Estimates of Autozygosity Through Runs of Homozygosity in Farmed Coho Salmon

 , ,
, ,
Abstract
1. Introduction
2. Methods
2.1. Coho Salmon Populations and Genotypes
2.2. Principal Components and Admixture Analysis
2.3. Runs of Homozygosity
2.4. Inbreeding Coefficient
3. Results
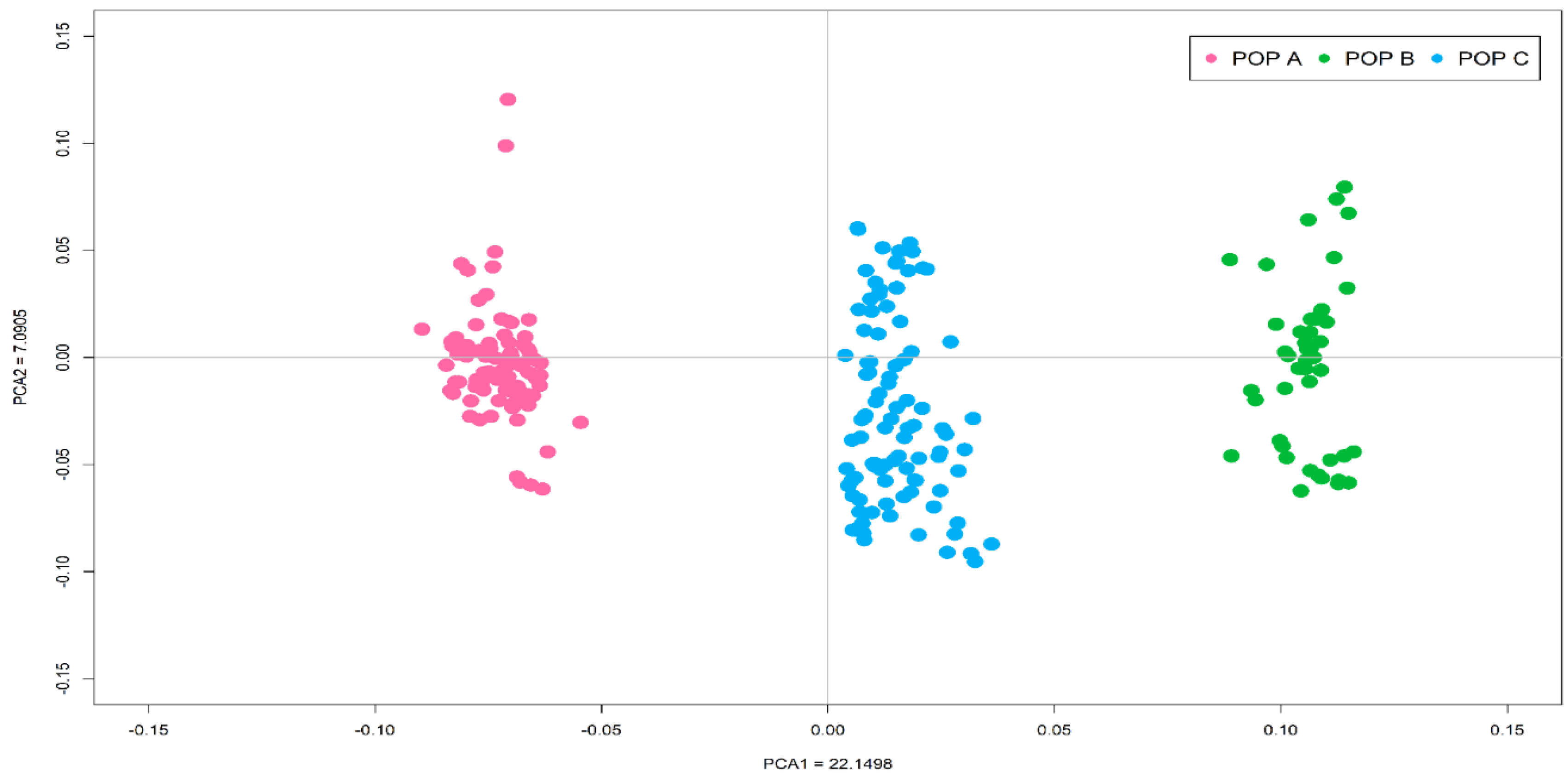
3.1. Quality Control and Genomic Population Structure
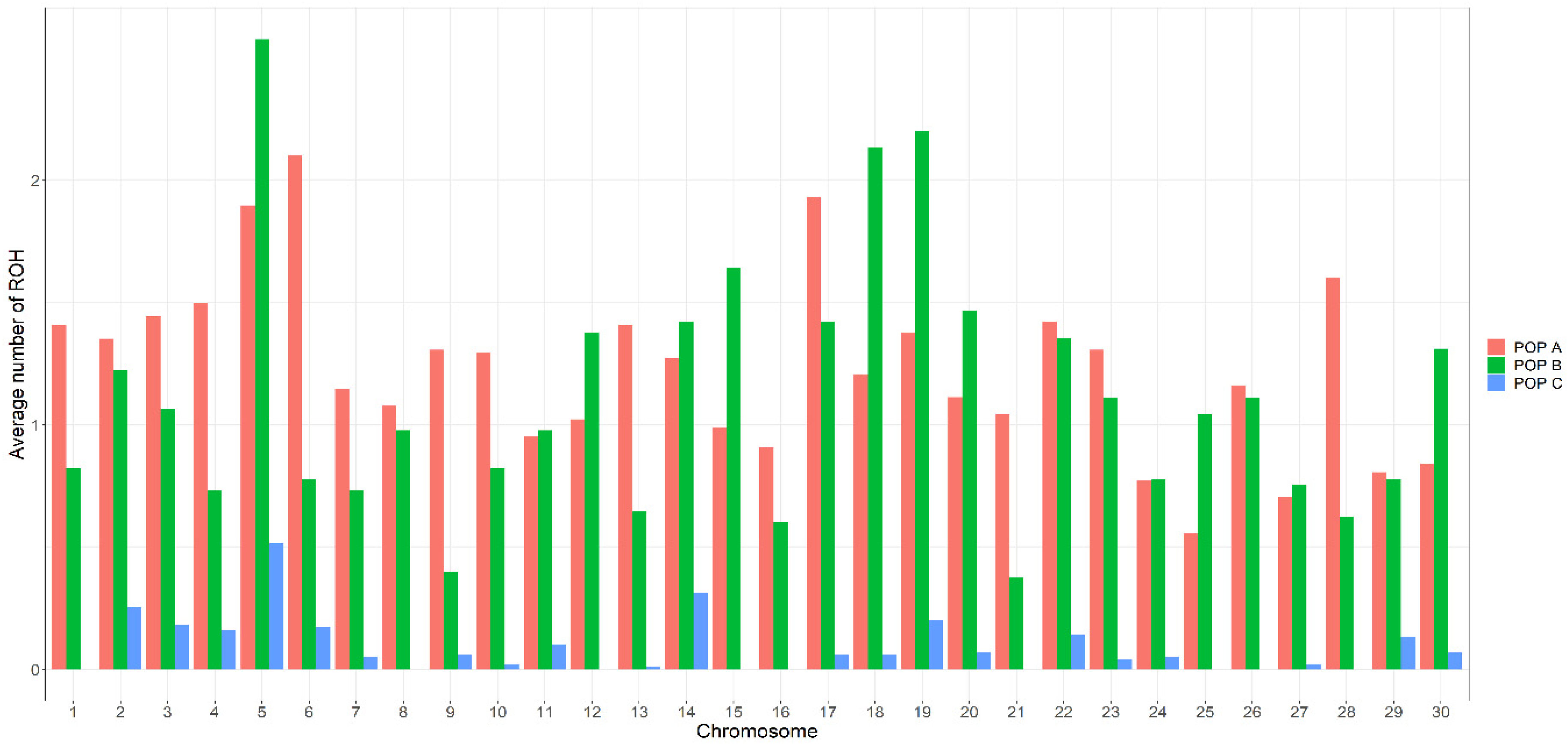
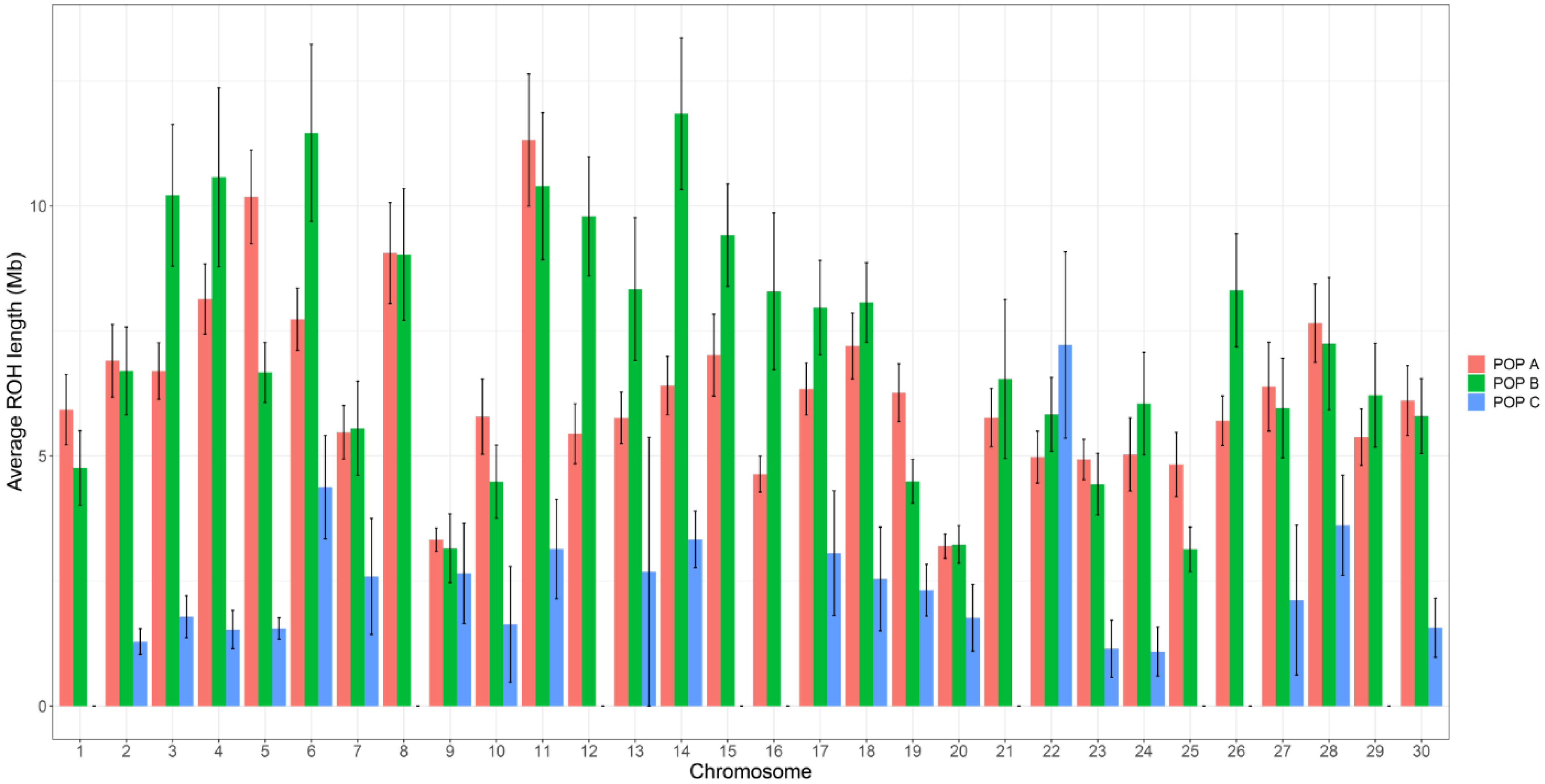
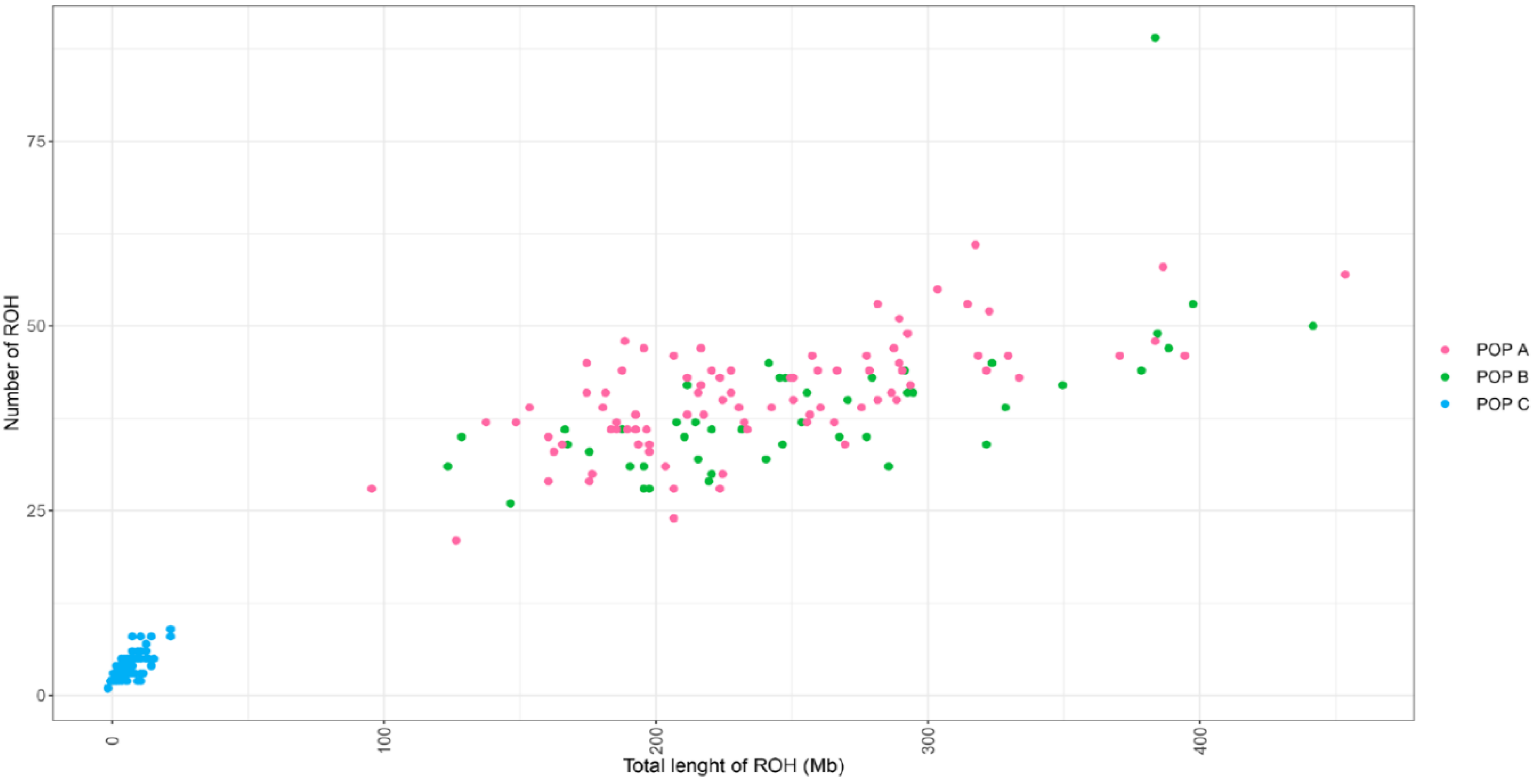
3.2. Distribution of Runs of Homozygosity
3.3. Genomic- and Pedigree-Based Inbreeding
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Groot, C.; Margolis, L. Pacific Salmon Life Histories; UBC Press: Vancouver, BC, Canada, 1991; ISBN 9780774803595. [Google Scholar]
- Neira, R.; Lhorente, J.P.; Yáñez, J.M.; Araneda, M.; Filp, M. Evolution of Coho Salmon (Oncorhynchus kisutch) Breeding Programs. In Proceedings of the 10th World Congress of Genetics Applied to Livestock Production, Vancouver, BC, Canada, 17–22 August 2014; pp. 1–6. [Google Scholar]
- Lhorente, J.P.; Araneda, M.; Neira, R.; Yáñez, J.M. Advances in genetic improvement for salmon and trout aquaculture: The Chilean situation and prospects. Rev. Aquac. 2019, 11, 340–353. [Google Scholar] [CrossRef]
- Gjedrem, T. Genetic improvement of cold-water fish species. Aquac. Res. 2000, 31, 25–33. [Google Scholar] [CrossRef]
- Gjedrem, T.; Robinson, N.; Rye, M. The importance of selective breeding in aquaculture to meet future demands for animal protein: A review. Aquaculture 2012, 350–353, 117–129. [Google Scholar] [CrossRef]
- Howard, J.T.; Pryce, J.E.; Baes, C.; Maltecca, C. Invited review: Inbreeding in the genomics era: Inbreeding, inbreeding depression, and management of genomic variability. J. Dairy Sci. 2017, 100, 6009–6024. [Google Scholar] [CrossRef] [PubMed]
- Gallardo, A.; Garcı, X.; Paul, J.; Neira, R. Inbreeding and inbreeding depression of female reproductive traits in two populations of Coho salmon selected using BLUP predictors of breeding values. Aquaculture 2004, 234, 111–122. [Google Scholar] [CrossRef]
- Yáñez, J.M.; Bassini, L.N.; Filp, M.; Lhorente, J.P.; Ponzoni, R.W.; Neira, R. Inbreeding and effective population size in a coho salmon (Oncorhynchus kisutch) breeding nucleus in Chile. Aquaculture 2014, 420–421, S15–S19. [Google Scholar] [CrossRef]
