NIPT Technique Based on the Use of Long Chimeric DNA Reads

 , ,
, ,  , , ,
, , , 
Abstract
1. Introduction
2. Materials and Methods
2.1. Ethics Statement
2.2. Sample Processing and Extraction of Blood Plasma and cfDNA
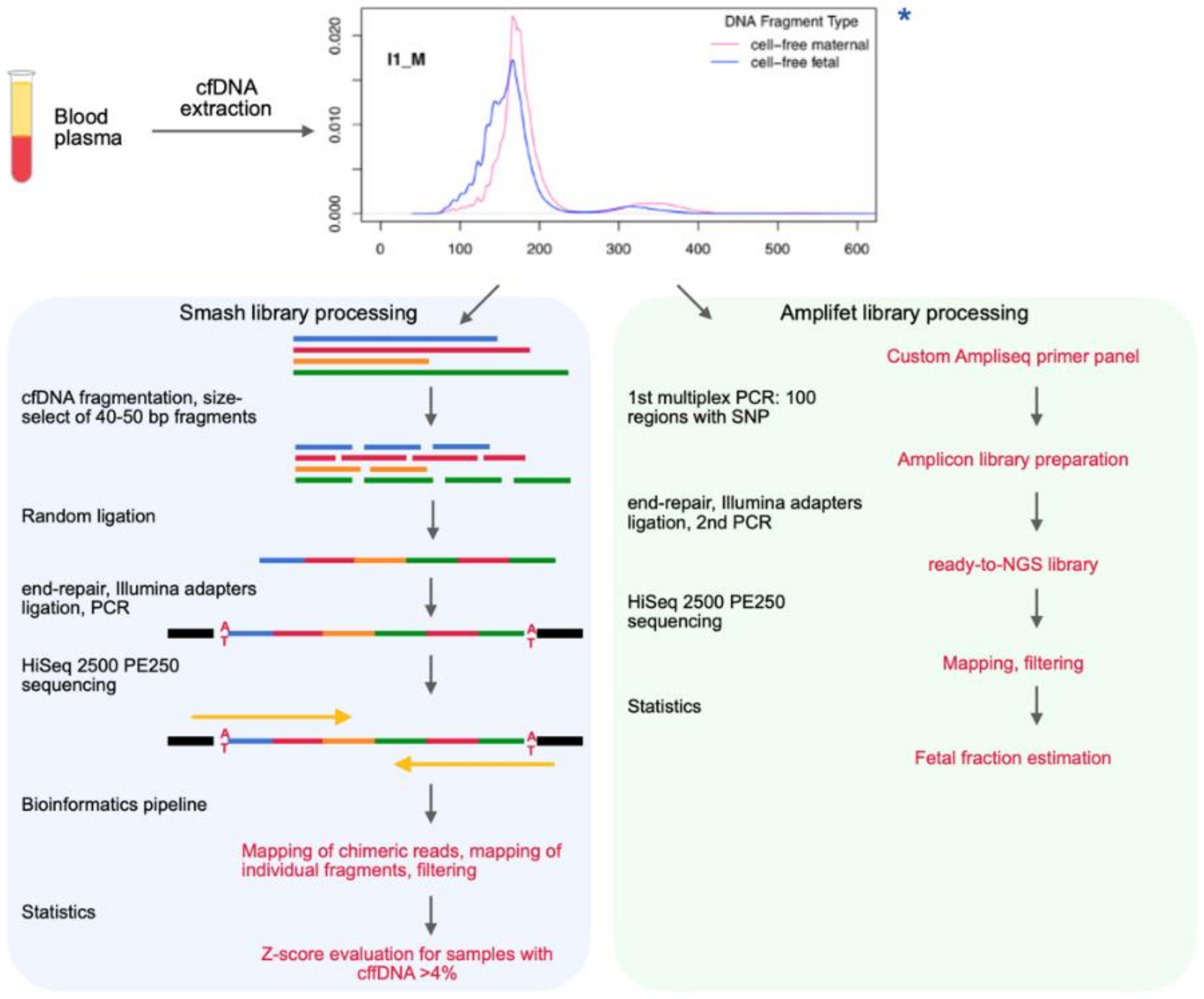
2.3. Chimeric DNA Library Preparation (“Smash”)
2.3.1. Fragmentation
2.3.2. Double Size Selection to Obtain 40–50 bp Fragments
2.3.3. End-Repair
2.3.4. Self-Ligation
2.3.5. A-Tailing
2.3.6. Adapter Ligation
2.3.7. Size Selection
2.3.8. Indexing PCR
2.3.9. Cleanup
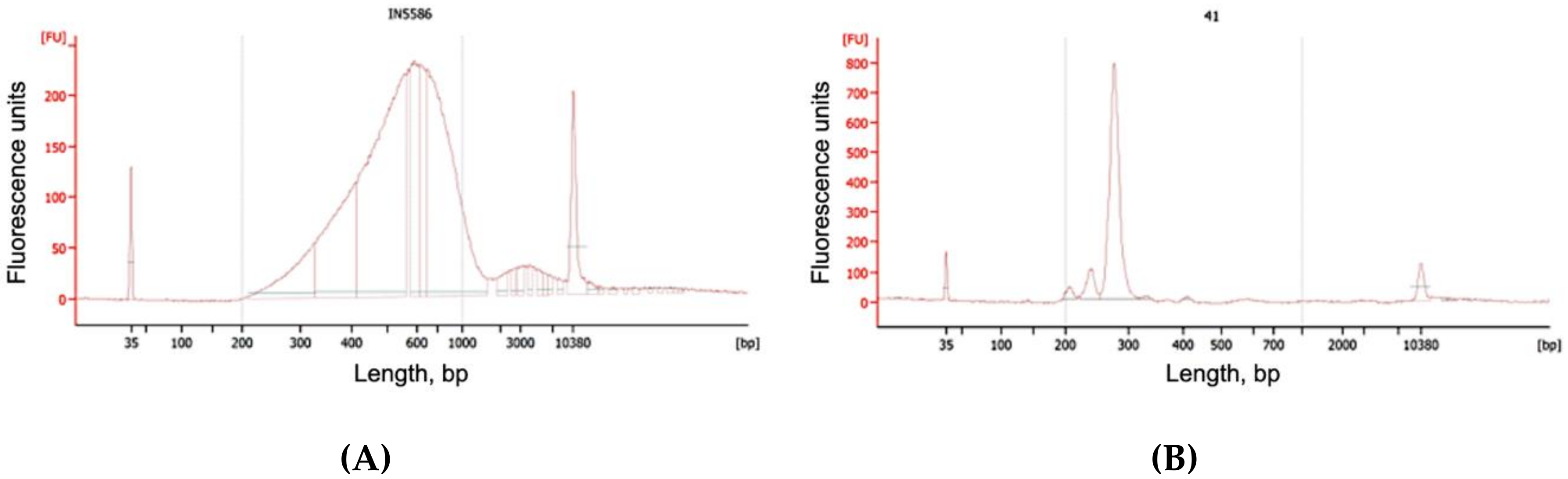
2.3.10. QC
2.4. Choice of SNPs
- Insertions, deletions, chromosomes X and Y, mitochondrial DNA and the p-arm of chromosome 6 (HLA region) were not included;
- Only diallelic polymorphisms were considered;
- Only SNP with identifiers “rs” were considered;
- Only alleles with a minimum allele frequency (MAF) of 0.4 were considered;
- We considered SNPs with a minimum probability of selection according to Hardy–Weinberg of p 0.00001 (i.e., the selection has almost no effect);
- Only unlinked SNPs were considered. They were determined using the sliding window method with the following parameters: correlation coefficient r2 < 0.5, sliding window size = 50 SNP, step = 5 SNP.
