, I.; Piro, G.; Ciuffreda, L.; Carbone, C.; Tortora, G.; Scarpa, A.; Bria, E.; Milella, M.; et al. PTEN in Lung Cancer: Dealing with the Problem, Building on New Knowledge and Turning the Game Around. Cancers 2019, 11, 1141. [Google Scholar] [CrossRef]
- Zhang, H.Y.; Liang, F.; Jia, Z.L.; Song, S.T.; Jiang, Z.F. mutation, methylation and expression in breast cancer patients. Oncol. Lett. 2013, 6, 161–168. [Google Scholar] [CrossRef]
- Mirmohammadsadegh, A.; Marini, A.; Nambiar, S.; Hassan, M.; Tannapfel, A.; Ruzicka, T.; Hengge, U.R. Epigenetic silencing of the PTEN gene in melanoma. Cancer Res. 2006, 66, 6546–6552. [Google Scholar] [CrossRef] [PubMed]
- Zhou, X.P.; Loukola, A.; Salovaara, R.; Nystrom-Lahti, M.; Peltomäki, P.; de la Chapelle, A.; Aaltonen, L.A.; Eng, C. PTEN mutational spectra, expression levels, and subcellular localization in microsatellite stable and unstable colorectal cancers. Am. J. Pathol. 2002, 161, 439–447. [Google Scholar] [CrossRef]
- Frattini, M.; Signoroni, S.; Pilotti, S.; Bertario, L.; Benvenuti, S.; Zanon, C.; Bardelli, A.; Pierotti, M.A. Phosphatase protein homologue to tensin expression and phosphatidylinositol-3 phosphate kinase mutations in colorectal cancer. Cancer Res. 2005, 65, 11227. [Google Scholar] [CrossRef][Green Version]
- Naguib, A.; Cooke, J.C.; Happerfield, L.; Kerr, L.; Gay, L.J.; Luben, R.N.; Ball, R.Y.; Mitrou, P.N.; McTaggart, A.; Arends, M.J. Alterations in PTEN and PIK3CA in colorectal cancers in the EPIC Norfolk study: Associations with clinicopathological and dietary factors. BMC Cancer 2011, 11, 123. [Google Scholar] [CrossRef] [PubMed]
- Molinari, F.; Frattini, M. Functions and Regulation of the PTEN Gene in Colorectal Cancer. Front. Oncol. 2013, 3, 326. [Google Scholar] [CrossRef]
- Chalhoub, N.; Baker, S.J. PTEN and the PI3-kinase pathway in cancer. Annu. Rev. Pathol. 2009, 4, 127–150. [Google Scholar] [CrossRef]
- Tamguney, T.; Stokoe, D. New insights into PTEN. J. Cell Sci. 2007, 120, 4071–4079. [Google Scholar] [CrossRef]
- Hollander, M.C.; Blumenthal, G.M.; Dennis, P.A. PTEN loss in the continuum of common cancers, rare syndromes and mouse models. Nat. Rev. Cancer 2011, 11, 289–301. [Google Scholar] [CrossRef]
- Yu, Y.X.; Wang, Y.; Liu, H. Overexpression of PTEN suppresses non-small-cell lung carcinoma metastasis through inhibition of integrin alphaVbeta6 signaling. Am. J. Transl. Res. 2017, 9, 3304–3314. [Google Scholar]
- Liu, L.; Huang, L.; He, J.; Cai, S.; Weng, Y.; Huang, S.; Ma, S. PTEN inhibits non-small cell lung cancer cell growth by promoting G0/G1 arrest and cell apoptosis. Oncol. Lett. 2019, 17, 1333–1340. [Google Scholar] [CrossRef]
- Li, B.; Zhang, J.; Su, Y.; Hou, Y.; Wang, Z.; Zhao, L.; Sun, S.; Fu, H. Overexpression of PTEN may increase the effect of pemetrexed on A549 cells via inhibition of the PI3K/AKT/mTOR pathway and carbohydrate metabolism. Mol. Med. Rep. 2019, 20, 3793–3801. [Google Scholar] [CrossRef] [PubMed]
- Bao, L.; Li, X.; Lin, Z. PTEN overexpression promotes glioblastoma death through triggering mitochondrial division and inactivating the Akt pathway. J. Recept. Signal Transduct. Res. 2019, 39, 215–225. [Google Scholar] [CrossRef] [PubMed]
- Li, M.F.; Guan, H.; Zhang, D.D. Effect of overexpression of PTEN on apoptosis of liver cancer cells. Genet. Mol. Res. GMR 2016, 15. [Google Scholar] [CrossRef] [PubMed]
- Wu, H.; Wang, K.; Liu, W.; Hao, Q. PTEN overexpression improves cisplatin-resistance of human ovarian cancer cells through upregulating KRT10 expression. Biochem. Biophys. Res. Commun. 2014, 444, 141–146. [Google Scholar] [CrossRef] [PubMed]