- Ponzoni, R.W.; Khaw, H.L.; Nguyen, N.H.; Hamzah, A. Inbreeding and effective population size in the Malaysian nucleus of the GIFT strain of Nile tilapia (Oreochromis niloticus). Aquaculture 2010, 302, 42–48. [Google Scholar] [CrossRef]
- Yoshida, G.M.; Yáñez, J.M.; de Oliveira, C.A.L.; Ribeiro, R.P.; Lhorente, J.P.; de Queiroz, S.A.; Carvalheiro, R. Mate selection in aquaculture breeding using differential evolution algorithm. Aquac. Res. 2017, 48, 5490–5497. [Google Scholar] [CrossRef]
- Forutan, M.; Ansari Mahyari, S.; Baes, C.; Melzer, N.; Schenkel, F.S.; Sargolzaei, M. Inbreeding and runs of homozygosity before and after genomic selection in North American Holstein cattle. BMC Genom. 2018, 19, 98. [Google Scholar] [CrossRef]
- Cassell, B.G.; Adamec, V.; Pearson, R.E. Effect of Incomplete Pedigrees on Estimates of Inbreeding and Inbreeding Depression for Days to First Service and Summit Milk Yield in Holsteins and Jerseys. J. Dairy Sci. 2003, 86, 2967–2976. [Google Scholar] [CrossRef]
- Curik, I.; Ferenčaković, M.; Sölkner, J. Inbreeding and runs of homozygosity: A possible solution to an old problem. Livest. Sci. 2014, 166, 26–34. [Google Scholar] [CrossRef]
- VanRaden, P.M. Efficient Methods to Compute Genomic Predictions. J. Dairy Sci. 2008, 91, 4414–4423. [Google Scholar] [CrossRef] [PubMed]
- Marras, G.; Gaspa, G.; Sorbolini, S.; Dimauro, C.; Ajmone-Marsan, P.; Valentini, A.; Williams, J.L.; Macciotta, N.P.P. Analysis of runs of homozygosity and their relationship with inbreeding in five cattle breeds farmed in Italy. Anim. Genet. 2015, 46, 110–121. [Google Scholar] [CrossRef] [PubMed]
- McQuillan, R.; Leutenegger, A.-L.; Abdel-Rahman, R.; Franklin, C.S.; Pericic, M.; Barac-Lauc, L.; Smolej-Narancic, N.; Janicijevic, B.; Polasek, O.; Tenesa, A.; et al. Runs of Homozygosity in European Populations. Am. J. Hum. Genet. 2008, 83, 359–372. [Google Scholar] [CrossRef]
- Brüniche-Olsen, A.; Kellner, K.F.; Anderson, C.J.; DeWoody, J.A. Runs of homozygosity have utility in mammalian conservation and evolutionary studies. Conserv. Genet. 2018, 19, 1295–1307. [Google Scholar] [CrossRef]