- absence of homopolymers in the primer (four or more identical nucleotides in a row);
- no repetitions (e.g., “ATATATA...”);
- absence of a large number of GC at the 3′ end;
- GC composition of the primer ranging from 0.4 to 0.6.
2.5. Fetal Fraction Estimation Library Preparation (“Amplifet”)
2.5.1. Multiplex PCR
2.5.2. QC
2.5.3. Cleanup
2.5.4. Adapter Ligation
2.5.5. Cleanup
2.5.6. Indexing PCR
2.5.7. Cleanup
2.5.8. QC
2.6. Sequencing
2.7. Bioinformatics
2.7.1. Mapping
2.7.2. Filtering
- those that were imperfectly mapped onto the genome (MAPQ < 60);
- those that fell into regions of known repeats (RepeatMasker in Genome Browser track);
- those that fell into amplicon regions.
2.8. Statistics
Filtering
2.9. Determination of the Sex of the Fetus
3. Results
3.1. Clinical Characteristics
3.2. Extraction of cfDNA
3.3. Smash Protocol
- Fragmentation of cfDNA to short fragments;
- Size selection of the fragmented cfDNA from two sides, leading to a number of fragments with an average length of 40–50 bp remaining in the sample;
- The random ligation of short cfDNA fragments and thereby the formation of long (more than 300 bp) chimeric DNA molecules to which adapters for NGS are already ligated.
