Transcriptome Profiling of Haloxylon persicum (Bunge ex Boiss and Buhse) an Endangered Plant Species under PEG-Induced Drought Stress

 , ,
, ,
Abstract
1. Introduction
2. Materials and Methods
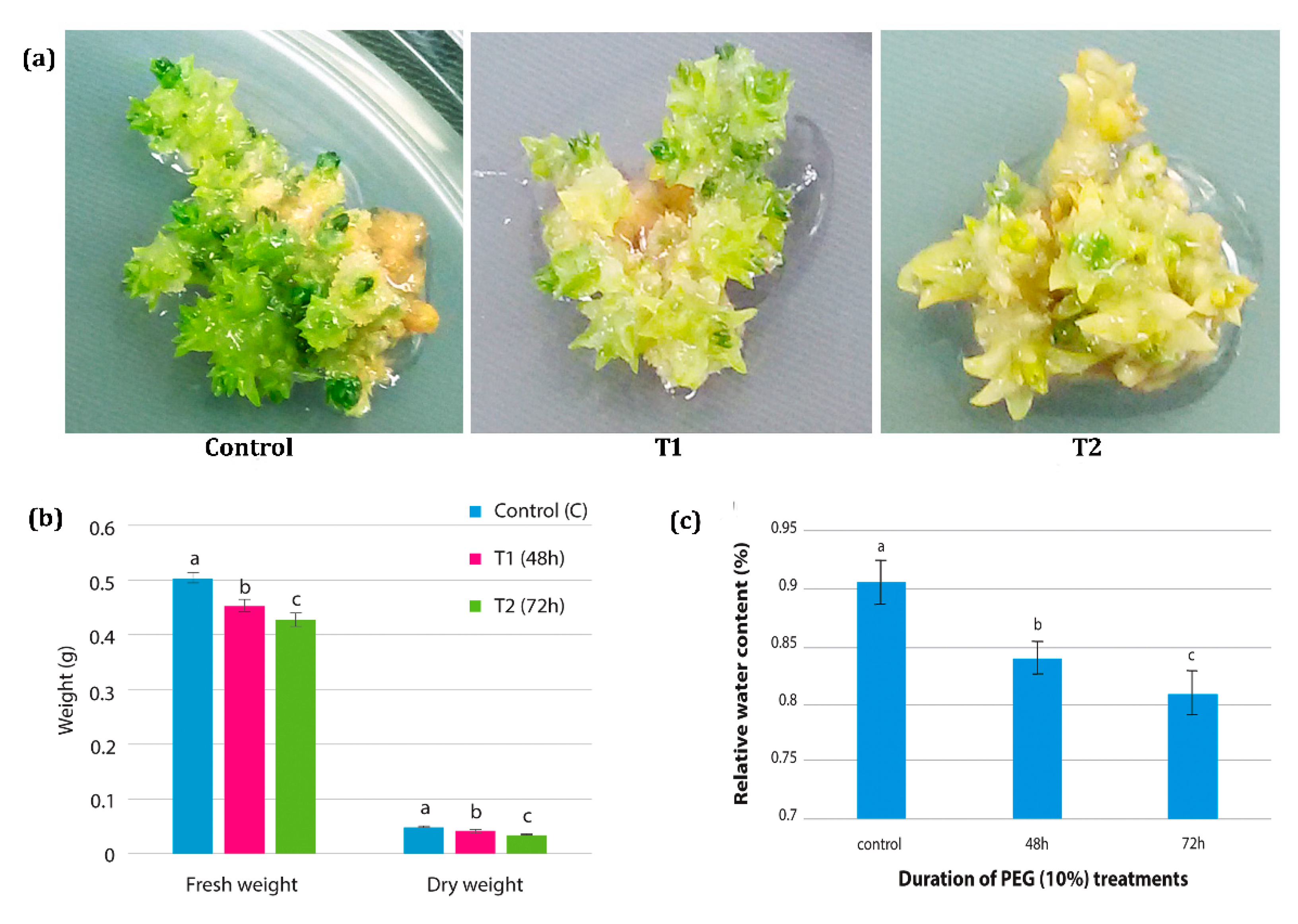
2.1. Plant Material and Drought Treatment
2.2. RNA Isolation and Transcriptome Library Preparation
2.3. Illumina Raw Data Processing and Transcriptome Assembly
2.4. Transcriptome Functional Annotation
2.5. Differential Gene Expression (DGE) Analysis
2.6. qPCR Validation
3. Results
3.1. Physiological Changes in H. persicum
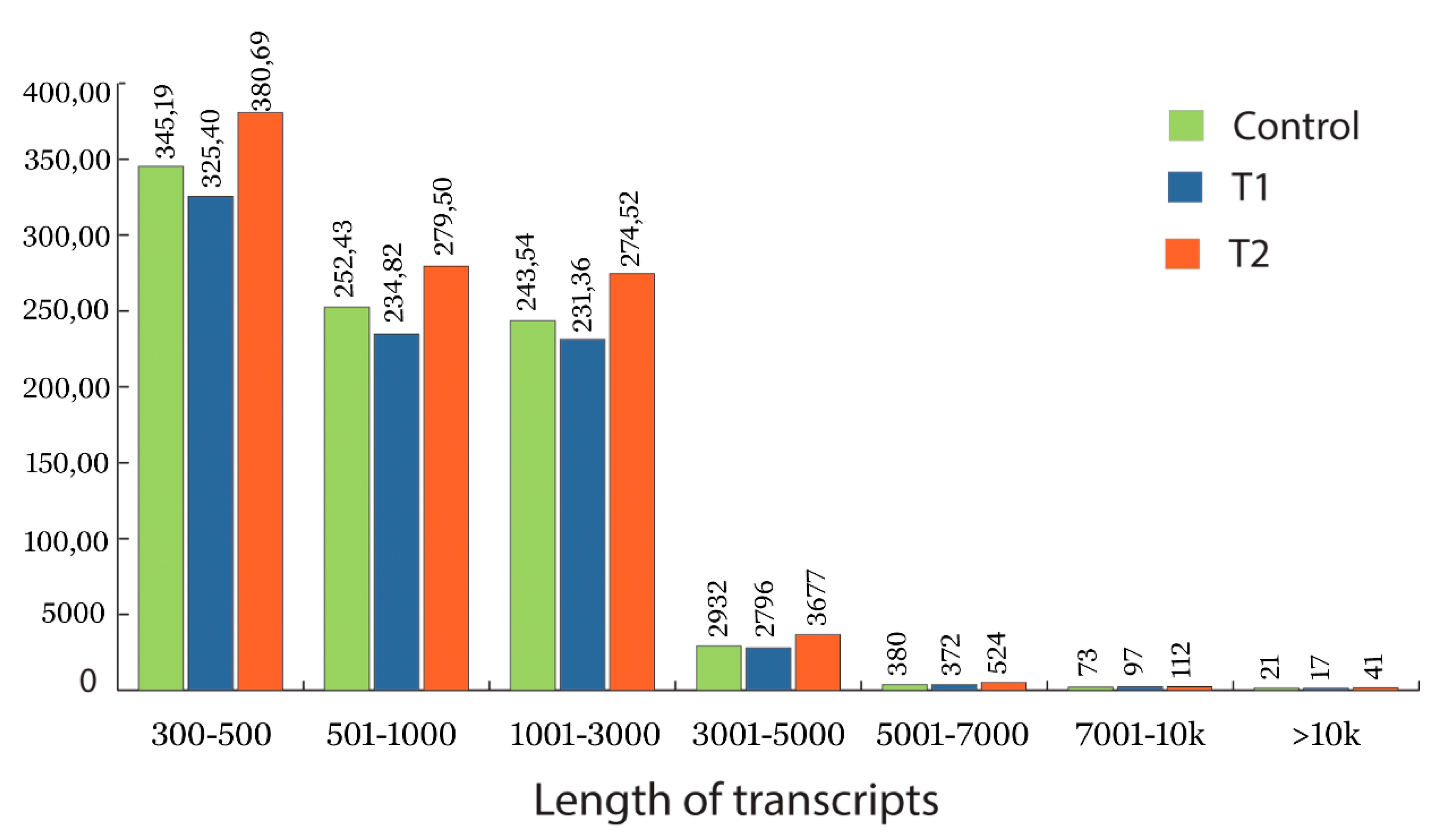
3.2. Data Processing and Transcriptome Assembly
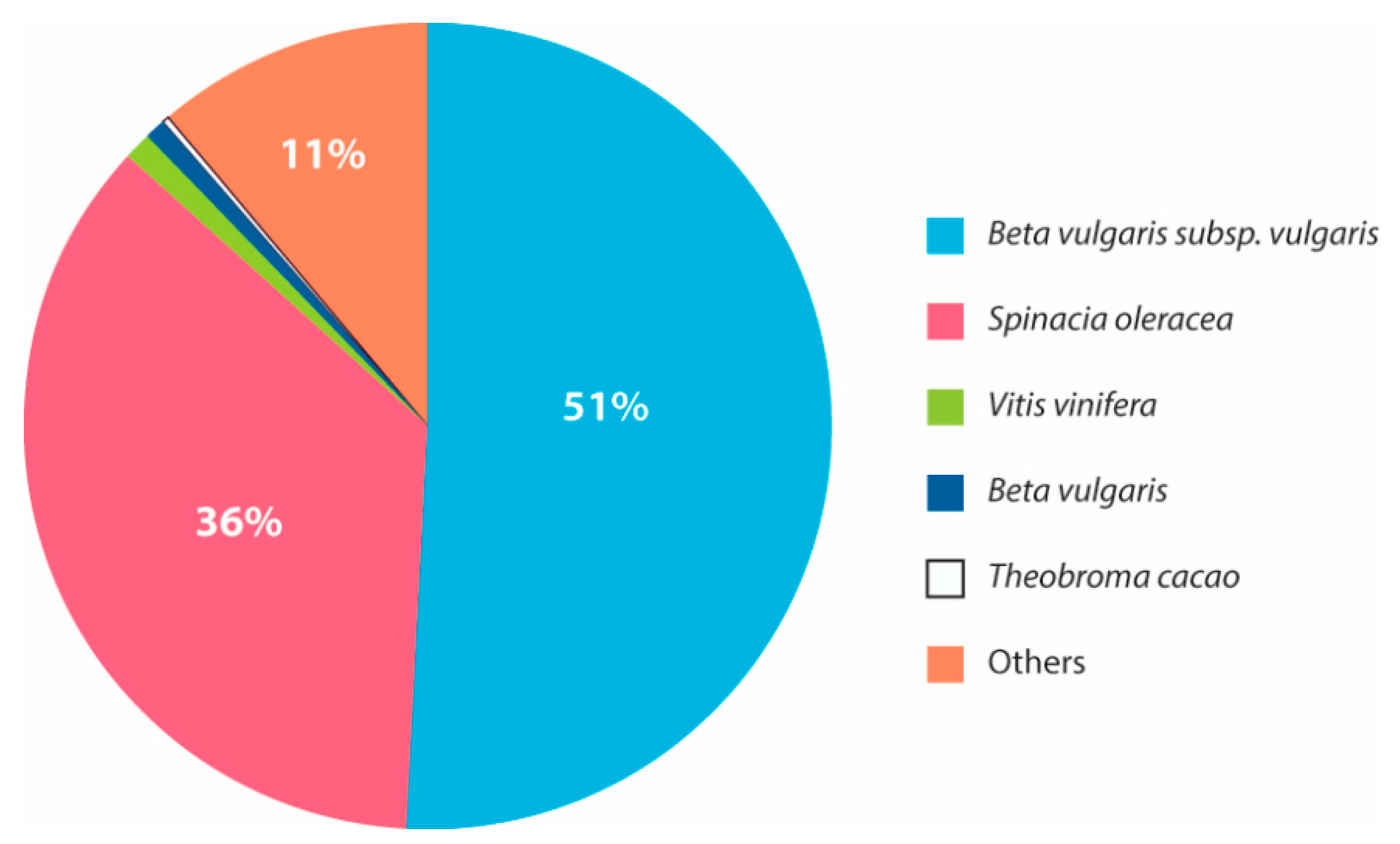
3.3. Transcriptome Annotation
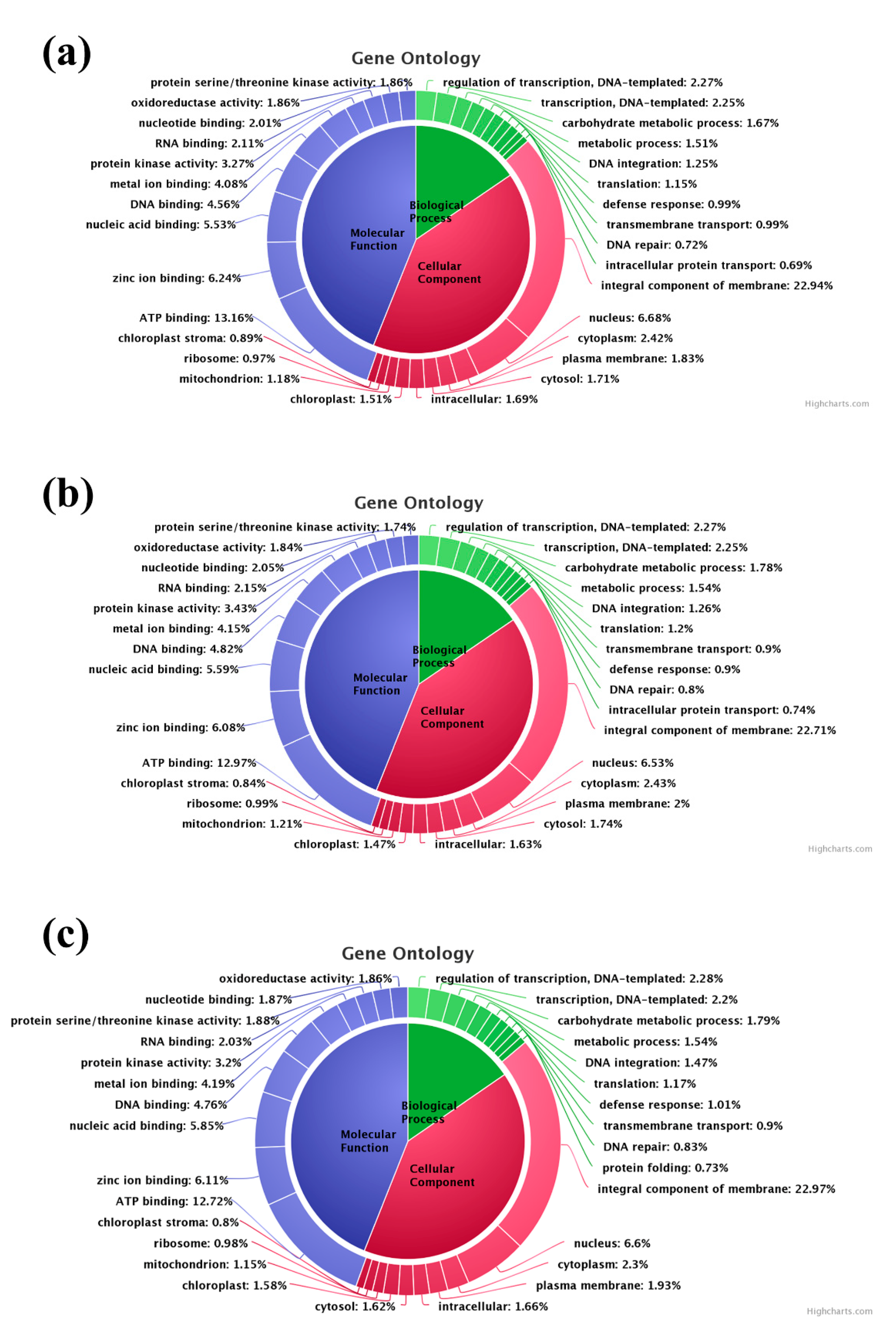
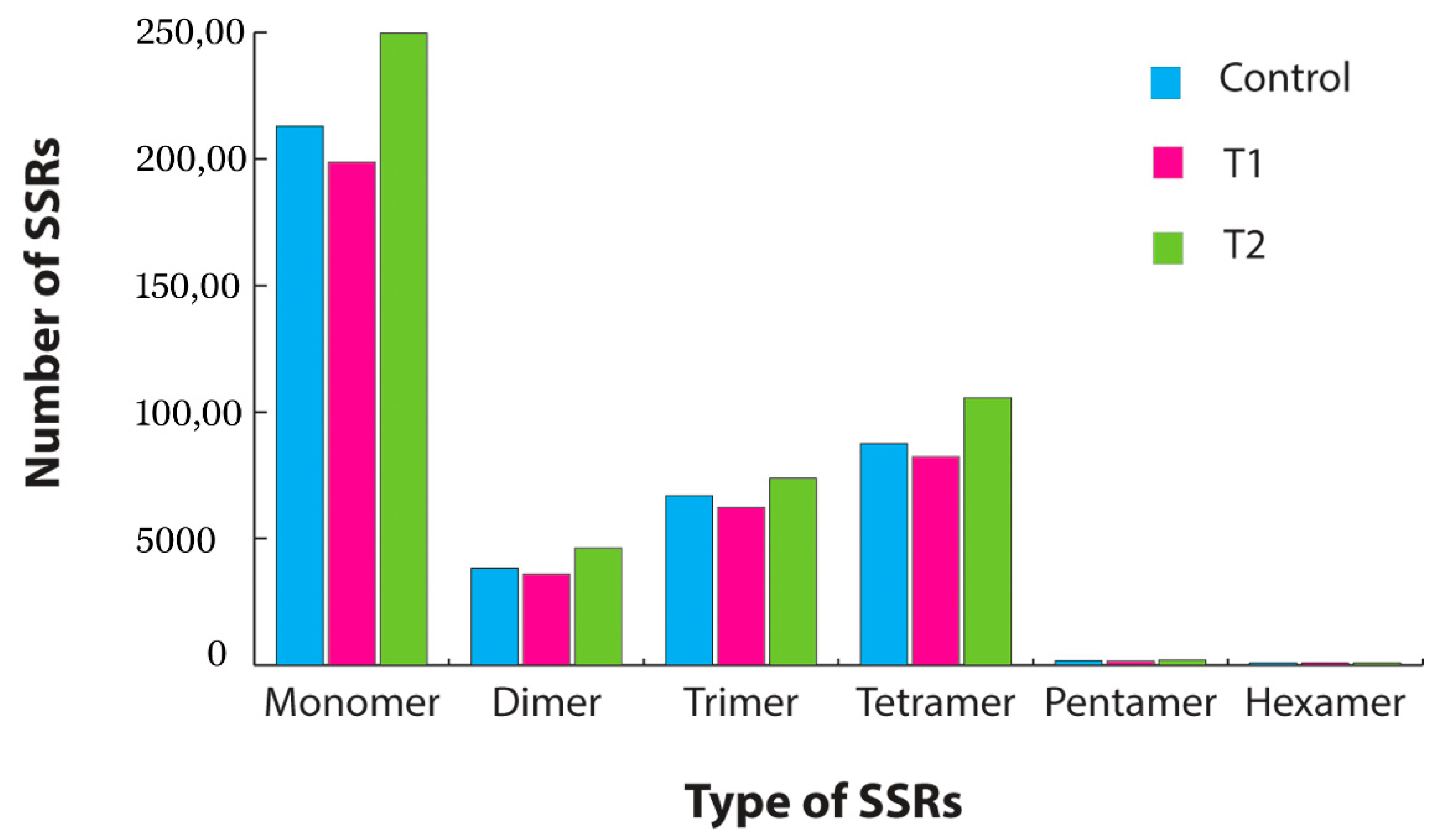
3.4. Gene Ontology (GO) Classification, SSR Mining and Pathway Analysis
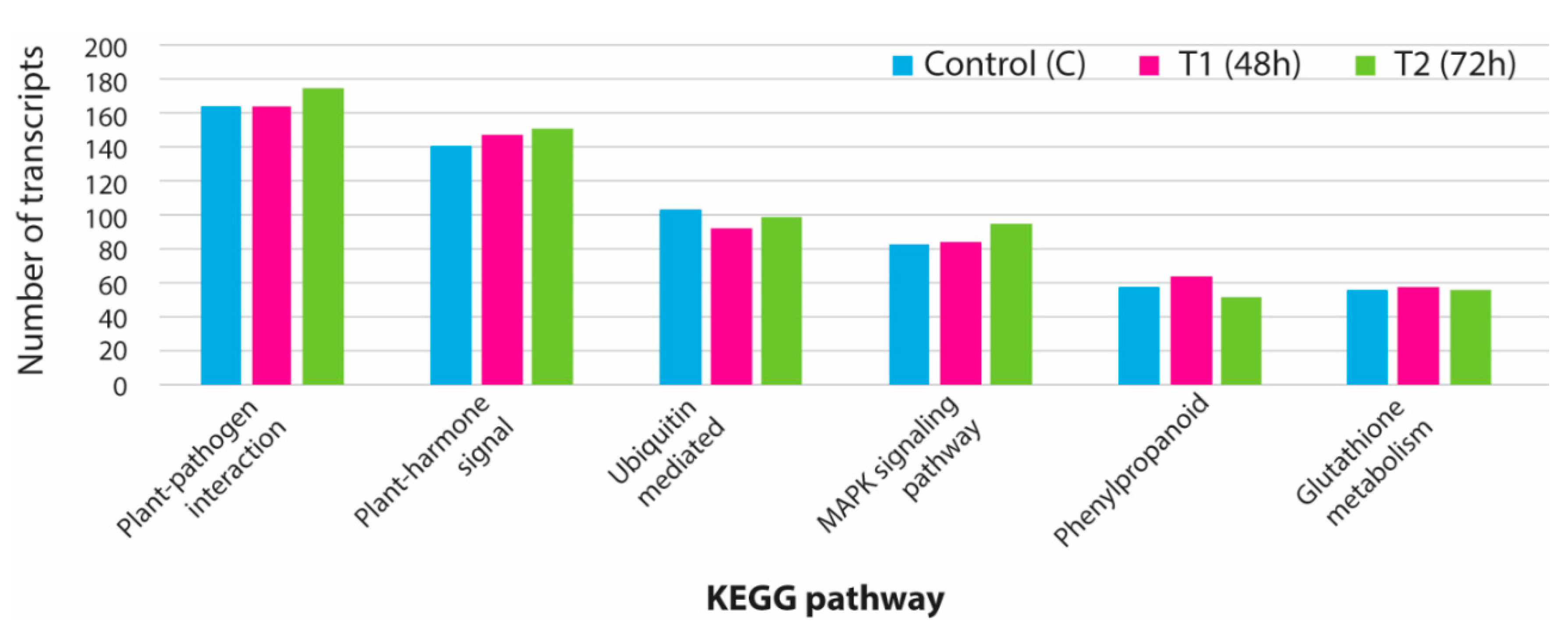
3.5. KEGG Analysis for Pathway Identification
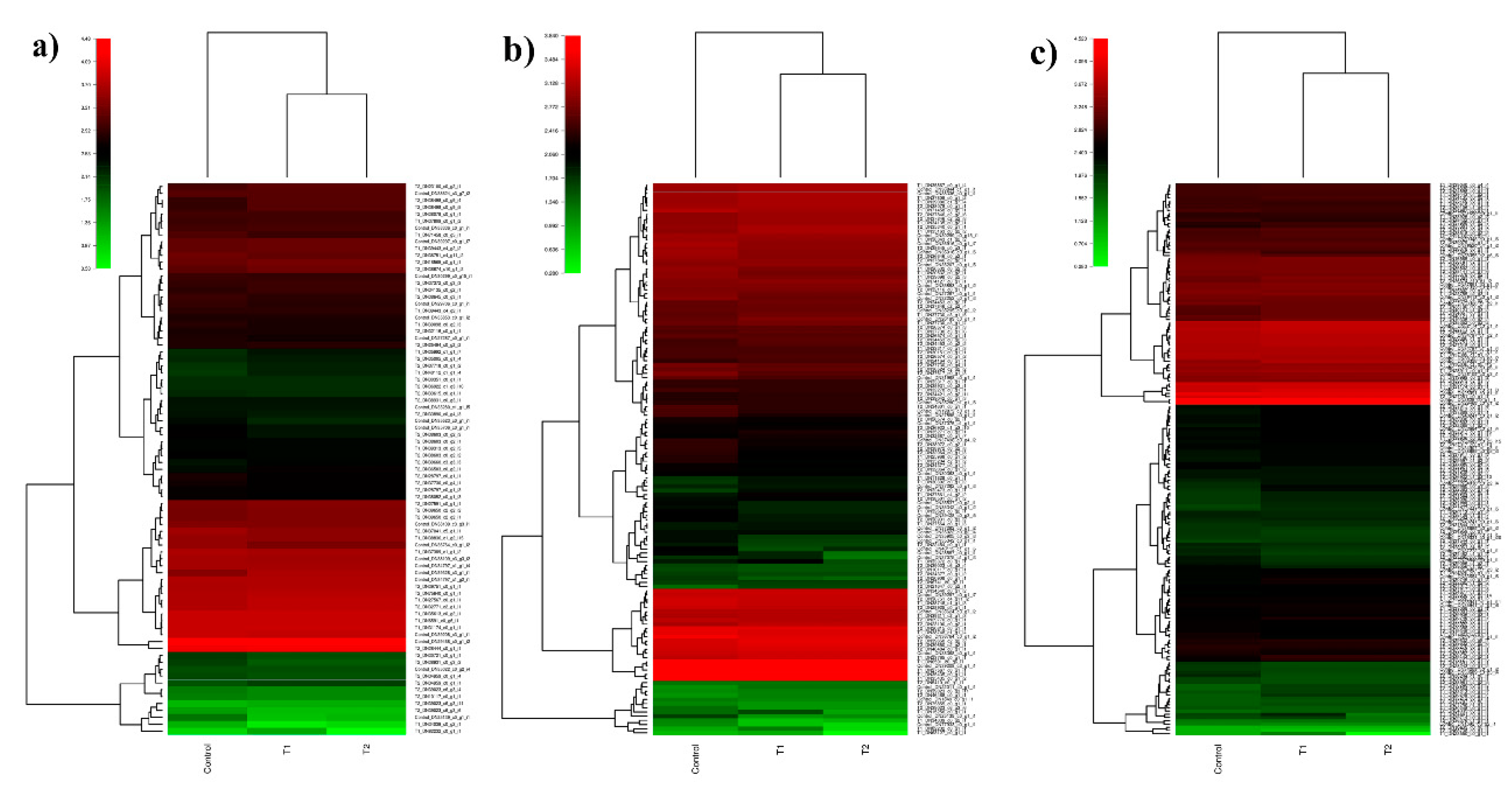
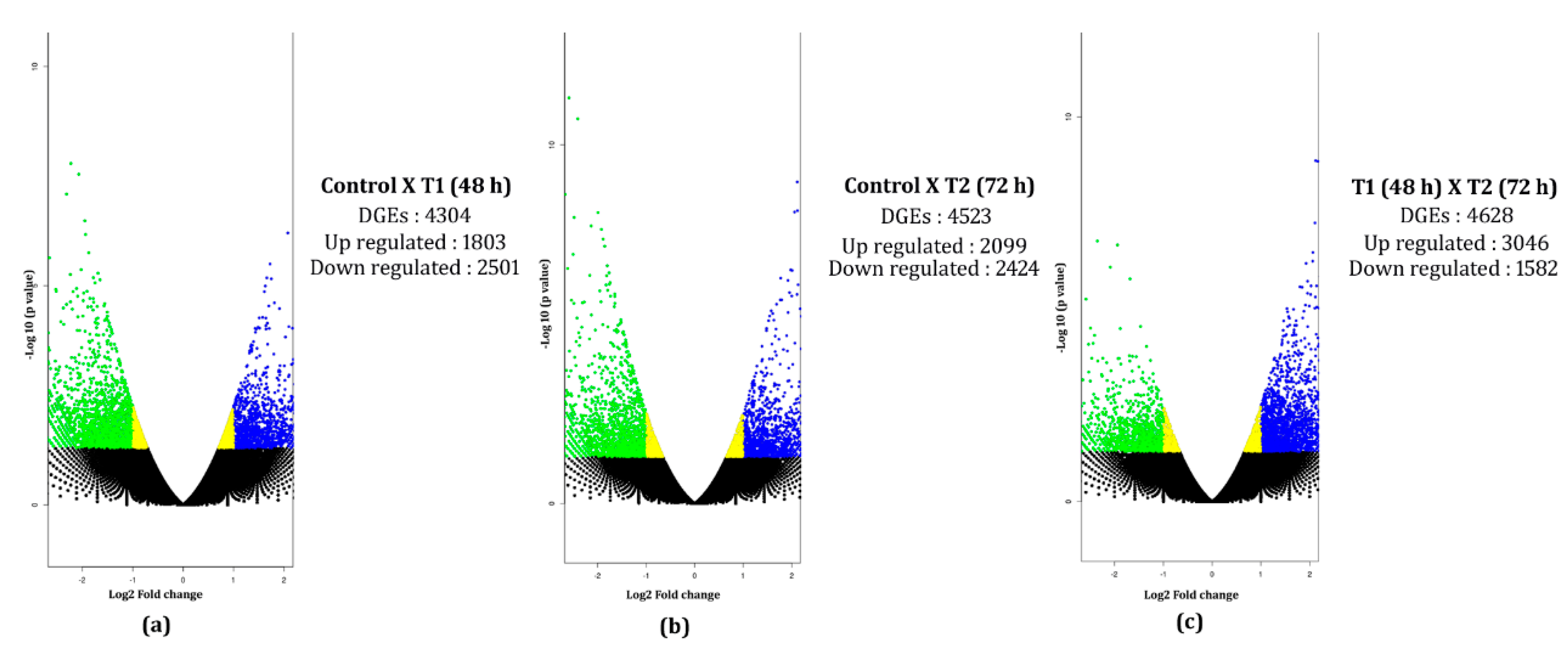
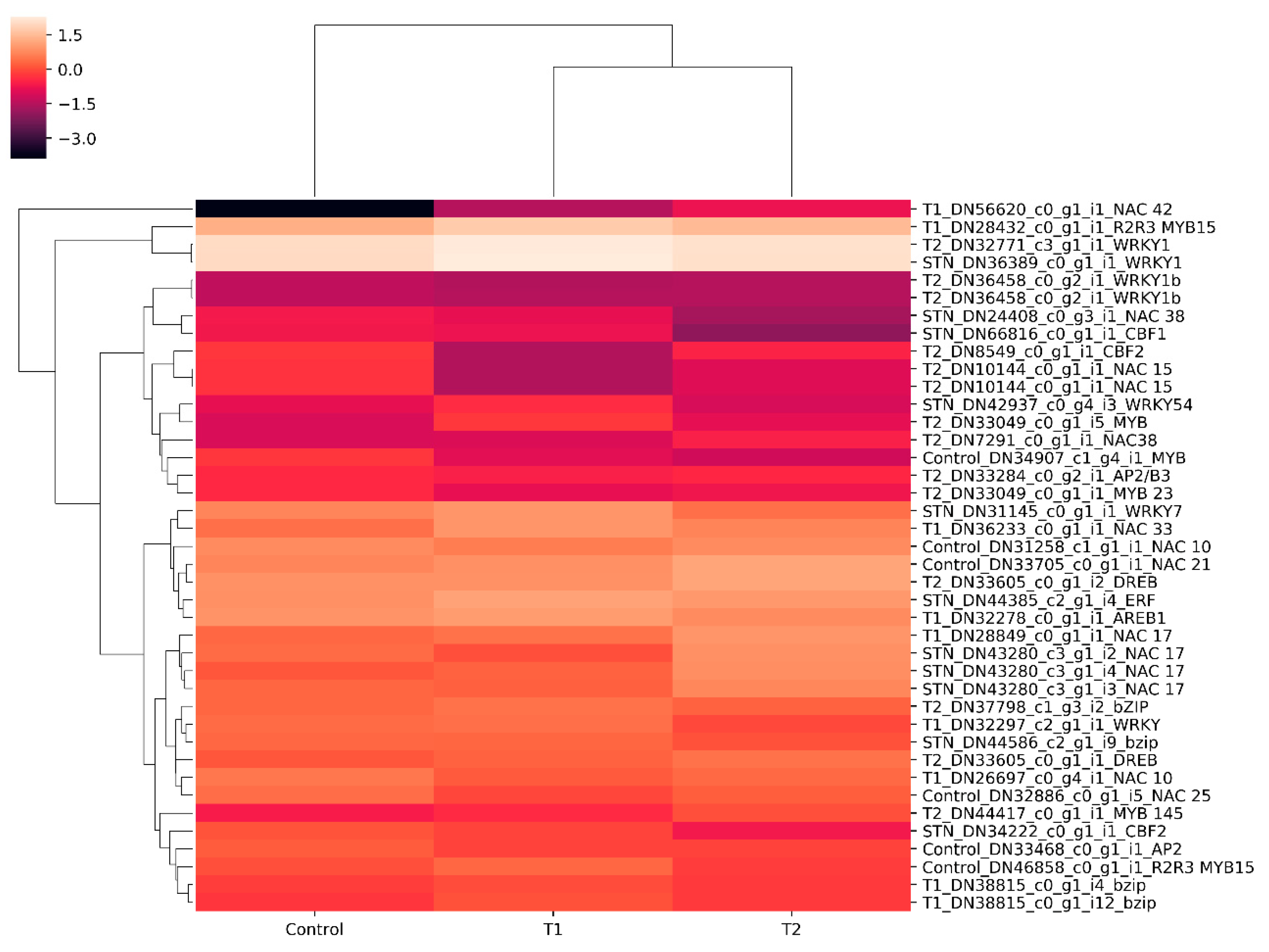
3.6. In Silico Differential Gene Expression (DGE) Analysis
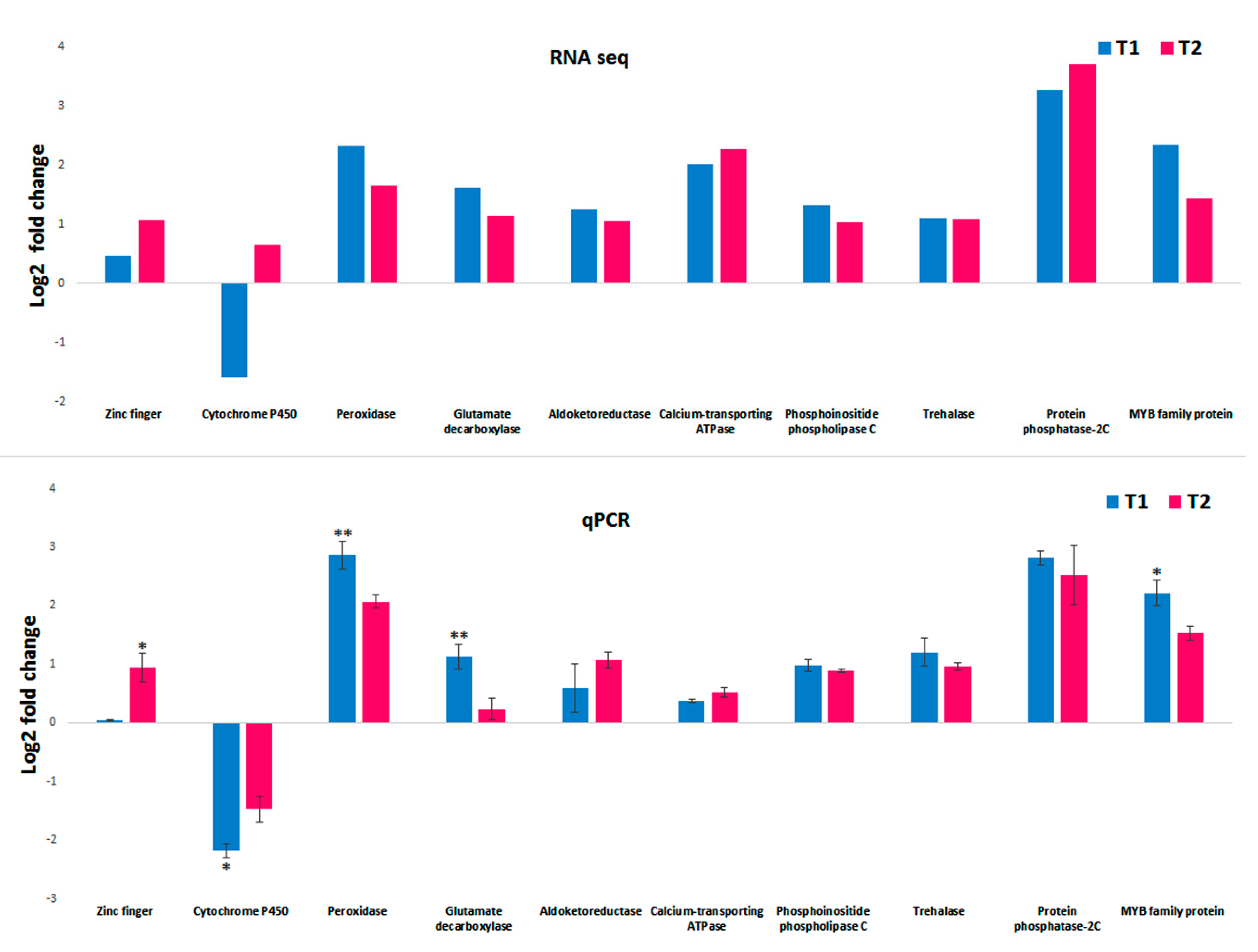
3.7. RT-PCR
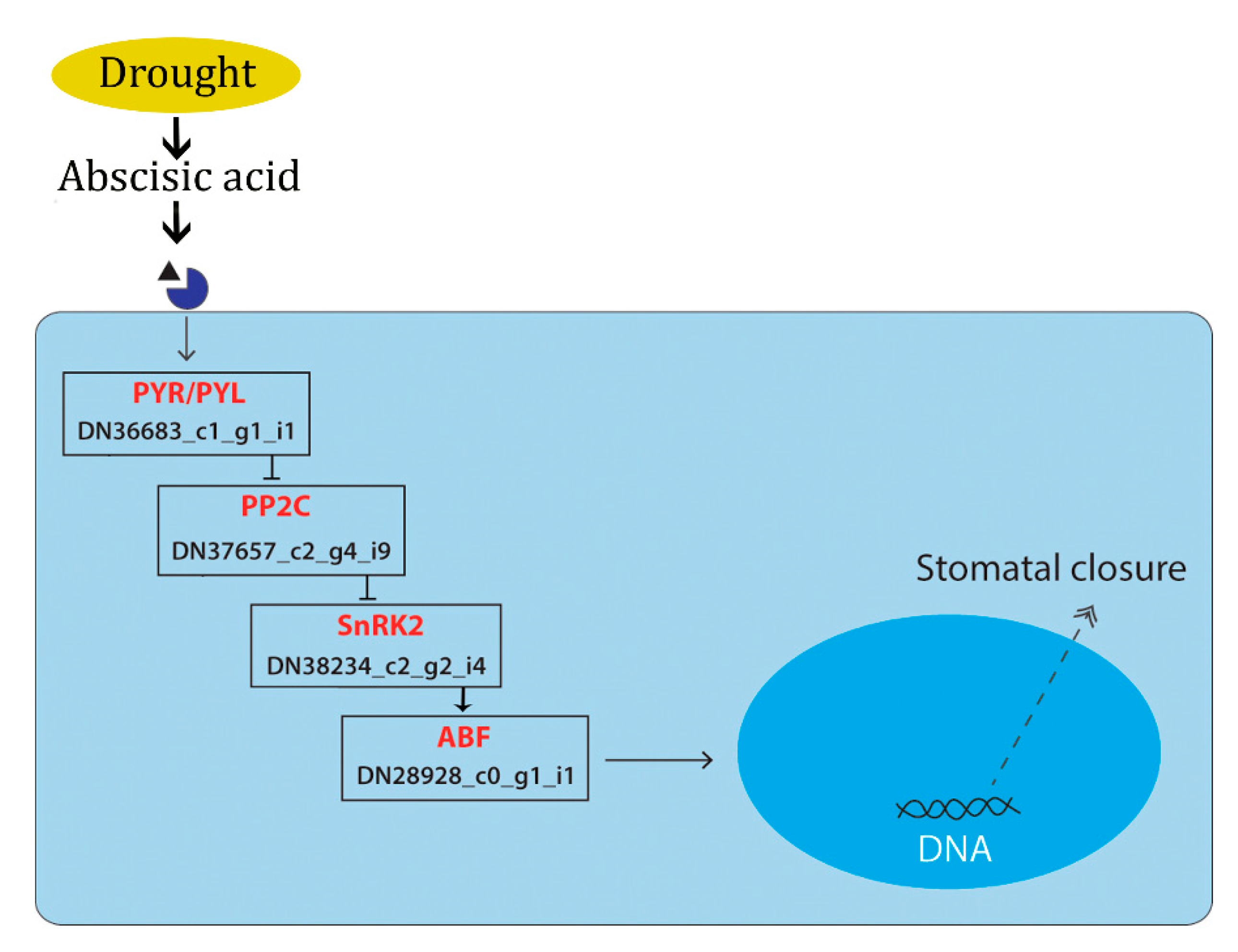
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Data availability
References
- Hadi, M.; Kharazipour, R. The effect of various moisture treatments on water use efficiency (WUE) in Haloxylon plant. In Review of Forests, Wood Products and Wood Biotechnology of Iran and Germany—Part II; Schöpper, A.R., Müller, C., Eds.; Universitätsverlag: Göttingen, Germany, 2008. [Google Scholar]
- Zohary, M. Geobotanical foundations of the Middle East; Gustav Fischer Verlag: Stuttgart, Germany, 1973; Volume 1. [Google Scholar]
- Lawson, T.; Oxborough, K.; Morison, J.I.L.; Baker, N.R. The responses of guard and mesophyll cell photosynthesis to CO2, O2, light and water stress in a range of species are similar. J. Exp. Bot. 2003, 54, 1743–1752. [Google Scholar] [CrossRef] [PubMed]