- Falconer, D.S.; Mackay, T.F.C. Introduction to Quantitative Genetics; Pearson, P.H., Ed.; Longman: Harlow, UK, 1996. [Google Scholar]
- Peripolli, E.; Munari, D.P.; Silva, M.V.G.B.; Lima, A.L.F.; Irgang, R.; Baldi, F. Runs of homozygosity: Current knowledge and applications in livestock. Anim. Genet. 2017, 48, 255–271. [Google Scholar] [CrossRef]
- MacLeod, I.M.; Larkin, D.M.; Lewin, H.A.; Hayes, B.J.; Goddard, M.E. Inferring demography from runs of homozygosity in whole-genome sequence, with correction for sequence errors. Mol. Biol. Evol. 2013, 30, 2209–2223. [Google Scholar] [CrossRef]
- Sams, A.J.; Boyko, A.R. Fine-Scale Resolution of Runs of Homozygosity Reveal Patterns of Inbreeding and Substantial Overlap with Recessive Disease Genotypes in Domestic Dogs. G3 2019, 9, 117–123. [Google Scholar] [CrossRef]
- Kirin, M.; McQuillan, R.; Franklin, C.S.; Campbell, H.; McKeigue, P.M.; Wilson, J.F. Genomic Runs of Homozygosity Record Population History and Consanguinity. PLoS ONE 2010, 5, e13996. [Google Scholar] [CrossRef]
- Nothnagel, M.; Lu, T.T.; Kayser, M.; Krawczak, M. Genomic and geographic distribution of SNP-defined runs of homozygosity in Europeans. Hum. Mol. Genet. 2010, 19, 2927–2935. [Google Scholar] [CrossRef]
- Gurgul, A.; Szmatoła, T.; Topolski, P.; Jasielczuk, I.; Żukowski, K.; Bugno-Poniewierska, M. The use of runs of homozygosity for estimation of recent inbreeding in Holstein cattle. J. Appl. Genet. 2016, 57, 527–530. [Google Scholar] [CrossRef] [PubMed]
- Mészáros, G.; Boison, S.A.; Pérez O’Brien, A.M.; Ferenčaković, M.; Curik, I.; Da Silva, M.V.B.; Utsunomiya, Y.T.; Garcia, J.F.; Sölkner, J. Genomic analysis for managing small and endangered populations: A case study in Tyrol Grey cattle. Front. Genet. 2015, 6, 173. [Google Scholar] [PubMed]
- Signer-Hasler, H.; Burren, A.; Neuditschko, M.; Frischknecht, M.; Garrick, D.; Stricker, C.; Gredler, B.; Bapst, B.; Flury, C. Population structure and genomic inbreeding in nine Swiss dairy cattle populations. Genet. Sel. Evol. 2017, 49, 83. [Google Scholar] [CrossRef] [PubMed]
- Ferenčaković, M.; Hamzić, E.; Gredler, B.; Solberg, T.R.; Klemetsdal, G.; Curik, I.; Sölkner, J. Estimates of autozygosity derived from runs of homozygosity: Empirical evidence from selected cattle populations. J. Anim. Breed. Genet. 2013, 130, 286–293. [Google Scholar] [CrossRef] [PubMed]