3.4. Smash Sequencing Results
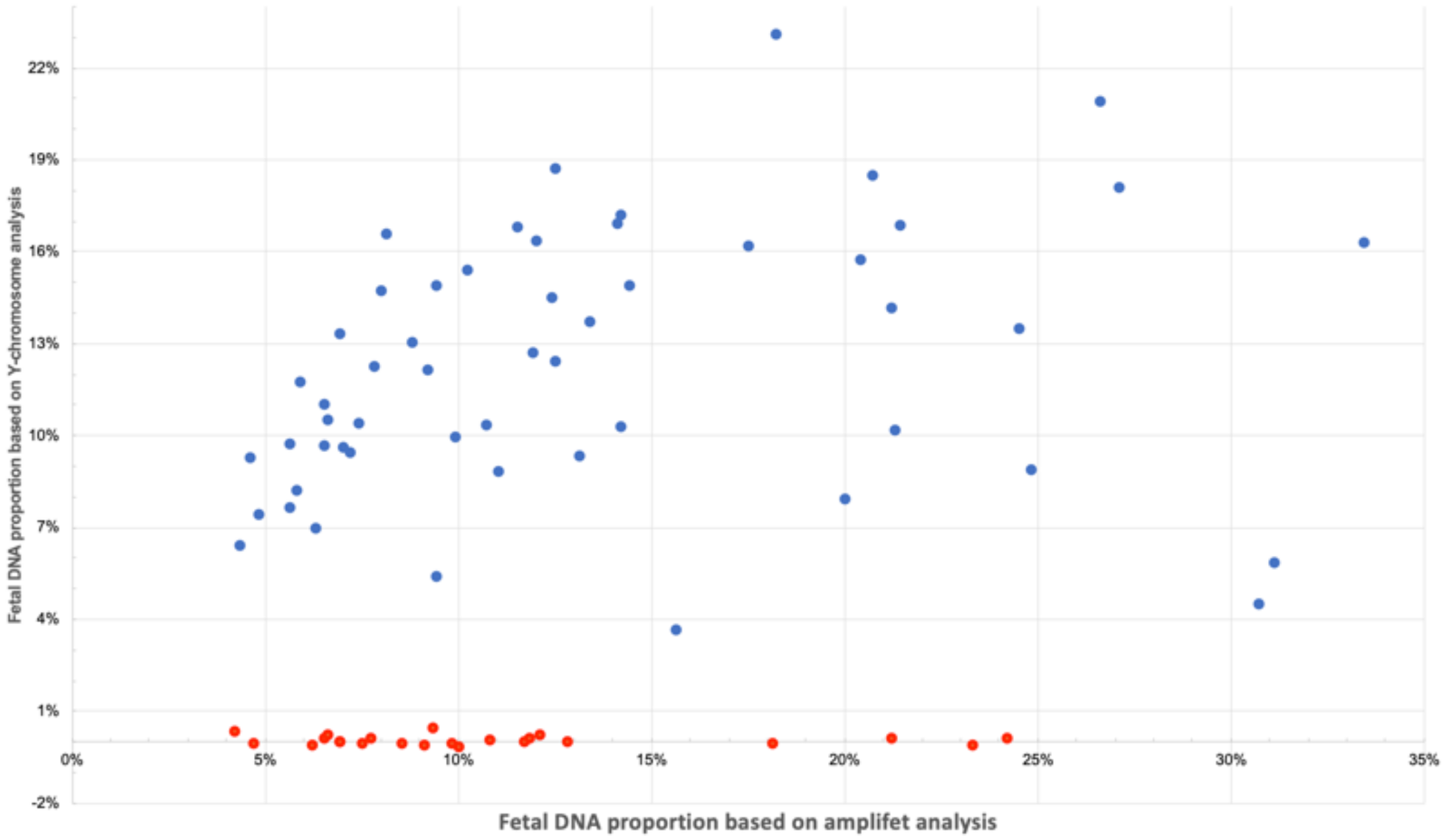
3.5. Amplifet Sample Sequencing Results
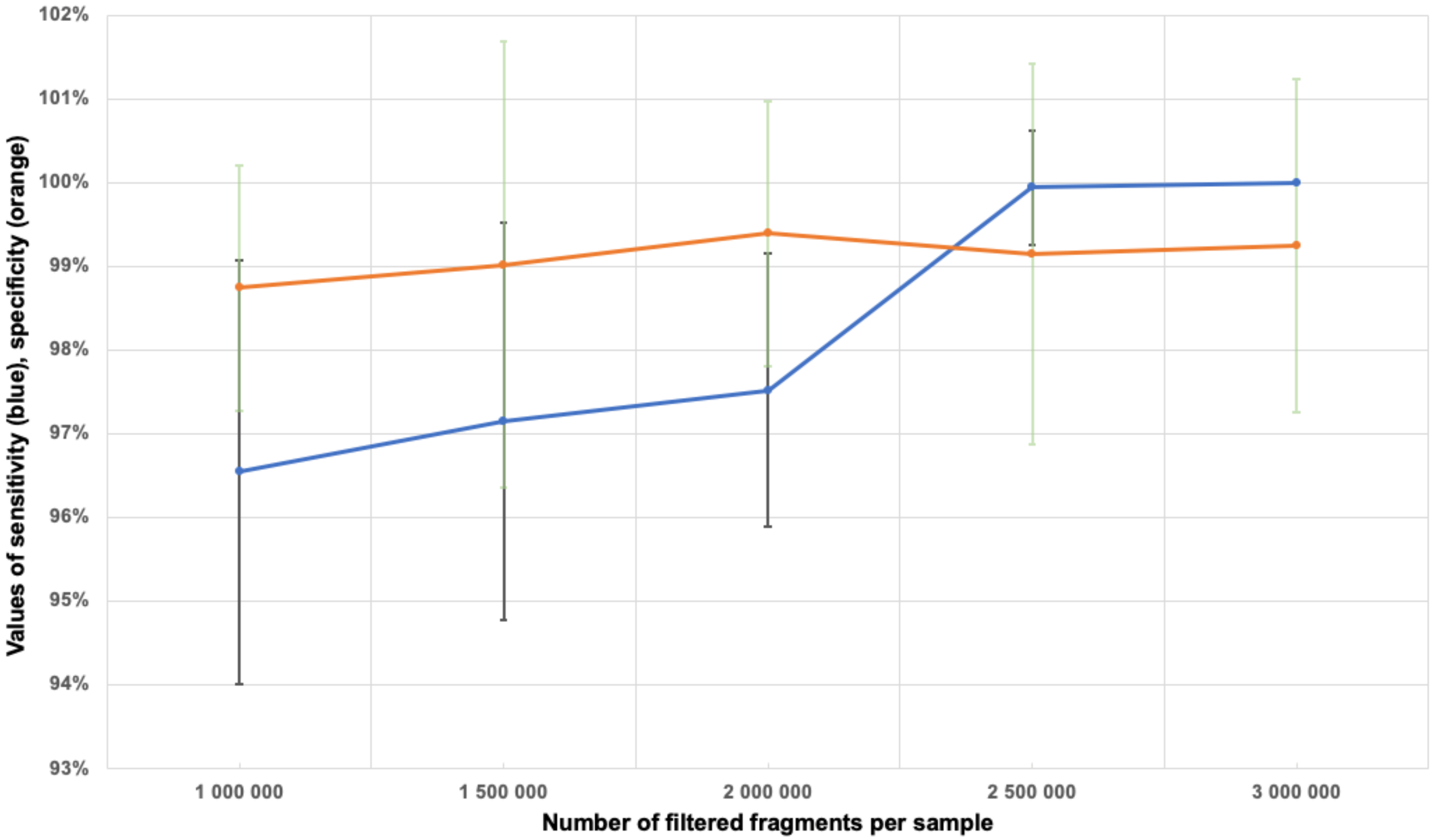
3.6. Determination of the Minimum Number of Fragments in the Sample
3.7. Sample Filtration
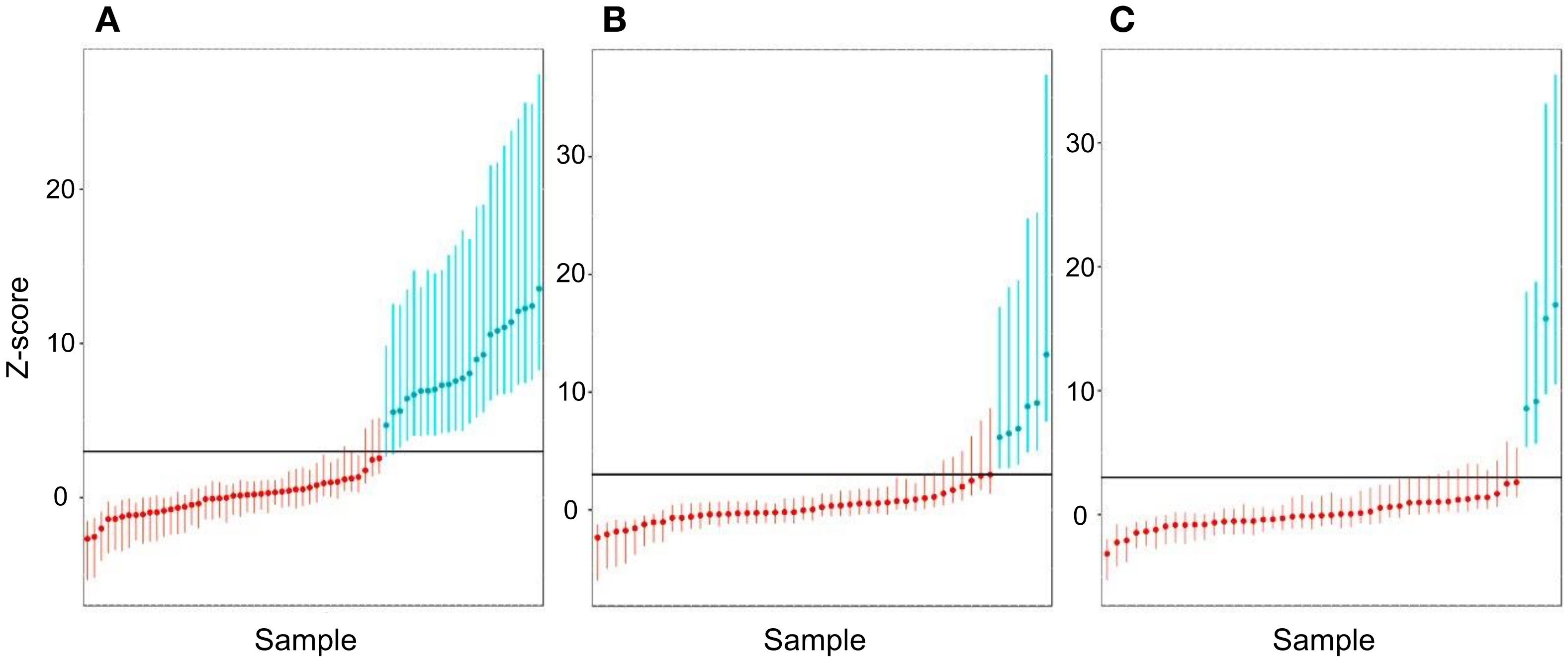
3.8. Sensitivity and Specificity Assessment for Aneuploidy
3.9. Calculation of PPV and NPV for Aneuploidy
3.10. Determination of the Sex of the Fetus
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Lo, Y.M.D.; Corbetta, N.; Chamberlain, P.F.; Rai, V.; Sargent, I.L.; Redman, C.W.; Wainscoat, J.S. Presence of fetal DNA in maternal plasma and serum. Lancet 1997, 350, 485–487. [Google Scholar] [CrossRef]
- Ishihara, N.; Matsuo, H.; Murakoshi, H.; Laoag-Fernandez, J.B.; Samoto, T.; Maruo, T. Increased apoptosis in the syncytiotrophoblast in human term placentas complicated by either preeclampsia or intrauterine growth retardation. Am. J. Obstet. Gynecol. 2002, 186, 158–166. [Google Scholar] [CrossRef]
- Farina, A.; Caramelli, E.; Concu, M.; Sekizawa, A.; Ruggeri, R.; Bovicelli, L.; Rizzo, N.; Carinci, P. Testing normality of fetal DNA concentration in maternal plasma at 10–12 completed weeks’ gestation: A preliminary approach to a new marker for genetic screening. Prenat. Diagnosis Publ. Affil. Int. Soc. Prenat. Diagn. 2002, 22, 148–152. [Google Scholar] [CrossRef]
- Lo, Y.M.D.; Chan, K.C.A.; Sun, H.; Chen, E.Z.; Jiang, P.; Lun, F.M.F.; Zheng, Y.W.; Leung, T.Y.; Lau, T.K.; Cantor, C.; et al. Maternal Plasma DNA Sequencing Reveals the Genome-Wide Genetic and Mutational Profile of the Fetus. Sci. Transl. Med. 2010, 2, 61ra91. [Google Scholar] [CrossRef]
- Ivanov, M.; Baranova, A.; Butler, T.; Spellman, P.T.; Mileyko, V. Non-random fragmentation patterns in circulating cell-free DNA reflect epigenetic regulation. BMC Genom. 2015, 16, S1. [Google Scholar] [CrossRef]
