Do Genes Associated with Dyslexia of Chinese Characters Evolve Neutrally?


Abstract
:1. Introduction
2. Materials and Methods
2.1. Examined SNPs
2.2. Study Populations
2.2.1. Study Populations for nSL
2.2.2. Study Populations for 2D SFS-Based Statistics
2.3. nSL
2.4. 2D SFS-Based Statistics
2.4.1. Overview of 2D SFS-Based Statistics
2.4.2. Simulations
2.4.3. Screening of the Candidate Core Regions under Selective Sweep
2.4.4. Searching for the Target Site of Natural Selection (“Target Site”)
3. Results
3.1. nSL
3.2. 2D SFS-Based Statistics
3.2.1. Screening of the Candidate Core Regions under Selective Sweep
3.2.2. Searching for the Target Site of Natural Selection
rs17031962 on GNPTAB
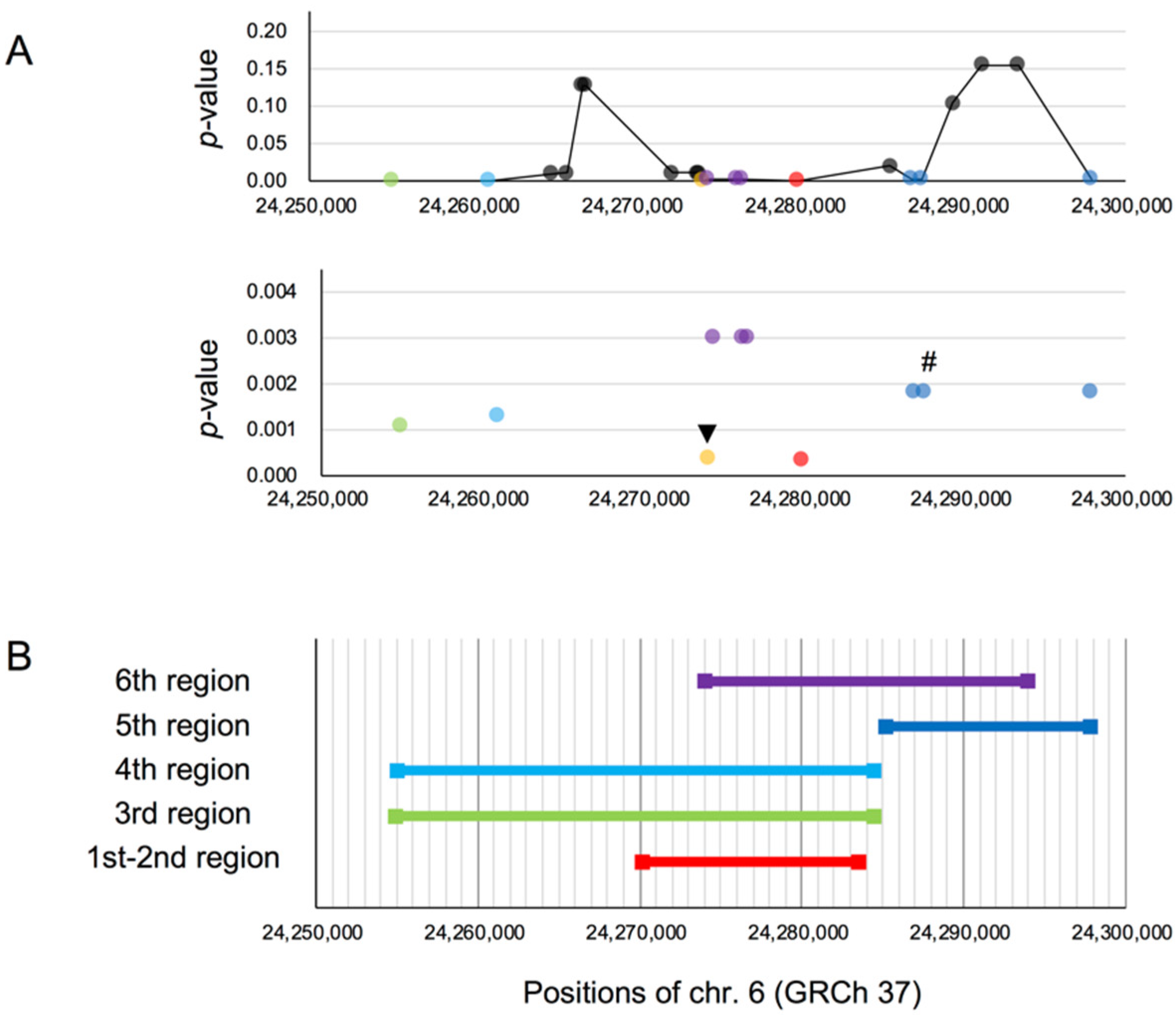
rs3789228 on DCDC2, as the Younger SNP of rs1091047
4. Discussion
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Paracchini, S.; Scerri, T.; Monaco, A.P. The Genetic Lexicon of Dyslexia. Annu. Rev. Genom. Hum. Genet. 2007, 8, 57–79. [Google Scholar] [CrossRef]
- Scerri, T.S.; Schulte-Körne, G. Genetics of developmental dyslexia. Eur. Child Adolesc. Psychiatry 2010, 19, 179–197. [Google Scholar] [CrossRef] [Green Version]
- Carrion-Castillo, A.; Franke, B.; Fisher, S.E. Molecular Genetics of Dyslexia: An Overview. Dyslexia 2013, 19, 214–240. [Google Scholar] [CrossRef]
- Kere, J. The molecular genetics and neurobiology of developmental dyslexia as model of a complex phenotype. Biochem. Biophys. Res. Commun. 2014, 452, 236–243. [Google Scholar] [CrossRef] [Green Version]
- Newbury, D.; Monaco, A.; Paracchini, S. Reading and Language Disorders: The Importance of Both Quantity and Quality. Genes 2014, 5, 285–309. [Google Scholar] [CrossRef]
- Peterson, R.L.; Pennington, B.F. Developmental Dyslexia. Annu. Rev. Clin. Psychol. 2015, 11, 283–307. [Google Scholar] [CrossRef]
- Daniels, P.T.; Share, D.L. Writing System Variation and Its Consequences for Reading and Dyslexia. Sci. Stud. Read. 2018, 22, 101–116. [Google Scholar] [CrossRef]
- Bishop, D.V.M. The interface between genetics and psychology: Lessons from developmental dyslexia. Proc. R. Soc. B Biol. Sci. 2015, 282, 20143139. [Google Scholar] [CrossRef] [Green Version]
- Protopapas, A.; Parrila, R. Is Dyslexia a Brain Disorder? Brain Sci. 2018, 8, 61. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fisher, S.E.; DeFries, J.C. Developmental dyslexia: Genetic dissection of a complex cognitive trait. Nat. Rev. Neurosci. 2002, 3, 767–780. [Google Scholar] [CrossRef] [Green Version]
- Plomin, R.; Haworth, C.M.A.; Davis, O.S.P. Common disorders are quantitative traits. Nat. Rev. Genet. 2009, 10, 872–878. [Google Scholar] [CrossRef] [PubMed]
- Su, M.; Wang, J.; Maurer, U.; Zhang, Y.; Li, J.; McBride, C.; Tardif, T.; Liu, Y.; Shu, H. Gene–environment interaction on neural mechanisms of orthographic processing in Chinese children. J. Neurolinguistics 2015, 33, 172–186. [Google Scholar] [CrossRef] [PubMed]