- Kiani, S.P.; Grieu, P.; Maury, P.; Hewezi, T.; Gentzbittel, L.; Sarrafi, A. Genetic variability for physiological traits under drought conditions and differential expression of water stress-associated genes in sunflower (Helianthus annuus L.). Theor. Appl. Genet. 2007, 114, 193–207. [Google Scholar] [CrossRef] [PubMed]
- Ramanjulu, S.; Bartels, D. Drought and desiccation induced modulation of gene expression in plants. Plant Cell Environ. 2002, 25, 141–151. [Google Scholar] [CrossRef] [PubMed]
- Valliyodan, B.; Nguyen, H.T. Understanding regulatory networks and engineering for enhanced drought tolerance in plants. Curr. Opin. Plant Biol. 2006, 9, 189–195. [Google Scholar] [CrossRef] [PubMed]
- Shinozaki, K.; Yamaguchi-Shinozaki, K. Gene networks involved in drought stress tolerance and response. J. Exp. Bot. 2007, 58, 221–227. [Google Scholar] [CrossRef] [PubMed]
- Golldack, D.; Li, C.; Mohan, H.; Probst, N. Tolerance to drought and salt stress in plants: Unravelling the signaling networks. Front Plant Sci. 2014, 5, 151. [Google Scholar] [CrossRef] [PubMed]
- Raghavendra, A.S.; Gonugunta, V.K.; Christmann, A.; Grill, E. ABA perception and signalling. Trends Plant Sci. 2010, 15, 395–401. [Google Scholar] [CrossRef] [PubMed]
- Kumar, M.; Kesawat, M.S.; Ali, A.; Lee, S.C.; Gill, S.S.; Kim, A.H.U. Integration of abscisic acid signaling with other signaling pathways in plant stress responses and development. Plants 2019, 8, 592. [Google Scholar] [CrossRef] [PubMed]
- Akpinar, B.A.; Avsar, B.; Lucas, S.J.; Budak, H. Plant abiotic stress signaling. Plant Signal. Behav. 2012, 7, 1450–1455. [Google Scholar] [CrossRef] [PubMed]
- Yu, W.; Zhao, R.; Wang, L.; Zhang, S.; Li, R.; Sheng, J.; Shen, L. ABA signaling rather than ABA metabolism is involved in trehalose-induced drought tolerance in tomato plants. Planta 2019, 250, 643–655. [Google Scholar] [CrossRef] [PubMed]
- Sinha, A.K.; Monika, J.; Badmi, R.; Narendra, T. Mitogen-activated protein kinase signaling in plants under abiotic stress. Plant Signal. Behav. 2011, 6, 196–203. [Google Scholar] [CrossRef] [PubMed]