- Kim, E.-S.; Cole, J.B.; Huson, H.; Wiggans, G.R.; Van Tassell, C.P.; Crooker, B.A.; Liu, G.; Da, Y.; Sonstegard, T.S. Effect of Artificial Selection on Runs of Homozygosity in U.S. Holstein Cattle. PLoS ONE 2013, 8, e80813. [Google Scholar] [CrossRef]
- Gomez-Raya, L.; Rodríguez, C.; Barragán, C.; Silió, L. Genomic inbreeding coefficients based on the distribution of the length of runs of homozygosity in a closed line of Iberian pigs. Genet. Sel. Evol. 2015, 47, 81. [Google Scholar] [CrossRef]
- Xu, Z.; Sun, H.; Zhang, Z.; Zhao, Q.; Olasege, B.S.; Li, Q.; Yue, Y.; Ma, P.; Zhang, X.; Wang, Q.; et al. Assessment of Autozygosity Derived From Runs of Homozygosity in Jinhua Pigs Disclosed by Sequencing Data. Front. Genet. 2019, 10, 274. [Google Scholar] [CrossRef]
- Ai, H.; Huang, L.; Ren, J. Genetic Diversity, Linkage Disequilibrium and Selection Signatures in Chinese and Western Pigs Revealed by Genome-Wide SNP Markers. PLoS ONE 2013, 8, e56001. [Google Scholar] [CrossRef]
- Beynon, S.E.; Slavov, G.T.; Farré, M.; Sunduimijid, B.; Waddams, K.; Davies, B.; Haresign, W.; Kijas, J.; MacLeod, I.M.; Newbold, C.J.; et al. Population structure and history of the Welsh sheep breeds determined by whole genome genotyping. BMC Genet. 2015, 16, 65. [Google Scholar] [CrossRef]
- Manunza, A.; Noce, A.; Serradilla, J.M.; Goyache, F.; Martínez, A.; Capote, J.; Delgado, J.V.; Jordana, J.; Muñoz, E.; Molina, A.; et al. A genome-wide perspective about the diversity and demographic history of seven Spanish goat breeds. Genet. Sel. Evol. 2016, 48, 52. [Google Scholar] [CrossRef]
- Ghoreishifar, S.M.; Moradi-Shahrbabak, H.; Fallahi, M.H.; Moradi-Shahrbabak, M.; Abdollahi-Arpanahi, R.; Khansefid, M. Genomic measures of inbreeding coefficients and genome-wide scan for runs of homozygosity islands in Iranian river buffalo, Bubalus bubalis. BMC Genet. 2020, 21, 16. [Google Scholar]
- Purfield, D.C.; Berry, D.P.; McParland, S.; Bradley, D.G. Runs of homozygosity and population history in cattle. BMC Genet. 2012, 13, 70. [Google Scholar] [CrossRef] [PubMed]