- Sharma, P.; Sagar, R. Unfolding the genetic pathways of dyslexia in Asian population: A review. Asian J. Psychiatr. 2017, 30, 225–229. [Google Scholar] [CrossRef] [PubMed]
- Sun, X.; Song, S.; Liang, X.; Xie, Y.; Zhao, C.; Zhang, Y.; Shu, H.; Gong, G. ROBO1 polymorphisms, callosal connectivity, and reading skills. Hum. Brain Mapp. 2017, 38, 2616–2626. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shao, S.; Kong, R.; Zou, L.; Zhong, R.; Lou, J.; Zhou, J.; Guo, S.; Wang, J.; Zhang, X.; Zhang, J.; et al. The Roles of Genes in the Neuronal Migration and Neurite Outgrowth Network in Developmental Dyslexia: Single- and Multiple-Risk Genetic Variants. Mol. Neurobiol. 2016, 53, 3967–3975. [Google Scholar] [CrossRef] [PubMed]
- Shao, S.; Niu, Y.; Zhang, X.; Kong, R.; Wang, J.; Liu, L.; Luo, X.; Zhang, J.; Song, R. Opposite Associations between Individual KIAA0319 Polymorphisms and Developmental Dyslexia Risk across Populations: A Stratified Meta-Analysis by the Study Population. Sci. Rep. 2016, 6, 30454. [Google Scholar] [CrossRef]
- Taipale, M.; Kaminen, N.; Nopola-Hemmi, J.; Haltia, T.; Myllyluoma, B.; Lyytinen, H.; Muller, K.; Kaaranen, M.; Lindsberg, P.J.; Hannula-Jouppi, K.; et al. A candidate gene for developmental dyslexia encodes a nuclear tetratricopeptide repeat domain protein dynamically regulated in brain. Proc. Natl. Acad. Sci. USA 2003, 100, 11553–11558. [Google Scholar] [CrossRef] [Green Version]
- Kaalund, S.S.; Newburn, E.N.; Ye, T.; Tao, R.; Li, C.; Deep-Soboslay, A.; Herman, M.M.; Hyde, T.M.; Weinberger, D.R.; Lipska, B.K.; et al. Contrasting changes in DRD1 and DRD2 splice variant expression in schizophrenia and affective disorders, and associations with SNPs in postmortem brain. Mol. Psychiatry 2014, 19, 1258–1266. [Google Scholar] [CrossRef]
- Balding, D.J. A tutorial on statistical methods for population association studies. Nat. Rev. Genet. 2006, 7, 781–791. [Google Scholar] [CrossRef]
- Zhang, Y.; Li, J.; Song, S.; Tardif, T.; Burmeister, M.; Villafuerte, S.M.; Su, M.; McBride, C.; Shu, H. Association of DCDC2 Polymorphisms with Normal Variations in Reading Abilities in a Chinese Population. PLoS ONE 2016, 11, e0153603. [Google Scholar] [CrossRef] [Green Version]
- Lim, C.K.-P.; Wong, A.M.-B.; Ho, C.S.-H.; Waye, M.M.-Y. A common haplotype of KIAA0319 contributes to the phonological awareness skill in Chinese children. Behav. Brain Funct. 2014, 10, 23. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, H.; Wang, G.; Xia, J.; Zhou, Y.; Gao, Y.; Xu, J.; Huen, M.S.Y.; Siok, W.; Jiang, Y.; Tan, L.; et al. Stuttering candidate genes DRD2 but not SLC6A3 is associated with developmental dyslexia in Chinese population. Behav. Brain Funct. 