- Bigeard, J.; Hirt, H. Nuclear Signaling of Plant MAPKs. Front. Plant Sci. 2018, 9, 469. [Google Scholar] [CrossRef] [PubMed]
- Mahmood, T.; Khalid, S.; Abdullah, M.; Ahmed, Z.; Shah, M.K.N.; Ghafoor, A.; Du, X. Insights into Drought Stress Signaling in Plants and the Molecular Genetic Basis of Cotton Drought Tolerance. Cells 2020, 9, 105. [Google Scholar] [CrossRef]
- Miller, G.; Suzuki, N.; Ciftci-Yilmaz, S.; Mittler, R. Reactive oxygen species homeostasis and signalling during drought and salinity stresses. Plant Cell Environ. 2010, 33, 453–467. [Google Scholar] [CrossRef] [PubMed]
- Malinich, E.A.; Wang, K.; Mukherjee, P.K.; Kolomiets, M.; Kenerley, C.M. Differential expression analysis of Trichoderma virens RNA reveals a dynamic transcriptome during colonization of Zea mays roots. BMC Genom. 2019, 20, 280. [Google Scholar] [CrossRef] [PubMed]
- Kumar, M.; Kim, I.; Kim, Y.K.; Heo, J.B.; Suh, M.C.; Kim, H.U. Strigolactone Signaling Genes Showing Differential Expression Patterns in Arabidopsis max Mutants. Plants 2019, 8, 352. [Google Scholar] [CrossRef] [PubMed]
- Zhua, J.; Xub, Q.; Zhaoa, S.; Xiaa, X.; Yana, X.; Ana, Y.; Mia, X.; Guoa, L.; Samarinac, L.; Wei, C. Comprehensive co-expression analysis provides novel insights into temporal variation of flavonoids in fresh leaves of the tea plant (Camellia sinensis). Plant Sci. 2020, 290, 110306. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Liu, M.; Li, X.; Cao, B.; Ma, X. Identification of Differentially Expressed Genes in Leaf of Reaumuria soongorica under PEG-Induced Drought Stress by Digital Gene Expression Profiling. PLoS ONE 2014, 9, e94277. [Google Scholar] [CrossRef] [PubMed]
- Ma, X.; Wang, P.; Zhou, S.; Sun, Y.; Liu, N.; Li, X.; Hou, Y. De novo transcriptome sequencing and comprehensive analysis of the drought-responsive genes in the desert plant Cynanchum komarovii. BMC Genom. 2015, 16, 753. [Google Scholar] [CrossRef] [PubMed]