- D’Ambrosio, J.; Phocas, F.; Haffray, P.; Bestin, A.; Brard-Fudulea, S.; Poncet, C.; Quillet, E.; Dechamp, N.; Fraslin, C.; Charles, M.; et al. Genome-wide estimates of genetic diversity, inbreeding and effective size of experimental and commercial rainbow trout lines undergoing selective breeding. Genet. Sel. Evol. 2019, 51, 26. [Google Scholar] [CrossRef] [PubMed]
- Yañez, J.M.; Bangera, R.; Lhorente, J.P.; Oyarzún, M.; Neira, R. Quantitative genetic variation of resistance against Piscirickettsia salmonis in Atlantic salmon (Salmo salar). Aquaculture 2013, 414, 155–159. [Google Scholar] [CrossRef]
- Yáñez, J.M.; Lhorente, J.P.; Bassini, L.N.; Oyarzún, M.; Neira, R.; Newman, S. Genetic co-variation between resistance against both Caligus rogercresseyi and Piscirickettsia salmonis, and body weight in Atlantic salmon (Salmo salar). Aquaculture 2014, 433, 295–298. [Google Scholar] [CrossRef]
- Barria, A.; Christensen, K.A.; Yoshida, G.; Jedlicki, A.; Leong, J.S.; Rondeau, E.B.; Lhorente, J.P.; Koop, B.F.; Davidson, W.S.; Yáñez, J.M. Whole genome linkage disequilibrium and effective population size in a coho salmon (Oncorhynchus kisutch) breeding population using a high density SNP array. Front. Genet. 2019, 10, 498. [Google Scholar] [CrossRef]
- Purcell, S.; Neale, B.; Todd-Brown, K.; Thomas, L.; Ferreira, M.A.R.; Bender, D.; Maller, J.; Sklar, P.; de Bakker, P.I.W.; Daly, M.J.; et al. PLINK: A tool set for whole-genome association and population-based linkage analyses. Am. J. Hum. Genet. 2007, 81, 559–575. [Google Scholar] [CrossRef]
- R Core Team. R: A Language and Environment for Statistical Computing; R Foundation for Statistical Computing: Vienna, Austria, 2015. [Google Scholar]
- Pritchard, J.K.; Stephens, M.; Rosenberg, N.A.; Donnelly, P. Association Mapping in Structured Populations. Am. J. Hum. Genet. 2000, 67, 170–181. [Google Scholar] [CrossRef]
- Evanno, G.; Regnaut, S.; Goudet, J. Detecting the number of clusters of individuals using the software structure: A simulation study. Mol. Ecol. 2005, 14, 2611–2620. [Google Scholar] [CrossRef]
- Biscarini, F.; Cozzi, P.; Gaspa, G.; Marras, G. Detect Runs of Homozygosity and Runs of Heterozygosity in Diploid Genomes. CRAN. Available online: https://cran.r-project.org/web/checks/check_results_detectRUNS.html (accessed on 24 October 2019).