2014, 10, 29. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, H.; Xu, J.; Zhou, Y.; Gao, Y.; Wang, G.; Xia, J.; Huen, M.; Siok, W.; Jiang, Y.; Tan, L.; et al. Association study of stuttering candidate genes GNPTAB, GNPTG and NAGPA with dyslexia in Chinese population. BMC Genet. 2015, 16, 7. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.; Li, J.; Tardif, T.; Burmeister, M.; Villafuerte, S.M.; McBride-Chang, C.; Li, H.; Shi, B.; Liang, W.; Zhang, Z.; et al. Association of the DYX1C1 Dyslexia Susceptibility Gene with Orthography in the Chinese Population. PLoS ONE 2012, 7, e42969. [Google Scholar] [CrossRef] [PubMed]
- Lim, C.K.P.; Ho, C.S.H.; Chou, C.H.N.; Waye, M.M.Y. Association of the rs3743205 variant of DYX1C1 with dyslexia in Chinese children. Behav. Brain Funct. 2011, 7, 16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, B.; Zhou, Y.; Leng, S.; Zheng, L.; Lv, H.; Wang, F.; Tan, L.-H.; Sun, Y. Genetic polymorphism of nonsyndromic cleft lip with or without cleft palate is associated with developmental dyslexia in Chinese school-aged populations. J. Hum. Genet. 2017, 62, 265–268. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kong, R.; Shao, S.; Wang, J.; Zhang, X.; Guo, S.; Zou, L.; Zhong, R.; Lou, J.; Zhou, J.; Zhang, J.; et al. Genetic variant in DIP2A gene is associated with developmental dyslexia in Chinese population. Am. J. Med. Genet. Part B Neuropsychiatr. Genet. 2016, 171, 203–208. [Google Scholar] [CrossRef]
- Dalby, J.T. Note: An ultimate view of reading ability. Int. J. Neurosci. 1986, 30, 227–230. [Google Scholar] [CrossRef]
- Dehaene, S.; Cohen, L. Cultural Recycling of Cortical Maps. Neuron 2007, 56, 384–398. [Google Scholar] [CrossRef] [Green Version]
- Christiansen, M.H.; Müller, R.-A. Cultural recycling of neural substrates during language evolution and development. In The Cognitive Neurosciences V; Gazzaniga, M.S., Mangun, G.R., Eds.; MIT Press: Cambridge, MA, USA, 2015; pp. 675–682. [Google Scholar]
- D’Errico, F.; Colagè, I. Cultural Exaptation and Cultural Neural Reuse: A Mechanism for the Emergence of Modern Culture and Behavior. Biol. Theory 2018, 13, 213–227. [Google Scholar] [CrossRef]
- Protopapas, A.; Parrila, R. Dyslexia: Still not a neurodevelopmental disorder. Brain Sci. 2019, 9, 9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dehaene, S. Evolution of human cortical circuits for reading and arithmetic: The “neuronal recycling” hypothesis. In From Monkey Brain to Human Brain; Dehaene, S., Duhamel, J.R., Hauser, M., Rizzolatti, G., Eds.; MIT Press: Cambridge, MA, USA, 2005; pp. 133–157. [Google Scholar]
- Colagè, I.; d’Errico, F. Culture: The Driving Force of Human Cognition. Top. Cogn. Sci. 2018, 12, 654–672. [Google Scholar] [CrossRef] [Green Version]