- Barrs, H.D.; Weatherley, P.E. A re-examinati on of the relative turgidity technique for estimating water deficit in leaves. Aust. J. Biol. Sci. 1962, 15, 413–428. [Google Scholar] [CrossRef]
- Andrews, S. FastQC: A Quality Control Tool for High Throughput Sequence Data. 2010. Available online: https://www.bioinformatics.babraham.ac.uk/projects/fastqc/ (accessed on 13 January 2020).
- Grabherr, M.G.; Haas, B.J.; Yassour, M.; Levin, J.Z.; Thompson, D.A.; Amit, I.; Adiconis, X.; Fan, L.; Raychowdhury, R.; Zeng, Q.; et al. Full-length transcriptome assembly from RNA-seq data without a reference genome. Nat. Biotechnol. 2011, 29, 644–652. [Google Scholar] [CrossRef] [PubMed]
- Altschul, S.F.; Madden, T.L.; Schäffer, A.A.; Zhang, J.; Zhang, Z.; Miller, W.; Lipman, D.J. Gapped BLAST and PSI-BLAST: A new generation of protein database search programs. Nucleic Acids Res. 1997, 25, 3389–3402. [Google Scholar] [CrossRef] [PubMed]
- Conesa, A.; Götz, S.; García-Gómez, J.M.; Terol, J.; Talón, M.; Robles, M. Blast2GO: A universal tool for annotation, visualization and analysis in functional genomics research. Bioinformatics 2005, 21, 3674–3676. [Google Scholar] [CrossRef] [PubMed]
- Tatusov, R.L.; Fedorova, N.D.; Jackson, J.D.; Jacobs, A.R.; Kiryutin, B.; Koonin, E.V.; Krylov, D.M.; Mazumder, R.; Mekhedov, S.L.; Nikolskaya, A.N.; et al. The COG database: An updated version includes eukaryotes. BMC Bioinform. 2003, 4, 41. [Google Scholar] [CrossRef] [PubMed]
- Moriya, Y.; Itoh, M.; Okuda, S.; Yoshizawa, A.C.; Kanehisa, M. KAAS: An automatic genome annotation and pathway reconstruction server. Nucleic Acids Res. 2007, 35, W182–W185. [Google Scholar] [CrossRef] [PubMed]
- Kanehisa, M.; Susumu, G. KEGG: Kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 2000, 28, 27–30. [Google Scholar] [CrossRef] [PubMed]
- Langmead, B.; Steven, L.S. Fast gapped-read alignment with Bowtie 2. Nat. Methods 2012, 9, 357–359. [Google Scholar] [CrossRef] [PubMed]