- Wright, S. Genetics of populations. Encycl. Br. 1948, 10, 111–112. [Google Scholar]
- Legarra, A.; Aguilar, I.; Misztal, I. A relationship matrix including full pedigree and genomic information. J. Dairy Sci. 2009, 92, 4656–4663. [Google Scholar] [CrossRef] [PubMed]
- Allendorf, F.W.; Phelps, S.R. Loss of Genetic Variation in a Hatchery Stock of Cutthroat Trout. Trans. Am. Fish. Soc. 1980, 109, 537–543. [Google Scholar] [CrossRef]
- Gibson, J.; Morton, N.E.; Collins, A. Extended tracts of homozygosity in outbred human populations. Hum. Mol. Genet. 2006, 15, 789–795. [Google Scholar] [CrossRef] [PubMed]
- Ceballos, F.C.; Joshi, P.K.; Clark, D.W.; Ramsay, M.; Wilson, J.F. Runs of homozygosity: Windows into population history and trait architecture. Nat. Rev. Genet. 2018, 19, 220–234. [Google Scholar] [CrossRef] [PubMed]
- Broman, K.W.; Weber, J.L. Long Homozygous Chromosomal Segments in Reference Families from the Centre d’Étude du Polymorphisme Humain. Am. J. Hum. Genet. 1999, 65, 1493–1500. [Google Scholar] [CrossRef]
- Zhang, Q.; Calus, M.P.; Guldbrandtsen, B.; Lund, M.S.; Sahana, G. Estimation of inbreeding using pedigree, 50k SNP chip genotypes and full sequence data in three cattle breeds. BMC Genet. 2015, 16, 88. [Google Scholar] [CrossRef] [PubMed]
- Peripolli, E.; Stafuzza, N.B.; Munari, D.P.; Lima, A.L.F.; Irgang, R.; Machado, M.A.; Panetto, J.C.d.C.; Ventura, R.V.; Baldi, F.; da Silva, M.V.G.B. Assessment of runs of homozygosity islands and estimates of genomic inbreeding in Gyr (Bos indicus) dairy cattle. BMC Genomics 2018, 19, 34. [Google Scholar] [CrossRef]
- Zhang, Z.; Zhang, Q.; Xiao, Q.; Sun, H.; Gao, H.; Yang, Y.; Chen, J.; Li, Z.; Xue, M.; Ma, P.; et al. Distribution of runs of homozygosity in Chinese and Western pig breeds evaluated by reduced-representation sequencing data. Anim. Genet. 2018, 49, 579–591. [Google Scholar] [CrossRef]
- Howard, J.T.; Tiezzi, F.; Huang, Y.; Gray, K.A.; Maltecca, C. Characterization and management of long runs of homozygosity in parental nucleus lines and their associated crossbred progeny. Genet. Sel. Evol. 2016, 48, 91. [Google Scholar] [CrossRef]
- Cardoso, T.F.; Amills, M.; Bertolini, F.; Rothschild, M.; Marras, G.; Boink, G.; Jordana, J.; Capote, J.; Carolan, S.; Hallsson, J.H.; et al. Patterns of homozygosity in insular and continental goat breeds. Genet. Sel. Evol. 2018, 50, 56. [Google Scholar] [CrossRef]
- Bertolini, F.; Cardoso, T.F.; Marras, G.; Nicolazzi, E.L.; Rothschild, M.F.; Amills, M. Genome-wide patterns of homozygosity provide clues about the population history and adaptation of goats. Genet. Sel. Evol. 2018, 50, 59. [Google Scholar] [CrossRef] [PubMed]
- Onzima, R.B.; Upadhyay, M.R.; Doekes, H.P.; Brito, L.F.; Bosse, M.; Kanis, E.; Groenen, M.A.M.; Crooijmans, R.P.M.A. Genome-Wide Characterization of Selection Signatures and Runs of Homozygosity in Ugandan Goat Breeds. Front. Genet. 2018, 9, 318. [Google Scholar] [CrossRef] [PubMed]