- Stearns, F.W. One Hundred Years of Pleiotropy: A Retrospective. Genetics 2010, 186, 767–773. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Paaby, A.B.; Rockman, M.V. The many faces of pleiotropy. Trends Genet. 2013, 29, 66–73. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dediu, D.; Christiansen, M.H. Language Evolution: Constraints and Opportunities from Modern Genetics. Top. Cogn. Sci. 2016, 8, 361–370. [Google Scholar] [CrossRef] [Green Version]
- Mozzi, A.; Forni, D.; Clerici, M.; Pozzoli, U.; Mascheretti, S.; Guerini, F.R.; Riva, S.; Bresolin, N.; Cagliani, R.; Sironi, M. The evolutionary history of genes involved in spoken and written language: Beyond FOXP2. Sci. Rep. 2016, 6, 22157. [Google Scholar] [CrossRef] [Green Version]
- Hannula-Jouppi, K.; Kaminen-Ahola, N.; Taipale, M.; Eklund, R.; Nopola-Hemmi, J.; Kääriäinen, H.; Kere, J. The Axon Guidance Receptor Gene ROBO1 Is a Candidate Gene for Developmental Dyslexia. PLoS Genet. 2005, 1, e50. [Google Scholar] [CrossRef]
- Hansell, M. Chinese writing. In The Sino-Tibetan Languages; Thurgood, G., LaPolla, R.J., Eds.; Routledge: London, UK, 2003; pp. 156–165. [Google Scholar]
- McBride, C.A. Is Chinese Special? Four Aspects of Chinese Literacy Acquisition that Might Distinguish Learning Chinese from Learning Alphabetic Orthographies. Educ. Psychol. Rev. 2016, 28, 523–549. [Google Scholar] [CrossRef]
- Siok, W.T.; Perfetti, C.A.; Jin, Z.; Tan, L.H. Biological abnormality of impaired reading is constrained by culture. Nature 2004, 431, 71–76. [Google Scholar] [CrossRef]
- Hoeft, F.; Meyler, A.; Hernandez, A.; Juel, C.; Taylor-Hill, H.; Martindale, J.L.; McMillon, G.; Kolchugina, G.; Black, J.M.; Faizi, A.; et al. Functional and morphometric brain dissociation between dyslexia and reading ability. Proc. Natl. Acad. Sci. USA 2007, 104, 4234–4239. [Google Scholar] [CrossRef] [Green Version]
- Siok, W.T.; Niu, Z.; Jin, Z.; Perfetti, C.A.; Tan, L.H. A structural-functional basis for dyslexia in the cortex of Chinese readers. Proc. Natl. Acad. Sci. USA 2008, 105, 5561–5566. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sabeti, P.C. Positive Natural Selection in the Human Lineage. Science 2006, 312, 1614–1620. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ferrer-Admetlla, A.; Liang, M.; Korneliussen, T.; Nielsen, R. On Detecting Incomplete Soft or Hard Selective Sweeps Using Haplotype Structure. Mol. Biol. Evol. 2014, 31, 1275–1291. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fujito, N.T.; Satta, Y.; Hayakawa, T.; Takahata, N. A new inference method for detecting an ongoing selective sweep. Genes Genet. Syst. 2018, 93, 149–161. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Satta, Y.; Zheng, W.; Nishiyama, K.V.; Iwasaki, R.L.; Hayakawa, T.; Fujito, N.T.; Takahata, N. Two-dimensional site frequency spectrum for detecting, classifying and dating incomplete selective sweeps. Genes Genet. Syst. 2019, 94, 283–300. [Google Scholar] [CrossRef] [Green Version]