- de Araujo, W.M.; Robbs, B.K.; Bastos, L.G.; de Souza, W.F.; Vidal, F.C.; Viola, J.P.; Morgado-Diaz, J.A. PTEN Overexpression Cooperates With Lithium to Reduce the Malignancy and to Increase Cell Death by Apoptosis via PI3K/Akt Suppression in Colorectal Cancer Cells. J. Cell. Biochem. 2016, 117, 458–469. [Google Scholar] [CrossRef]
- Pal, A.; Barber, T.M.; Van de Bunt, M.; Rudge, S.A.; Zhang, Q.; Lachlan, K.L.; Cooper, N.S.; Linden, H.; Levy, J.C.; Wakelam, M.J.; et al. PTEN mutations as a cause of constitutive insulin sensitivity and obesity. N. Engl. J. Med. 2012, 367, 1002–1011. [Google Scholar] [CrossRef]
- Leslie, N.R.; Longy, M. Inherited PTEN mutations and the prediction of phenotype. Semin. Cell Dev. Biol. 2016, 52, 30–38. [Google Scholar] [CrossRef]
- Pilarski, R.; Burt, R.; Kohlman, W.; Pho, L.; Shannon, K.M.; Swisher, E. Cowden syndrome and the PTEN hamartoma tumor syndrome: Systematic review and revised diagnostic criteria. J. Natl. Cancer Inst. 2013, 105, 1607–1616. [Google Scholar] [CrossRef]
- Haddadi, N.; Travis, G.; Nassif, N.T.; Simpson, A.M.; Marsh, D.J. Toward Systems Pathology for PTEN Diagnostics. Cold Spring Harb. Perspect. Med. 2020, 10. [Google Scholar] [CrossRef]
- Tan, M.H.; Mester, J.L.; Ngeow, J.; Rybicki, L.A.; Orloff, M.S.; Eng, C. Lifetime cancer risks in individuals with germline PTEN mutations. Clin. Cancer Res. 2012, 18, 400–407. [Google Scholar] [CrossRef]
- Bubien, V.; Bonnet, F.; Brouste, V.; Hoppe, S.; Barouk-Simonet, E.; David, A.; Edery, P.; Bottani, A.; Layet, V.; Caron, O.; et al. High cumulative risks of cancer in patients with PTEN hamartoma tumour syndrome. J. Med. Genet. 2013, 50, 255–263. [Google Scholar] [CrossRef]
- Hoang, L.N.; Gilks, B.C. Hereditary Breast and Ovarian Cancer Syndrome: Moving Beyond BRCA1 and BRCA2. Adv. Anat. Pathol. 2018, 25, 85–95. [Google Scholar] [CrossRef] [PubMed]
- Keeney, M.G.; Couch, F.J.; Visscher, D.W.; Lindor, N.M. Non-BRCA familial breast cancer: Review of reported pathology and molecular findings. Pathology 2017, 49, 363–370. [Google Scholar] [CrossRef] [PubMed]
- Zhou, X.P.; Marsh, D.J.; Morrison, C.D.; Chaudhury, A.R.; Maxwell, M.; Reifenberger, G.; Eng, C. Germline inactivation of PTEN and dysregulation of the phosphoinositol-3-kinase/Akt pathway cause human Lhermitte-Duclos disease in adults. Am. J. Hum. Genet. 2003, 73, 1191–1198. [Google Scholar] [CrossRef] [PubMed]
- Abel, T.W.; Baker, S.J.; Fraser, M.M.; Tihan, T.; Nelson, J.S.; Yachnis, A.T.; Bouffard, J.P.; Mena, H.; Burger, P.C.; Eberhart, C.G. Lhermitte-Duclos disease: A report of 31 cases with immunohistochemical analysis of the PTEN/AKT/mTOR pathway. J. Neuropathol. Exp. Neurol. 2005, 64, 341–349. [Google Scholar] [CrossRef] [PubMed]
- Zhou, X.; Hampel, H.; Thiele, H.; Gorlin, R.J.; Hennekam, R.C.; Parisi, M.; Winter, R.M.; Eng, C. Association of germline mutation in the PTEN tumour suppressor gene and Proteus and Proteus-like syndromes. Lancet 2001, 358, 210–211. [Google Scholar] [CrossRef]
- Happle, R. Linear Cowden nevus: A new distinct epidermal nevus. Eur. J. Dermatol. 2007, 17, 133–136. [Google Scholar] [CrossRef]
- Caux, F.; Plauchu, H.; Chibon, F.; Faivre, L.; Fain, O.; Vabres, P.; Bonnet, F.; Selma, Z.B.; Laroche, L.; Gérard, M.; et al. Segmental overgrowth, lipomatosis, arteriovenous malformation and epidermal nevus (SOLAMEN) syndrome is related to mosaic PTEN nullizygosity. Eur. J. Hum. Genet. 2007, 15, 767–773. [Google Scholar] [CrossRef]
- Butler, M.G.; Dasouki, M.J.; Zhou, X.P.; Talebizadeh, Z.; Brown, M.; Takahashi, T.N.; Miles, J.H.; Wang, C.H.; Stratton, R.; Pilarski, R.; et al. Subset of individuals with autism spectrum disorders and extreme macrocephaly associated with germline PTEN tumour suppressor gene mutations. J. Med. Genet. 2005, 42, 318–321. [Google Scholar] [CrossRef]