- Anders, S.; Wolfgang, H. Differential Expression of RNA-Seq Data at the Gene Level–the DESeq Package; European Molecular Biology Laboratory (EMBL): Heidelberg, Germany, 2012. [Google Scholar]
- Mayra, R.; Eduardo, C.; Orlando, B. Molecular aspects of abiotic stress in plants. Biotecnol. Appl. 2005, 22, 10. [Google Scholar]
- Caruso, A.; Chefdor, F.; Carpin, S.; Depierreux, C.; Delmotte, F.M.; Kahlem, G.; Morabito, D. Physiological characterization and identification of genes differentially expressed in response to drought induced by PEG 6000 in Populus canadensis leaves. J. Plant Physiol. 2008, 165, 932–941. [Google Scholar] [CrossRef] [PubMed]
- Long, Y.; Zhang, J.; Tian, X.; Wu, S.; Zhang, Q.; Zhang, J.; Dang, Z.; Pei, X.W. De novo assembly of the desert tree Haloxylon ammodendron (CA Mey.) based on RNA-Seq data provides insight into drought response, gene discovery and marker identification. BMC Genom. 2014, 15, 1111. [Google Scholar] [CrossRef] [PubMed]
- Shanker, A.K.; Maheswari, M.; Yadav, S.K.; Desai, S.; Bhanu, D.; Attal, N.B.; Venkateswarlu, B. Drought stress responses in crops. Funct. Integr. Genom. 2014, 14, 11. [Google Scholar] [CrossRef] [PubMed]
- Fu, L.; Ding, Z.; Han, B.; Hu, W.; Li, Y.; Zhang, J. Physiological investigation and transcriptome analysis of polyethylene glycol (PEG)-induced dehydration stress in Cassava. Int. J. Mol. Sci. 2016, 17, 283. [Google Scholar] [CrossRef] [PubMed]
- Joshi, R.; Wani, S.H.; Singh, B.; Bohra, A.; Dar, Z.A.; Lone, A.A.; Pareek, A.; Singla-Pareek, S.L. Transcription factors and plants response to drought stress: Current understanding and future directions. Front Plant Sci. 2016, 7, 1029. [Google Scholar] [CrossRef] [PubMed]
- Nakashima, K.; Takasaki, H.; Mizoi, J.; Shinozaki, K.; Yamaguchi-Shinozaki, K. NAC transcription factors in plant abiotic stress responses. Biochim. Biophys. Acta 2012, 1819, 97–103. [Google Scholar] [CrossRef] [PubMed]
- Shao, H.; Wang, H.; Tang, X. NAC transcription factors in plant multiple abiotic stress responses: Progress and prospects. Front Plant Sci. 2015, 6, 902. [Google Scholar] [CrossRef] [PubMed]