- Zimmermann, B.; Hill, M.; Gemelos, G.; Demko, Z.; Banjevic, M.; Baner, J.; Ryan, A.; Sigurjonsson, S.; Chopra, N.; Dodd, M.; et al. Noninvasive prenatal aneuploidy testing of chromosomes 13, 18, 21, X, and Y, using targeted sequencing of polymorphic loci. Prenat. Diagn. 2012, 32, 1233–1241. [Google Scholar] [CrossRef]
- Palomaki, G.E.; Kloza, E.M.; Lambert-Messerlian, G.; Haddow, J.E.; Neveux, L.M.; Ehrich, M.; Boom, D.V.D.; Bombard, A.T.; Deciu, C.; Grody, W.W.; et al. DNA sequencing of maternal plasma to detect Down syndrome: An international clinical validation study. Genet. Med. 2011, 13, 913–920. [Google Scholar] [CrossRef]
- Lo, K.K.; Boustred, C.; Chitty, L.S.; Plagnol, V. RAPIDR: An analysis package for non-invasive prenatal testing of aneuploidy. Bioinformatics 2014, 30, 2965–2967. [Google Scholar] [CrossRef][Green Version]
- Liao, G.J.W.; Lun, F.M.F.; Zheng, Y.W.L.; Chan, K.C.A.; Leung, T.Y.; Lau, T.K.; Chiu, R.W.K.; Lo, Y.D. Targeted Massively Parallel Sequencing of Maternal Plasma DNA Permits Efficient and Unbiased Detection of Fetal Alleles. Clin. Chem. 2011, 57, 92–101. [Google Scholar] [CrossRef]
- Sparks, A.B.; Struble, C.A.; Wang, E.; Song, K.; Oliphant, A. Noninvasive prenatal detection and selective analysis of cell-free DNA obtained from maternal blood: Evaluation for trisomy 21 and trisomy 18. Am. J. Obstet. Gynecol. 2012, 206, 319.e1–319.e9. [Google Scholar] [CrossRef]
- Yu, S.C.Y.; Chan, K.C.A.; Zheng, Y.W.L.; Jiang, P.; Liao, G.J.W.; Sun, H.; Akolekar, R.; Leung, T.Y.; Go, A.T.J.I.; Van Vugt, J.M.G.; et al. Size-based molecular diagnostics using plasma DNA for noninvasive prenatal testing. Proc. Natl. Acad. Sci. USA 2014, 111, 8583–8588. [Google Scholar] [CrossRef]
- Cirigliano, V.; Ordoñez, E.; Rueda, L.; Syngelaki, A.; Nicolaides, K.H. Performance of the neoBona test: A new paired-end massively parallel shotgun sequencing approach for cell-free DNA-based aneuploidy screening. Ultrasound Obstet. Gynecol. 2017, 49, 460–464. [Google Scholar] [CrossRef]