- Morales-González, E.; Saura, M.; Fernández, A.; Fernández, J.; Pong-Wong, R.; Cabaleiro, S.; Martínez, P.; Martín-García, A.; Villanueva, B. Evaluating different genomic coancestry matrices for managing genetic variability in turbot. Aquaculture 2020, 520, 734985. [Google Scholar] [CrossRef]
- Wang, J. Marker-based estimates of relatedness and inbreeding coefficients: An assessment of current methods. J. Evol. Biol. 2014, 27, 518–530. [Google Scholar] [CrossRef] [PubMed]
- Kardos, M.; Luikart, G.; Allendorf, F.W. Measuring individual inbreeding in the age of genomics: Marker-based measures are better than pedigrees. Heredity 2015, 115, 63–72. [Google Scholar] [CrossRef] [PubMed]
- Keller, M.C.; Visscher, P.M.; Goddard, M.E. Quantification of inbreeding due to distant ancestors and its detection using dense single nucleotide polymorphism data. Genetics 2011, 189, 237–249. [Google Scholar] [CrossRef]
- Mastrangelo, S.; Tolone, M.; Di Gerlando, R.; Fontanesi, L.; Sardina, M.T.; Portolano, B. Genomic inbreeding estimation in small populations: Evaluation of runs of homozygosity in three local dairy cattle breeds. Animal 2016, 10, 746–754. [Google Scholar] [CrossRef]
- Purfield, D.C.; McParland, S.; Wall, E.; Berry, D.P. The distribution of runs of homozygosity and selection signatures in six commercial meat sheep breeds. PLoS ONE 2017, 12, e0176780. [Google Scholar] [CrossRef]
- Sumreddee, P.; Toghiani, S.; Hay, E.H.; Roberts, A.; Agrrey, S.E.; Rekaya, R. Inbreeding depression in line Hereford cattle population using pedigree and genomic information. J. Anim. Sci. 2019, 97, 1–18. [Google Scholar] [CrossRef]
- Zavarez, L.B.; Utsunomiya, Y.T.; Carmo, A.S.; Neves, H.H.R.; Brien, A.M.P.O.; Curik, I.; Cole, J.B.; Carvalheiro, R.; Feren, M.; Tassell, V.; et al. Assessment of autozygosity in Nellore cows (Bos indicus) through high-density SNP genotypes. Front. Genet. 2015, 6, 1–8. [Google Scholar] [CrossRef]
- Lencz, T.; Lambert, C.; DeRosse, P.; Burdick, K.E.; Morgan, T.V.; Kane, J.M.; Kucherlapati, R.; Malhotra, A.K. Runs of homozygosity reveal highly penetrant recessive loci in schizophrenia. Proc. Natl. Acad. Sci. USA 2007, 104, 19942–19947. [Google Scholar] [CrossRef] [PubMed]
- Bosse, M.; Megens, H.-J.; Madsen, O.; Paudel, Y.; Frantz, L.A.F.; Schook, L.B.; Crooijmans, R.P.M.A.; Groenen, M.A.M. Regions of Homozygosity in the Porcine Genome: Consequence of Demography and the Recombination Landscape. PLoS Genet. 2012, 8, e1003100. [Google Scholar] [CrossRef] [PubMed]
- Traspov, A.; Deng, W.; Kostyunina, O.; Ji, J.; Shatokhin, K.; Lugovoy, S.; Zinovieva, N.; Yang, B.; Huang, L. Population structure and genome characterization of local pig breeds in Russia, Belorussia, Kazakhstan and Ukraine. Genet. Sel. Evol. 2016, 48, 16. [Google Scholar] [CrossRef] [PubMed]
- Goszczynski, D.; Molina, A.; Terán, E.; Morales-Durand, H.; Ross, P.; Cheng, H.; Giovambattista, G.; Demyda-Peyrás, S. Runs of homozygosity in a selected cattle population with extremely inbred bulls: Descriptive and functional analyses revealed highly variable patterns. PLoS ONE 2018, 13, e0200069. [Google Scholar] [CrossRef] [PubMed]