- Auton, A.; Abecasis, G.R.; Altshuler, D.M.; Durbin, R.M.; Bentley, D.R.; Chakravarti, A.; Clark, A.G.; Donnelly, P.; Eichler, E.E.; Flicek, P.; et al. A global reference for human genetic variation. Nature 2015, 526, 68–74. [Google Scholar]
- Kim, J.; Weber, J.A.; Jho, S.; Jang, J.; Jun, J.; Cho, Y.S.; Kim, H.-M.; Kim, H.; Kim, Y.; Chung, O.; et al. KoVariome: Korean National Standard Reference Variome database of whole genomes with comprehensive SNV, indel, CNV, and SV analyses. Sci. Rep. 2018, 8, 5677. [Google Scholar] [CrossRef] [Green Version]
- Kim, J.; Jeon, S.; Choi, J.; Blazyte, A.; Jeon, Y.; Kim, J.; Ohashi, J.; Tokunaga, K.; Sugano, S.; Fucharoen, S.; et al. The origin and composition of Korean ethnicity analyzed by ancient and present-day genome sequences. Genome Biol. Evol. 2020, 12, 553–565. [Google Scholar] [CrossRef]
- Loh, P.-R.; Danecek, P.; Palamara, P.F.; Fuchsberger, C.; A Reshef, Y.; K Finucane, H.; Schoenherr, S.; Forer, L.; McCarthy, S.; Abecasis, G.R.; et al. Reference-based phasing using the Haplotype Reference Consortium panel. Nat. Genet. 2016, 48, 1443–1448. [Google Scholar] [CrossRef] [Green Version]
- Szpiech, Z.A.; Hernandez, R.D. selscan: An Efficient Multithreaded Program to Perform EHH-Based Scans for Positive Selection. Mol. Biol. Evol. 2014, 31, 2824–2827. [Google Scholar] [CrossRef] [Green Version]
- Slatkin, M.; Rannala, B. Estimating the age of alleles by use of intraallelic variability. Am. J. Hum. Genet. 1997, 60, 447–458. [Google Scholar] [PubMed]
- Slatkin, M.; Rannala, B. Estimating Allele Age. Annu. Rev. Genom. Hum. Genet. 2000, 1, 225–249. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kimura, M.; Ohta, T. The age of a neutral mutant persisting in a finite population. Genetics 1973, 75, 199–212. [Google Scholar] [PubMed]
- Griffiths, R.C.; Tavaré, S. The age of a mutation in a general coalescent tree. Commun. Stat. Stoch. Model. 1998, 14, 273–295. [Google Scholar] [CrossRef]
- Fu, Y.X. Statistical Properties of Segregating Sites. Theor. Popul. Biol. 1995, 48, 172–197. [Google Scholar] [CrossRef]
- Hudson, R.R. Generating samples under a Wright-Fisher neutral model of genetic variation. Bioinformatics 2002, 18, 337–338. [Google Scholar] [CrossRef]
- Schaffner, S.F. Calibrating a coalescent simulation of human genome sequence variation. Genome Res. 2005, 15, 1576–1583. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Henneberry, A.L.; Wistow, G.; McMaster, C.R. Cloning, Genomic Organization, and Characterization of a Human Cholinephosphotransferase. J. Biol. Chem. 2000, 275, 29808–29815. [Google Scholar] [CrossRef] [Green Version]