- Le, D.T.; Nishiyama, R.; Watanabe, Y.; Mochida, K.; Yamaguchi-Shinozaki, K.; Shinozaki, K.; Tran, L.S.P. Genome-wide survey and expression analysis of the plant-specific NAC transcription factor family in soybean during development and dehydration stress. DNA Res. 2011, 18, 263–276. [Google Scholar] [CrossRef] [PubMed]
- Sharoni, A.M.; Nuruzzaman, M.; Satoh, K.; Shimizu, T.; Kondoh, H.; Sasaya, T.; Choi, I.R.; Omura, T.; Kikuchi, S. Gene structures, classification and expression models of the AP2/EREBP transcription factor family in rice. Plant Cell Physiol. 2011, 52, 344–360. [Google Scholar] [CrossRef] [PubMed]
- Matsukura, S.; Mizoi, J.; Yoshida, T.; Todaka, D.; Ito, Y.; Maruyama, K.; Shinozaki, K.; Yamaguchi-Shinozaki, K. Comprehensive analysis of rice DREB2-type genes that encode transcription factors involved in the expression of abiotic stress-responsive genes. Mol. Genet. Genom. 2010, 283, 185–196. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z.; Dai, Z.; Lu, R.; Wu, B.; Tang, Q.; Xu, Y.; Cheng, C.; Su, J. Transcriptome Analysis of Two Species of Jute in Response to Polyethylene Glycol (PEG)-induced Drought Stress. Sci. Rep. 2017, 7, 16565. [Google Scholar] [CrossRef] [PubMed]
- Bai, Z.; Wang, T.; Wu, Y.; Wang, K.; Liang, Q.; Pan, Y.; Jiang, B.; Zhang, L.; Liu, G.; Jia, Y.; et al. Whole-transcriptome sequence analysis of differentially expressed genes in Phormium tenax under drought stress. Sci. Rep. 2017, 7, 41700. [Google Scholar] [CrossRef] [PubMed]
- Golan, I.; Dominguez, P.G.; Konrad, Z.; Shkolnik-Inbar, D.; Carrari, F.; Bar-Zvi, D. Tomato abscisic acid stress ripening (ASR) gene family revisited. PLoS ONE 2014, 9, e107117. [Google Scholar] [CrossRef] [PubMed]
- Yang, C.Y.; Chen, Y.C.; Jauh, G.Y.; Wang, C.S. A lily ASR protein involves abscisic acid signaling and confers drought and salt resistance in Arabidopsis. Plant Physiol. 2005, 139, 836–846. [Google Scholar] [CrossRef] [PubMed]