- Wang, Z.; Andrews, P.; Kendall, J.; Ma, B.; Hakker, I.; Rodgers, L.; Ronemus, M.; Wigler, M.; Levy, D. SMASH, a fragmentation and sequencing method for genomic copy number analysis. Genome Res. 2016, 26, 844–851. [Google Scholar] [CrossRef]
- Velculescu, V.E.; Zhang, L.; Vogelstein, B.; Kinzler, K.W. Serial Analysis of Gene Expression. Science 1995, 270, 484–487. [Google Scholar] [CrossRef]
- Matsumura, H.; Reich, S.; Ito, A.; Saitoh, H.; Kamoun, S.; Winter, P.; Kahl, G.; Reuter, M.; Krüger, D.H.; Terauchi, R. Gene expression analysis of plant host-pathogen interactions by SuperSAGE. Proc. Natl. Acad. Sci. USA 2003, 100, 15718–15723. [Google Scholar] [CrossRef]
- Chandrananda, D.; Thorne, N.P.; Bahlo, M. High-resolution characterization of sequence signatures due to non-random cleavage of cell-free DNA. BMC Med. Genom. 2015, 8, 29. [Google Scholar] [CrossRef]
- The 1000 Genomes Project Consortium An integrated map of genetic variation from 1092 human genomes. Nature 2012, 491, 56–65. [CrossRef]
- Lek, M.; Exome Aggregation Consortium; Karczewski, K.J.; Minikel, E.V.; Samocha, K.E.; Banks, E.; Fennell, T.; O’Donnell-Luria, A.H.; Ware, J.S.; Hill, A.J.; et al. Analysis of protein-coding genetic variation in 60,706 humans. Nature 2016, 536, 285–291. [Google Scholar] [CrossRef]
- Danecek, P.; Auton, A.; Abecasis, G.; Albers, C.A.; Banks, E.; Depristo, M.A.; Ha, G.; Lunter, G.; Marth, G.T.; Sherry, S.T.; et al. The variant call format and VCFtools. Bioinformatics 2011, 27, 2156–2158. [Google Scholar] [CrossRef]
- Li, H.; Durbin, R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 2009, 25, 1754–1760. [Google Scholar] [CrossRef]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R. The Sequence Alignment/Map format and SAMtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef]
- Magoč, T.; Salzberg, S. FLASH: Fast length adjustment of short reads to improve genome assemblies. Bioinformatics 2011, 27, 2957–2963. [Google Scholar] [CrossRef]