- Pilarski, R. Cowden syndrome: A critical review of the clinical literature. J. Genet. Couns. 2009, 18, 13–27. [Google Scholar] [CrossRef]
- Marsh, D.J.; Coulon, V.; Lunetta, K.L.; Rocca-Serra, P.; Dahia, P.L.; Zheng, Z.; Liaw, D.; Caron, S.; Duboué, B.; Lin, A.Y.; et al. Mutation spectrum and genotype-phenotype analyses in Cowden disease and Bannayan-Zonana syndrome, two hamartoma syndromes with germline PTEN mutation. Hum. Mol. Genet. 1998, 7, 507–515. [Google Scholar] [CrossRef] [PubMed]
- Eng, C. PTEN: One gene, many syndromes. Hum. Mutat. 2003, 22, 183–198. [Google Scholar] [CrossRef] [PubMed]
- Puc, J.; Keniry, M.; Li, H.S.; Pandita, T.K.; Choudhury, A.D.; Memeo, L.; Mansukhani, M.; Murty, V.V.; Gaciong, Z.; Meek, S.E.; et al. Lack of PTEN sequesters CHK1 and initiates genetic instability. Cancer Cell 2005, 7, 193–204. [Google Scholar] [CrossRef] [PubMed]
- Kong, D.; Suzuki, A.; Zou, T.T.; Sakurada, A.; Kemp, L.W.; Wakatsuki, S.; Yokoyama, T.; Yamakawa, H.; Furukawa, T.; Sato, M.; et al. PTEN1 is frequently mutated in primary endometrial carcinomas. Nat. Genet. 1997, 17, 143–144. [Google Scholar] [CrossRef]
- Zhang, Y.; Park, J.; Han, S.J.; Yang, S.Y.; Yoon, H.J.; Park, I.; Woo, H.A.; Lee, S.R. Redox regulation of tumor suppressor PTEN in cell signaling. Redox Biol. 2020, 34, 101553. [Google Scholar] [CrossRef]
- Chen, L.; Liu, S.; Tao, Y. Regulating tumor suppressor genes: Post-translational modifications. Signal Transduct. Target Ther. 2020, 5, 90. [Google Scholar] [CrossRef]
- Juric, D.; Castel, P.; Griffith, M.; Griffith, O.L.; Won, H.H.; Ellis, H.; Ebbesen, S.H.; Ainscough, B.J.; Ramu, A.; Iyer, G.; et al. Convergent loss of PTEN leads to clinical resistance to a PI(3)Kα inhibitor. Nature 2015, 518, 240–244. [Google Scholar] [CrossRef]
- Costa, C.; Wang, Y.; Ly, A.; Hosono, Y.; Murchie, E.; Walmsley, C.S.; Huynh, T.; Healy, C.; Peterson, R.; Yanase, S.; et al. PTEN Loss Mediates Clinical Cross-Resistance to CDK4/6 and PI3Kalpha Inhibitors in Breast Cancer. Cancer Discov. 2020, 10, 72–85. [Google Scholar] [CrossRef]
- Razavi, P.; Dickler, M.N.; Shah, P.D.; Toy, W.; Brown, D.N.; Won, H.H.; Li, B.T.; Shen, R.; Vasan, N.; Modi, S.; et al. Alterations in PTEN and ESR1 promote clinical resistance to alpelisib plus aromatase inhibitors. Nat. Cancer 2020, 1, 382–393. [Google Scholar] [CrossRef]








© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fusco, N.; Sajjadi, E.; Venetis, K.; Gaudioso, G.; Lopez, G.; Corti, C.; Guerini Rocco, E.; Criscitiello, C.; Malapelle, U.; Invernizzi, M. PTEN Alterations and Their Role in Cancer Management: Are We Making Headway on Precision Medicine? Genes 2020, 11, 719. https://doi.org/10.3390/genes11070719
Fusco N, Sajjadi E, Venetis K, Gaudioso G, Lopez G, Corti C, Guerini Rocco E, Criscitiello C, Malapelle U, Invernizzi M. PTEN Alterations and Their Role in Cancer Management: Are We Making Headway on Precision Medicine? Genes. 2020; 11(7):719. https://doi.org/10.3390/genes11070719
Chicago/Turabian StyleFusco, Nicola, Elham Sajjadi, Konstantinos Venetis, Gabriella Gaudioso, Gianluca Lopez, Chiara Corti, Elena Guerini Rocco, Carmen Criscitiello, Umberto Malapelle, and Marco Invernizzi. 2020. "PTEN Alterations and Their Role in Cancer Management: Are We Making Headway on Precision Medicine?" Genes 11, no. 7: 719. https://doi.org/10.3390/genes11070719
APA StyleFusco, N., Sajjadi, E., Venetis, K., Gaudioso, G., Lopez, G., Corti, C., Guerini Rocco, E., Criscitiello, C., Malapelle, U., & Invernizzi, M. (2020). PTEN Alterations and Their Role in Cancer Management: Are We Making Headway on Precision Medicine? Genes, 11(7), 719. https://doi.org/10.3390/genes11070719