- Dai, J.R.; Liu, B.; Feng, D.R.; Liu, H.; He, Y.; Qi, K.B.; Wang, H.B.; Wang, J.F. MpAsr encodes an intrinsically unstructured protein and enhances osmotic tolerance in transgenic Arabidopsis. Plant Cell Rep. 2011, 30, 1219–1230. [Google Scholar] [CrossRef] [PubMed]
- Hsu, Y.F.; Yu, S.C.; Yang, C.Y.; Wang, C.S. Lily ASR protein-conferred cold and freezing resistance in Arabidopsis. Plant Physiol. Biochem. 2011, 49, 937–945. [Google Scholar] [CrossRef] [PubMed]
- Jha, B.; Lal, S.; Tiwari, V.; Yadav, S.K.; Agarwal, P.K. The SbASR-1 gene cloned from an extreme halophyte Salicornia brachiata enhances salt tolerance in transgenic tobacco. Mar. Biotechnol. 2012, 14, 782–792. [Google Scholar] [CrossRef] [PubMed]
- Ecker, J.R. The ethylene signal transduction pathway in plants. Science 1995, 268, 667–675. [Google Scholar] [CrossRef] [PubMed]
- Borisjuk, N.; Hrmova, M.; Lopato, S. Transcriptional regulation of cuticle biosynthesis. Biotechnol. Adv. 2014, 32, 526–540. [Google Scholar] [CrossRef] [PubMed]
- Lorenzo, O.; Piqueras, R.; Sánchez-Serrano, J.J.; Solano, R. Ethylene response factor 1 integrates signal from ethylene and jasmonate pathway in plant defense. Plant Cell. 2002, 15, 165–178. [Google Scholar] [CrossRef] [PubMed]
- Zhu, J.; Jianping, N.I. Abiotic Stress Signalling and Responses in Plants. Cell 2016, 167, 313. [Google Scholar] [CrossRef] [PubMed]
- Fini, A.; Guidi, L.; Ferrini, F.; Brunettia, C.; Di Ferdinando, M.; Biricolti, S.; Pollastri, S.; Calamaia, L.; Tattini, M. Drought stress has contrasting effects on antioxidant enzymes activity and phenylpropanoid biosynthesis in Fraxinusornus leaves: An excess light stress affair? J. Plant. Physiol. 2012, 169, 929–939. [Google Scholar] [CrossRef] [PubMed]
- Baldoni, E.; Monica, M.; Franca, L.; Roberto, C.; Laura, R.C.; Valentina, P.; Pamela, A.; Annamaria, G. Analysis of transcript and metabolite levels in Italian rice (Oryza sativa L.) cultivars subjected to osmotic stress or benzothiadiazole treatment. Plant Physiol. Biochem. 2013, 70, 492e503. [Google Scholar] [CrossRef] [PubMed]