- Jiang, P.; Chan, A.K.; Liao, G.J.W.; Zheng, Y.W.L.; Leung, T.Y.; Chiu, R.W.K.; Lo, Y.M.D.; Sun, H. FetalQuant: Deducing fractional fetal DNA concentration from massively parallel sequencing of DNA in maternal plasma. Bioinformatics 2012, 28, 2883–2890. [Google Scholar] [CrossRef]
- Johansson, L.; De Weerd, H.A.; De Boer, E.N.; Van Dijk, F.; Meerman, G.J.T.; Sijmons, R.; Sikkema-Raddatz, B.; Swertz, M.A. NIPTeR: An R package for fast and accurate trisomy prediction in non-invasive prenatal testing. BMC Bioinform. 2018, 19, 531. [Google Scholar] [CrossRef]
- Wang, T.; He, Q.; Li, H.; Ding, J.; Wen, P.; Zhang, Q.; Xiang, J.; Li, Q.; Xuan, L.; Kong, L.; et al. An Optimized Method for Accurate Fetal Sex Prediction and Sex Chromosome Aneuploidy Detection in Non-Invasive Prenatal Testing. PLoS ONE 2016, 11, e0159648. [Google Scholar] [CrossRef]
- Hudecova, I.; Sahota, D.; Heung, M.M.S.; Jin, Y.; Lee, W.S.; Leung, T.Y.; Lo, Y.M.D.; Chiu, R.W.K. Maternal Plasma Fetal DNA Fractions in Pregnancies with Low and High Risks for Fetal Chromosomal Aneuploidies. PLoS ONE 2014, 9, e88484. [Google Scholar] [CrossRef]
- Xu, X.-P.; Gan, H.-Y.; Li, F.-X.; Tian, Q.; Zhang, J.; Liang, R.-L.; Li, M.; Yang, X.; Wu, Y.-S. A Method to Quantify Cell-Free Fetal DNA Fraction in Maternal Plasma Using Next Generation Sequencing: Its Application in Non-Invasive Prenatal Chromosomal Aneuploidy Detection. PLoS ONE 2016, 11, e0146997. [Google Scholar] [CrossRef]
- Breitbach, S.; Tug, S.; Helmig, S.; Zahn, D.; Kubiak, T.; Michal, M.; Gori, T.; Ehlert, T.; Beiter, T.; Simon, P. Direct Quantification of Cell-Free, Circulating DNA from Unpurified Plasma. PLoS ONE 2014, 9, e87838. [Google Scholar] [CrossRef]
- Wang, E.; Batey, A.; Struble, C.; Musci, T.; Song, K.; Oliphant, A. Gestational age and maternal weight effects on fetal cell-free DNA in maternal plasma. Prenat. Diagn. 2013, 33, 662–666. [Google Scholar] [CrossRef]