- Yuan, L.; Liu, J.-G.; Zhao, J.; Brundell, E.; Daneholt, B.; Höög, C. The Murine SCP3 Gene Is Required for Synaptonemal Complex Assembly, Chromosome Synapsis, and Male Fertility. Mol. Cell 2000, 5, 73–83. [Google Scholar] [CrossRef]
- McLaren, W.; Gil, L.; Hunt, S.E.; Riat, H.S.; Ritchie, G.R.S.; Thormann, A.; Flicek, P.; Cunningham, F. The Ensembl Variant Effect Predictor. Genome Biol. 2016, 17, 122. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Polimanti, R.; Gelernter, J. Widespread signatures of positive selection in common risk alleles associated to autism spectrum disorder. PLoS Genet. 2017, 13, e1006618. [Google Scholar] [CrossRef] [PubMed]
- Fujito, N.T.; Satta, Y.; Hane, M.; Matsui, A.; Yashima, K.; Kitajima, K.; Sato, C.; Takahata, N.; Hayakawa, T. Positive selection on schizophrenia-associated ST8SIA2 gene in post-glacial Asia. PLoS ONE 2018, 13, e0200278. [Google Scholar] [CrossRef] [PubMed]
- Kang, C.; Riazuddin, S.; Mundorff, J.; Krasnewich, D.; Friedman, P.; Mullikin, J.C.; Drayna, D. Mutations in the Lysosomal Enzyme–Targeting Pathway and Persistent Stuttering. N. Engl. J. Med. 2010, 362, 677–685. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Drayna, D.; Kang, C. Genetic approaches to understanding the causes of stuttering. J. Neurodev. Disord. 2011, 3, 374–380. [Google Scholar] [CrossRef] [Green Version]
- Kang, C.; Drayna, D. A role for inherited metabolic deficits in persistent developmental stuttering. Mol. Genet. Metab. 2012, 107, 276–280. [Google Scholar] [CrossRef] [Green Version]
- Flint, M.; Chatterjee, P.; Lin, D.L.; McMullan, L.K.; Shrivastava-Ranjan, P.; Bergeron, É.; Lo, M.K.; Welch, S.R.; Nichol, S.T.; Tai, A.W.; et al. A genome-wide CRISPR screen identifies N-acetylglucosamine-1-phosphate transferase as a potential antiviral target for Ebola virus. Nat. Commun. 2019, 10, 285. [Google Scholar] [CrossRef] [Green Version]
- Lind, P.A.; Luciano, M.; Wright, M.J.; Montgomery, G.W.; Martin, N.G.; Bates, T.C. Dyslexia and DCDC2: Normal variation in reading and spelling is associated with DCDC2 polymorphisms in an Australian population sample. Eur. J. Hum. Genet. 2010, 18, 668–673. [Google Scholar] [CrossRef]
- DeMille, M.M.C.; Tang, K.; Mehta, C.M.; Geissler, C.; Malins, J.G.; Powers, N.R.; Bowen, B.M.; Adams, A.K.; Truong, D.T.; Frijters, J.C.; et al. Worldwide distribution of the DCDC2 READ1 regulatory element and its relationship with phoneme variation across languages. Proc. Natl. Acad. Sci. USA 2018, 115, 4951–4956. [Google Scholar] [CrossRef] [Green Version]
- Dediu, D.; Ladd, D.R. Linguistic tone is related to the population frequency of the adaptive haplogroups of two brain size genes, ASPM and Microcephalin. Proc. Natl. Acad. Sci. USA 2007, 104, 10944–10949. [Google Scholar] [CrossRef] [Green Version]