- Smitherman, R.O.; Tave, D. Maintenance of genetic quality in cultured tilapia. Asian Fish. Sci. 1987, 1, 75–82. [Google Scholar]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
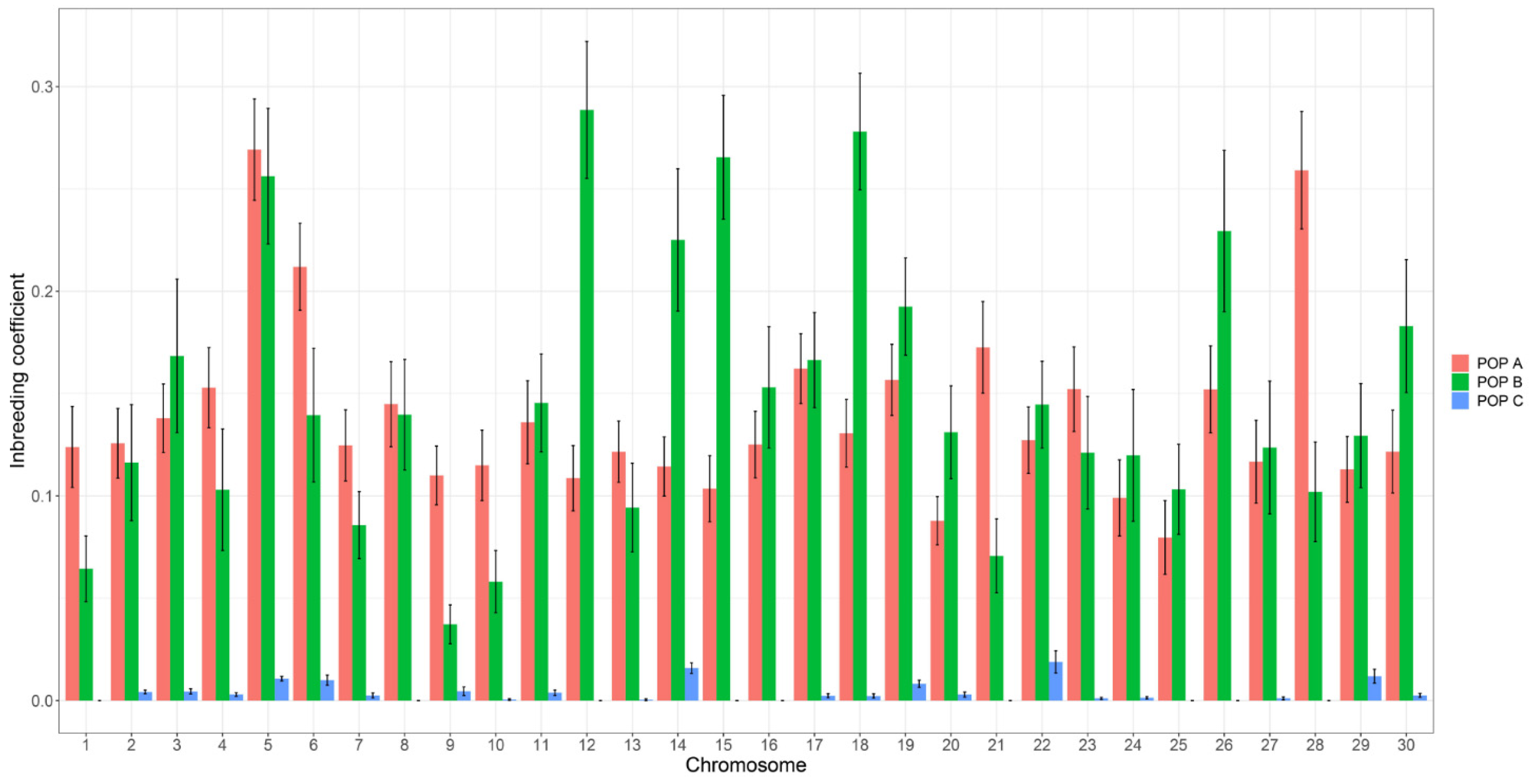{kind=link}
| Class | POP A | POP B | POP C | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| N | N Mean | Length | N | N Mean | Length | N | N Mean | Length | |||
| ROHALL | 3250 | 7.39 (3.75) | 6.47 (7.39) | 1497 | 6.65 (3.84) | 7.17 (7.69) | 266 | 0.54 (0.85) | 2.58 (2.07) | ||
| ROH1–2 Mb | 847 | 9.63 (3.16) | 1.46 (0.29) | 341 | 7.58 (3.60) | 1.46 (0.29) | 158 | 1.60 (1.06) | 1.40 (0.26) | ||
| ROH2–4 Mb | 937 | 10.65 (3.28) | 2.89 (0.58) | 400 | 8.89 (4.33) | 2.84 (0.53) | 58 | 0.59 (0.69) | 2.86 (0.58) | ||
| ROH4–8 Mb | 680 | 7.73 (2.73) | 5.59 (1.07) | 310 | 6.89 (3.74) | 5.56 (1.03) | 43 | 0.43 (0.59) | 5.11 (1.14) | ||
| ROH8–16 Mb | 463 | 5.26 (2.26) | 11.31 (2.32) | 260 | 5.78 (3.06) | 11.45 (2.25) | 7 | 0.07 (0.26) | 11.54 (0.04) | ||
| ROH>16 Mb | 323 | 3.67 (1.75) | 24.93 (7.68) | 186 | 4.13 (2.58) | 23.69 (8.08) | 0 | 0 | - | ||
| Class | POP A | POP B | POP C | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| N | Mean | SD | N | Mean | SD | N | Mean | SD | |||
| FROH1–2 Mb | 88 | 0.142 a | 0.038 | 45 | 0.152 a | 0.044 | 103 | 0.004 b | 0.003 | ||
| FROH2–4 Mb | 88 | 0.133 a | 0.038 | 45 | 0.145 a | 0.044 | 73 | 0.003 b | 0.003 | ||
| FROH4–8 Mb | 88 | 0.115 a | 0.037 | 45 | 0.129 a | 0.043 | 47 | 0.002 b | 0.002 | ||
| FROH8–16 Mb | 88 | 0.090 a | 0.036 | 45 | 0.104 a | 0.043 | 7 | 0.000 b | 0.002 | ||
| FROH>16 Mb | 85 | 0.054 a | 0.028 | 45 | 0.062 a | 0.036 | - | - | - | ||
| FROHALL | 88 | 0.142 a | 0.038 | 45 | 0.152 a | 0.044 | 103 | 0.004 b | 0.003 | ||
| FHOM | 88 | −0.036 b | 0.048 | 45 | −0.069 a | 0.056 | 104 | −0.105 c | 0.011 | ||
| FGRM | 88 | 0.145 b | 0.037 | 45 | 0.193 a | 0.040 | 104 | 0.051 c | 0.009 | ||
| FPED | 88 | 0.071 a | 0.021 | 45 | 0.076 a | 0.027 | 104 | 0.002 b | 0.012 | ||
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yoshida, G.M.; Cáceres, P.; Marín-Nahuelpi, R.; Koop, B.F.; Yáñez, J.M. Estimates of Autozygosity Through Runs of Homozygosity in Farmed Coho Salmon. Genes 2020, 11, 490. https://doi.org/10.3390/genes11050490
Yoshida GM, Cáceres P, Marín-Nahuelpi R, Koop BF, Yáñez JM. Estimates of Autozygosity Through Runs of Homozygosity in Farmed Coho Salmon. Genes. 2020; 11(5):490. https://doi.org/10.3390/genes11050490
Chicago/Turabian StyleYoshida, Grazyella M., Pablo Cáceres, Rodrigo Marín-Nahuelpi, Ben F. Koop, and José M. Yáñez. 2020. "Estimates of Autozygosity Through Runs of Homozygosity in Farmed Coho Salmon" Genes 11, no. 5: 490. https://doi.org/10.3390/genes11050490
APA StyleYoshida, G. M., Cáceres, P., Marín-Nahuelpi, R., Koop, B. F., & Yáñez, J. M. (2020). Estimates of Autozygosity Through Runs of Homozygosity in Farmed Coho Salmon. Genes, 11(5), 490. https://doi.org/10.3390/genes11050490