- Gil, M.M.; Quezada, M.S.; Revello, R.; Akolekar, R.; Nicolaides, K.H. Analysis of cell-free DNA in maternal blood in screening for fetal aneuploidies: Updated meta-analysis. Ultrasound Obstet. Gynecol. 2015, 45, 249–266. [Google Scholar] [CrossRef]
- Bianchi, D.W.; Platt, L.D.; Goldberg, J.D.; Abuhamad, A.Z.; Sehnert, A.J.; Rava, R.P. Genome-Wide Fetal Aneuploidy Detection by Maternal Plasma DNA Sequencing. Obstet. Gynecol. 2012, 119, 890–901. [Google Scholar] [CrossRef] [PubMed]
- Norton, M.E.; Jacobsson, B.; Swamy, G.K.; Laurent, L.C.; Ranzini, A.C.; Brar, H.; Tomlinson, M.W.; Pereira, L.; Spitz, J.L.; Hollemon, D.; et al. Cell-free DNA Analysis for Noninvasive Examination of Trisomy. N. Engl. J. Med. 2015, 372, 1589–1597. [Google Scholar] [CrossRef] [PubMed]
- McCullough, R.M.; Almasri, E.A.; Guan, X.; Geis, J.A.; Hicks, S.C.; Mazloom, A.R.; Deciu, C.; Oeth, P.; Bombard, A.T.; Paxton, B.; et al. Non-Invasive Prenatal Chromosomal Aneuploidy Testing—Clinical Experience: 100,000 Clinical Samples. PLoS ONE 2014, 9, e109173. [Google Scholar] [CrossRef] [PubMed]
- Loane, M.; Morris, J.K.; Addor, M.-C.; Arriola, L.; Budd, J.; Doray, B.; Garne, E.; Gatt, M.; Haeusler, M.; Khoshnood, B.; et al. Twenty-year trends in the prevalence of Down syndrome and other trisomies in Europe: Impact of maternal age and prenatal screening. Eur. J. Hum. Genet. 2012, 21, 27–33. [Google Scholar] [CrossRef]
- Prabakar, R.K.; Xu, L.; Hicks, J.; Smith, A. SMURF-seq: Efficient copy number profiling on long-read sequencers. Genome Biol. 2019, 20, 134. [Google Scholar] [CrossRef]
- Cheng, S.H.; Jiang, P.; Sun, K.; Cheng, Y.K.Y.; Chan, K.C.A.; Leung, T.Y.; Chiu, R.W.K.; Lo, Y.M.D. Noninvasive Prenatal Testing by Nanopore Sequencing of Maternal Plasma DNA: Feasibility Assessment. Clin. Chem. 2015, 61, 1305–1306. [Google Scholar] [CrossRef]
- Illumina Official Site. Available online: https://www.illumina.com/products/by-type/ivd-products/veriseq-nipt.html (accessed on 14 May 2020).