- Evans, P.D.; Gilbert, S.L.; Mekel-Bobrov, N.; Vallender, E.J.; Anderson, J.R.; Vaez-Azizi, L.M.; Tishkoff, S.A.; Hudson, R.R.; Lahn, B.T. Microcephalin, a gene regulating brain size, continues to evolve adaptively in humans. Science 2005, 309, 1717–1720. [Google Scholar] [CrossRef]
- Mekel-Bobrov, N.; Gilbert, S.L.; Evans, P.D.; Vallender, E.J.; Anderson, J.R.; Hudson, R.R.; Tishkoff, S.A.; Lahn, B.T. Ongoing adaptive evolution of ASPM, a brain size determinant in Homo sapiens. Science 2005, 309, 1720–1722. [Google Scholar] [CrossRef]
- Currat, M.; Excoffier, L.; Maddison, W.; Otto, S.P.; Ray, N.; Whitlock, M.C.; Yeaman, S. Comment on “Ongoing Adaptive Evolution of ASPM, a Brain Size Determinant in Homo sapiens” and “Microcephalin, a Gene Regulating Brain Size, Continues to Evolve Adaptively in Humans”. Science 2006, 313, 172a. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, F.; Hill, R.S.; Schaffner, S.F.; Sabeti, P.C.; Wang, E.T.; Mignault, A.A.; Ferland, R.J.; Moyzis, R.K.; Walsh, C.A.; Reich, D. Comment on “Ongoing Adaptive Evolution of ASPM, a Brain Size Determinant in Homo sapiens”. Science 2007, 316, 370b. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vatsiou, A.I.; Bazin, E.; Gaggiotti, O.E. Detection of selective sweeps in structured populations: A comparison of recent methods. Mol. Ecol. 2016, 25, 89–103. [Google Scholar] [CrossRef] [PubMed] [Green Version]
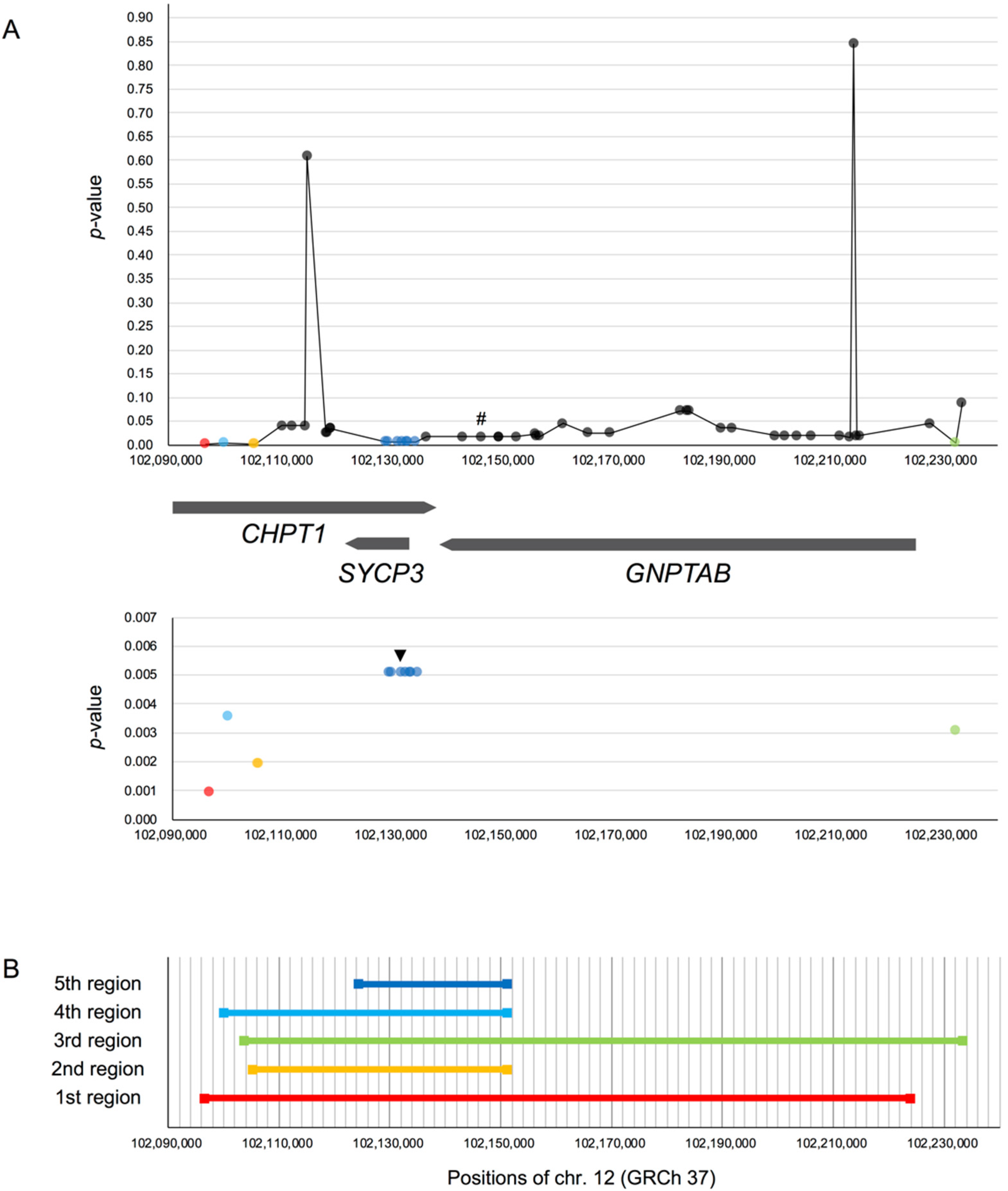

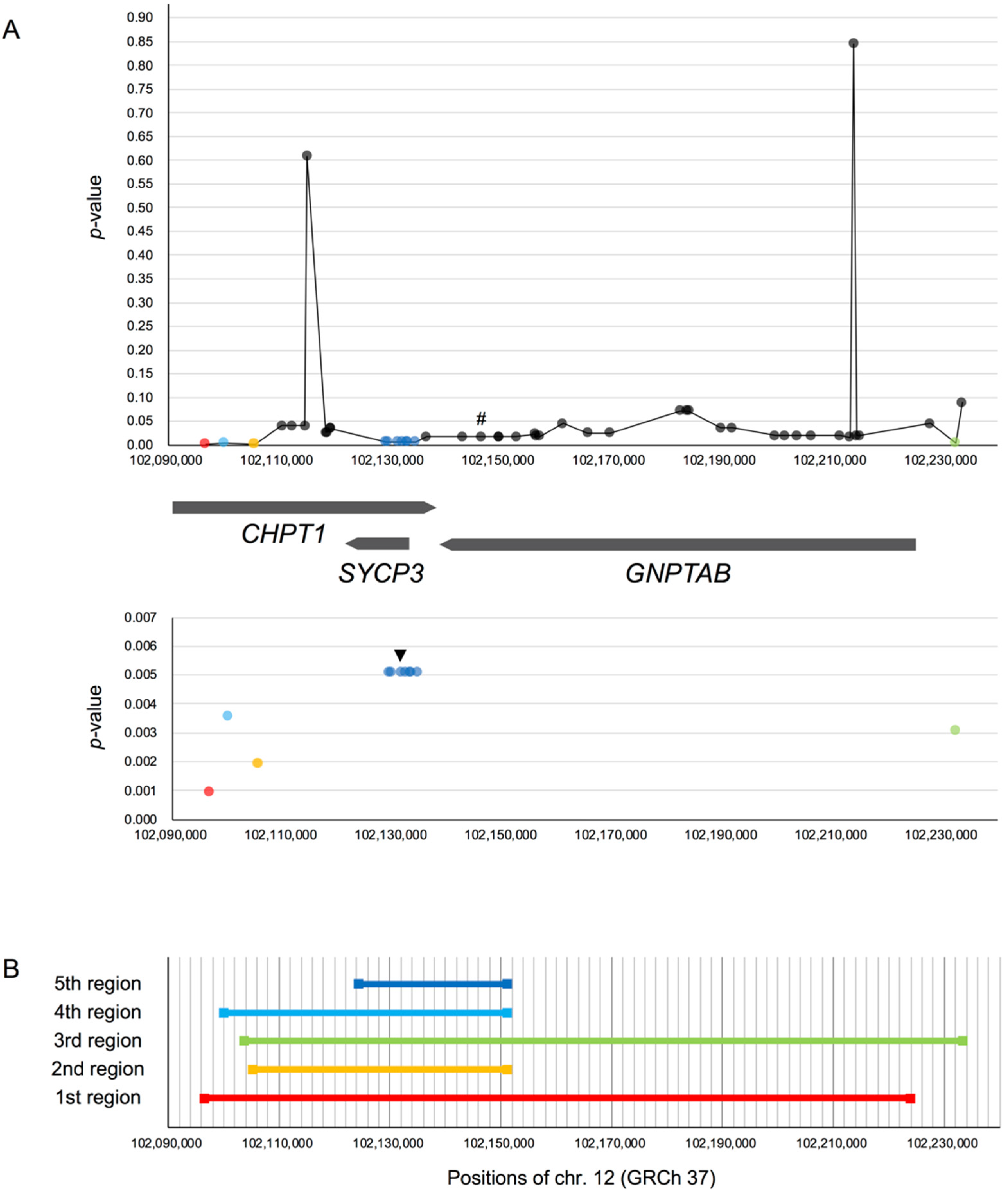{kind=link}
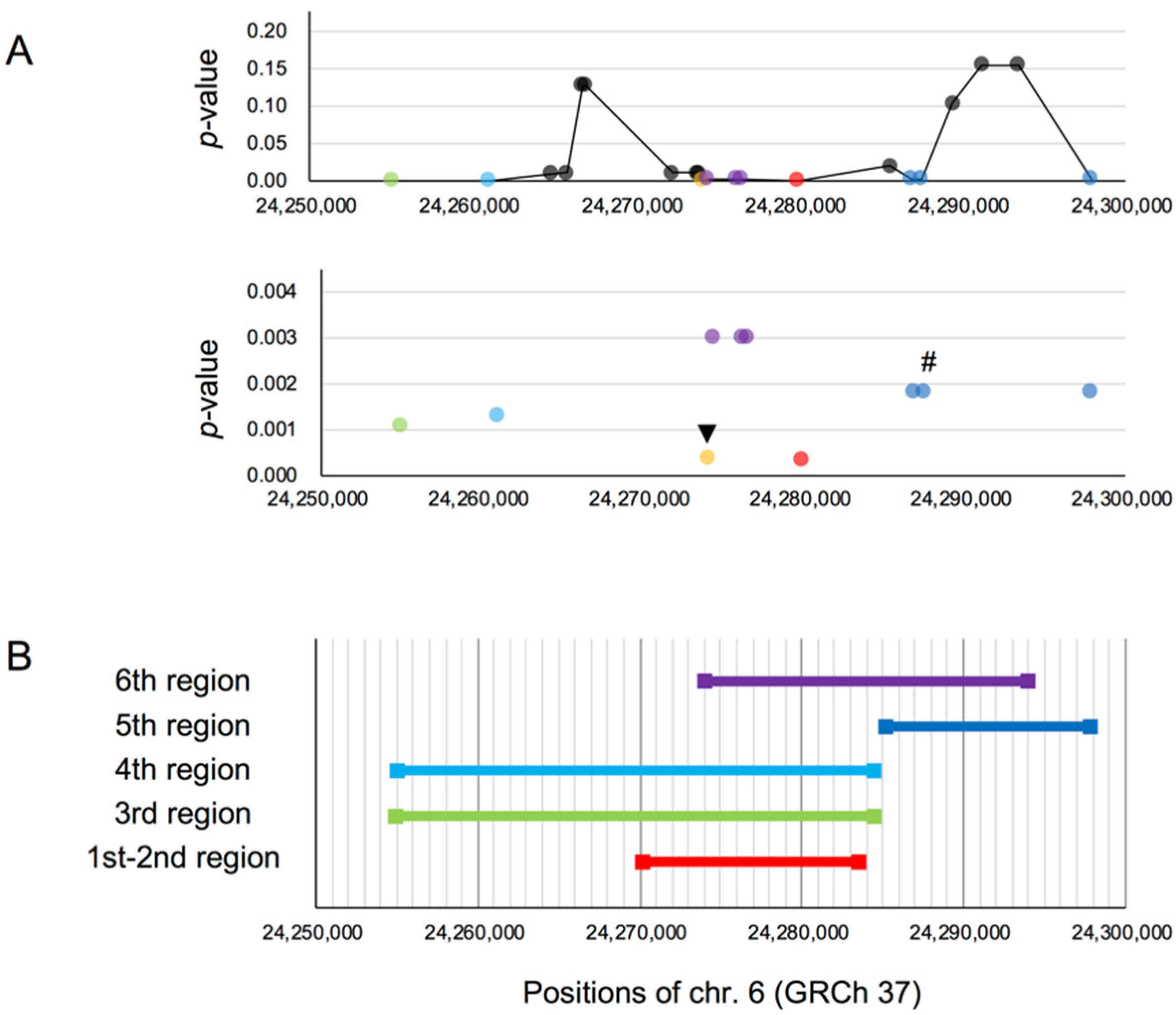{kind=link}
| Gene | Core SNP | Chr. | Position | Risk Allele | Derived Allele Frequency | References | |
|---|---|---|---|---|---|---|---|
| (GRCh37/hg19) | EAS | (EAS and KPGP) | |||||
| KIAA0319L | rs28366021 | 1 | 36,022,859 | Ancestral | 0.234 | (0.227) | [15] |
| ROBO1 | rs4535189 | 3 | 79,489,971 | Derived | 0.366 | (0.373) | [14] |
| DCDC2 | rs807724 | 6 | 24,278,869 | Ancestral | 0.957 | (0.956) | [20] |
| DCDC2 | rs1091047 | 6 | 24,295,256 | Ancestral | 0.817 | (0.823) | [12] |
| KIAA0319 | rs2760157 | 6 | 24,578,272 | Ancestral | 0.456 | (0.470) | [21] |
| KIAA0319 | rs807507 | 6 | 24,579,867 | Derived | 0.188 | (0.187) | [21] |
| KIAA0319 | rs4504469 | 6 | 24,588,884 | Derived | 0.112 | (0.122) | [15] |
| DOCK4 | rs2074130 | 7 | 111,487,098 | Derived | 0.101 | (0.115) | [15] |
| DRD2 | rs1079727 | 11 | 113,289,182 | Derived | 0.416 | (0.420) | [22] |
| GNPTAB | rs17031962 | 12 | 102,146,558 | Ancestral | 0.294 | (0.297) | [23] |
| DYX1C1 | rs11629841 | 15 | 55,777,638 | Derived | 0.058 | (0.056) | [24] |
| DYX1C1 | rs3743205 | 15 | 55,790,530 | Derived | 0.035 | (0.037) | [25] |
| intergenic region | rs8049367 | 16 | 3,980,445 | Derived | 0.339 | (0.340) | [26] |
| NAGPA | rs882294 | 16 | 5,092,118 | Derived | 0.189 | (0.188) | [23] |