{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Control | T1 | T2 | |
|---|---|---|---|
| Total raw reads (PE reads) | 19,007,972 | 22,409,657 | 21,771,386 |
| Total trimmed reads (PE reads) in Million | 15.24 | 18.04 | 17.12 |
| Read length (bp) | 151 | 151 | 151 |
| Total transcripts | 87522 | 82440 | 97825 |
| Total bases | 85,611,629 | 81,373,840 | 98,916,209 |
| (G + C) % | 38.71 | 38.92 | 38.54 |
| N% | 0 | 0 | 0 |
| Minimum sequence length | 300 | 300 | 300 |
| Maximum sequence length | 16,107 | 16,067 | 16,736 |
| Average sequence length | 978.17 | 987.07 | 1011.15 |
| N50 length | 1437 | 1467 | 1524 |
| L50 number | 17,792 | 16,679 | 19,400 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Thayale Purayil, F.; Rajashekar, B.; S. Kurup, S.; Cheruth, A.J.; Subramaniam, S.; Hassan Tawfik, N.; M.A. Amiri, K. Transcriptome Profiling of Haloxylon persicum (Bunge ex Boiss and Buhse) an Endangered Plant Species under PEG-Induced Drought Stress. Genes 2020, 11, 640. https://doi.org/10.3390/genes11060640
Thayale Purayil F, Rajashekar B, S. Kurup S, Cheruth AJ, Subramaniam S, Hassan Tawfik N, M.A. Amiri K. Transcriptome Profiling of Haloxylon persicum (Bunge ex Boiss and Buhse) an Endangered Plant Species under PEG-Induced Drought Stress. Genes. 2020; 11(6):640. https://doi.org/10.3390/genes11060640
Chicago/Turabian StyleThayale Purayil, Fayas, Balaji Rajashekar, Shyam S. Kurup, Abdul Jaleel Cheruth, Sreeramanan Subramaniam, Nadia Hassan Tawfik, and Khaled M.A. Amiri. 2020. "Transcriptome Profiling of Haloxylon persicum (Bunge ex Boiss and Buhse) an Endangered Plant Species under PEG-Induced Drought Stress" Genes 11, no. 6: 640. https://doi.org/10.3390/genes11060640
APA StyleThayale Purayil, F., Rajashekar, B., S. Kurup, S., Cheruth, A. J., Subramaniam, S., Hassan Tawfik, N., & M.A. Amiri, K. (2020). Transcriptome Profiling of Haloxylon persicum (Bunge ex Boiss and Buhse) an Endangered Plant Species under PEG-Induced Drought Stress. Genes, 11(6), 640. https://doi.org/10.3390/genes11060640