- Sun, K.; Jiang, P.; Wong, A.I.C.; Cheng, Y.K.Y.; Cheng, S.H.; Zhang, H.; Chan, K.C.A.; Leung, T.Y.; Chiu, R.W.K.; Lo, Y.M.D. Size-tagged preferred ends in maternal plasma DNA shed light on the production mechanism and show utility in noninvasive prenatal testing. Proc. Natl. Acad. Sci. USA 2018, 115, E5106–E5114. [Google Scholar] [CrossRef]
- Kim, S.K.; Hannum, G.; Geis, J.; Tynan, J.; Hogg, G.; Zhao, C.; Jensen, T.J.; Mazloom, A.R.; Oeth, P.; Ehrich, M.; et al. Determination of Fetal DNA Fraction from the Plasma of Pregnant Women using Sequence Read Counts. Prenat. Diagn. 2015, 35, 810–815. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Characteristic | Value |
|---|---|
| Number of patients | 145 |
| Average age (range) (years) | 35.6 (20–48) |
| Mean gestational age (weeks) | 15 (11–25) |
| Median gestational age (weeks) | 14 |
| Median weight of the pregnant women (range) (kg) | 65 (47–108) |
| Median height of the pregnant women (range) (cm) | 165 (150–185) |
| Race or ethnic group, number (proportion in %) | |
| white | 138 (95.2) |
| of which Slavonian | 131 (94.9) |
| Asian | 7 (4.8) |
| not indicated | 7 (4.8) |
| Result of the invasive diagnostic, number (proportion in %) | |
| normal karyotype | 82 (56.6) |
| chromosome trisomy 13 | 4 (2.8) |
| chromosome trisomy 18 | 14 (9.7) |
| chromosome trisomy 21 | 45 (31) |
| Chromosome | Minimal Filtered Fragment Threshold | Sensitivity | Sensitivity SD | Specificity | Specificity SD |
|---|---|---|---|---|---|
| 13 | 1,000,000 | 1 | 0 | 0.99536 | 0.011605 |
| 13 | 1,500,000 | 1 | 0 | 0.98883 | 0.030454 |
| 13 | 2,000,000 | 1 | 0 | 0.9952 | 0.020135 |
| 13 | 2,500,000 | 1 | 0 | 0.99169 | 0.032054 |
| 13 | 3,000,000 | 1 | 0 | 0.99556 | 0.012202 |
| 18 | 1,000,000 | 1 | 0 | 0.98154 | 0.023683 |
| 18 | 1,500,000 | 1 | 0 | 0.97256 | 0.04133 |
| 18 | 2,000,000 | 1 | 0 | 0.98368 | 0.020579 |
| 18 | 2,500,000 | 1 | 0 | 0.98342 | 0.031326 |
| 18 | 3,000,000 | 1 | 0 | 0.9925 | 0.01991 |
| 21 | 1,000,000 | 0.96537 | 0.025367 | 0.98735 | 0.014686 |
| 21 | 1,500,000 | 0.97143 | 0.023793 | 0.99017 | 0.026724 |
| 21 | 2,000,000 | 0.97517 | 0.016273 | 0.99388 | 0.015848 |
| 21 | 2,500,000 | 0.99935 | 0.0068607 | 0.9914 | 0.022761 |
| 21 | 3,000,000 | 1 | 0 | 0.9925 | 0.01991 |
| Trisomy 21 | Trisomy 18 | Trisomy 13 | |
|---|---|---|---|
| Sensitivity | 99.93% | 100% | 100% |
| 2-sided 95% CI | (85.58–99.99%) | (60.96–100%) | (51.01–100%) |
| Specificity | 99.14% | 98.34% | 99.17% |
| 2-sided 95% CI | (90.29–99.93%) | (91.16–99.7%) | (92.7–99.91%) |
| Aneuploidy | Prevalence | PPV | NPV |
|---|---|---|---|
| Trisomy 21 | 0.05% | 5.491% | 100.000% |
| 0.10% | 10.415% | 100.000% | |
| 0.20% | 18.880% | 100.000% | |
| 0.50% | 36.853% | 100.000% | |
| 1.00% | 53.983% | 99.999% | |
| 1.50% | 63.881% | 99.999% | |
| 2.00% | 70.328% | 99.999% | |
| Trisomy 18 | 0.03% | 1.777% | 100.000% |
| 0.05% | 2.928% | 100.000% | |
| 0.10% | 5.693% | 100.000% | |
| 0.20% | 10.782% | 100.000% | |
| 0.30% | 15.358% | 100.000% | |
| 0.40% | 19.496% | 100.000% | |
| 0.50% | 23.255% | 100.000% | |
| Trisomy 13 | 0.01% | 1.190% | 100.000% |
| 0.02% | 2.352% | 100.000% | |
| 0.05% | 5.680% | 100.000% | |
| 0.10% | 10.755% | 100.000% | |
| 0.20% | 19.437% | 100.000% |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Belova, V.; Plakhina, D.; Evfratov, S.; Tsukanov, K.; Khvorykh, G.; Rakitko, A.; Konoplyannikov, A.; Ilinsky, V.; Rebrikov, D.; Korostin, D. NIPT Technique Based on the Use of Long Chimeric DNA Reads. Genes 2020, 11, 590. https://doi.org/10.3390/genes11060590
Belova V, Plakhina D, Evfratov S, Tsukanov K, Khvorykh G, Rakitko A, Konoplyannikov A, Ilinsky V, Rebrikov D, Korostin D. NIPT Technique Based on the Use of Long Chimeric DNA Reads. Genes. 2020; 11(6):590. https://doi.org/10.3390/genes11060590
Chicago/Turabian StyleBelova, Vera, Daria Plakhina, Sergey Evfratov, Kirill Tsukanov, Gennady Khvorykh, Alexander Rakitko, Alexander Konoplyannikov, Valery Ilinsky, Denis Rebrikov, and Dmitriy Korostin. 2020. "NIPT Technique Based on the Use of Long Chimeric DNA Reads" Genes 11, no. 6: 590. https://doi.org/10.3390/genes11060590
APA StyleBelova, V., Plakhina, D., Evfratov, S., Tsukanov, K., Khvorykh, G., Rakitko, A., Konoplyannikov, A., Ilinsky, V., Rebrikov, D., & Korostin, D. (2020). NIPT Technique Based on the Use of Long Chimeric DNA Reads. Genes, 11(6), 590. https://doi.org/10.3390/genes11060590