| DIP2A | rs2255526 | 21 | 47,971,539 | Derived | 0.264 | (0.262) | [27] |
| Gene | Core SNP | Normalized nSL | p-Value |
|---|---|---|---|
| KIAA0319L | rs28366021 a | 0.0771 | 0.469 |
| ROBO1 | rs4535189 | −0.1882 | 0.575 |
| DCDC2 | rs807724 | 1.1328 | 0.129 |
| DCDC2 | rs1091047 | −0.5967 | 0.725 |
| KIAA0319 | rs2760157 | −2.1853 | 0.986 |
| KIAA0319 | rs807507 | 0.7329 | 0.232 |
| KIAA0319 | rs4504469 | 0.7098 | 0.239 |
| DOCK4 | rs2074130 | 0.3068 | 0.379 |
| DRD2 | rs1079727 | −0.1744 | 0.569 |
| GNPTAB | rs17031962 | 1.2369 | 0.108 |
| DYX1C1 | rs11629841 | −0.0922 | 0.537 |
| DYX1C1 | rs3743205 | −0.1939 | 0.577 |
| intergenic region | rs8049367 | −0.4421 | 0.671 |
| NAGPA | rs882294 | 0.2399 | 0.405 |
| DIP2A | rs2255526 | - | - |
| Gene | Core SNP | Number of Derived Alleles = 1008 | Length of the Core Region | Number of Segregating Sites | p-Value | |
|---|---|---|---|---|---|---|
| KIAA0319L | rs28366021 | 236 | 330,223 | 2204 | 0.1476 | 0.718 |
| ROBO1 | rs4535189 | 369 | 124,626 | 866 | 0.1287 | 0.316 |
| ROBO1 | rs73129039 a | 363 | 124,626 | 866 | 0.1232 | 0.303 |
| DCDC2 | rs807724 | 965 | 5910 | 53 | 0.6742 | 0.159 |
| DCDC2 | rs1091047 | 824 | 41,134 | 334 | 0.3044 | 0.111 |
| DCDC2 | rs3789228 b | 782 | 41,134 | 334 | 0.2020 | 0.068 * |
| KIAA0319 | rs2760157 | 460 | 7387 | 53 | 0.7765 | 0.939 |
| KIAA0319 | rs807507 | 189 | 11,475 | 81 | 0.0220 | 0.111 |
| KIAA03219 | rs4504469 | 113 | 32,025 | 241 | 0.0736 | 0.529 |
| DOCK4 | rs2074130 | 102 | - | - | - | - |
| DRD2 | rs1079727 | 419 | 38,525 | 372 | 0.1370 | 0.260 |
| GNPTAB | rs17031962 | 296 | 136,804 | 868 | 0.0400 | 0.038 * |
| DYX1C1 | rs11629841 | 58 | 130,280 | 1113 | 0.0589 | 0.769 |
| DYX1C1 | rs3743205 | 35 | 242,254 | 2024 | 0.0680 | 0.963 |
| DYX1C1 | rs79024225 c | 31 | 242,254 | 2024 | 0.0308 | 0.758 |
| intergenic region | rs8049367 | 342 | 14,513 | 177 | 0.1486 | 0.428 |
| NAGPA | rs882294 | 191 | 34,706 | 339 | 0.2875 | 0.905 |
| DIP2A | rs2255526 | 266 | 67,101 | 661 | 0.0899 | 0.361 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nishiyama, K.V.; Satta, Y.; Gojobori, J. Do Genes Associated with Dyslexia of Chinese Characters Evolve Neutrally? Genes 2020, 11, 658. https://doi.org/10.3390/genes11060658
Nishiyama KV, Satta Y, Gojobori J. Do Genes Associated with Dyslexia of Chinese Characters Evolve Neutrally? Genes. 2020; 11(6):658. https://doi.org/10.3390/genes11060658
Chicago/Turabian StyleNishiyama, Kumiko V., Yoko Satta, and Jun Gojobori. 2020. "Do Genes Associated with Dyslexia of Chinese Characters Evolve Neutrally?" Genes 11, no. 6: 658. https://doi.org/10.3390/genes11060658
APA StyleNishiyama, K. V., Satta, Y., & Gojobori, J. (2020). Do Genes Associated with Dyslexia of Chinese Characters Evolve Neutrally? Genes, 11(6), 658. https://doi.org/10.3390/genes11060658