MCPH1 Lack of Function Enhances Mitotic Cell Sensitivity Caused by Catalytic Inhibitors of Topo II

 ,
, 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Ethic Statement
2.2. Cell Culture and Treatments
2.3. Live-Cell Microscopy
2.4. Immunofluorescence
2.5. Western Blot
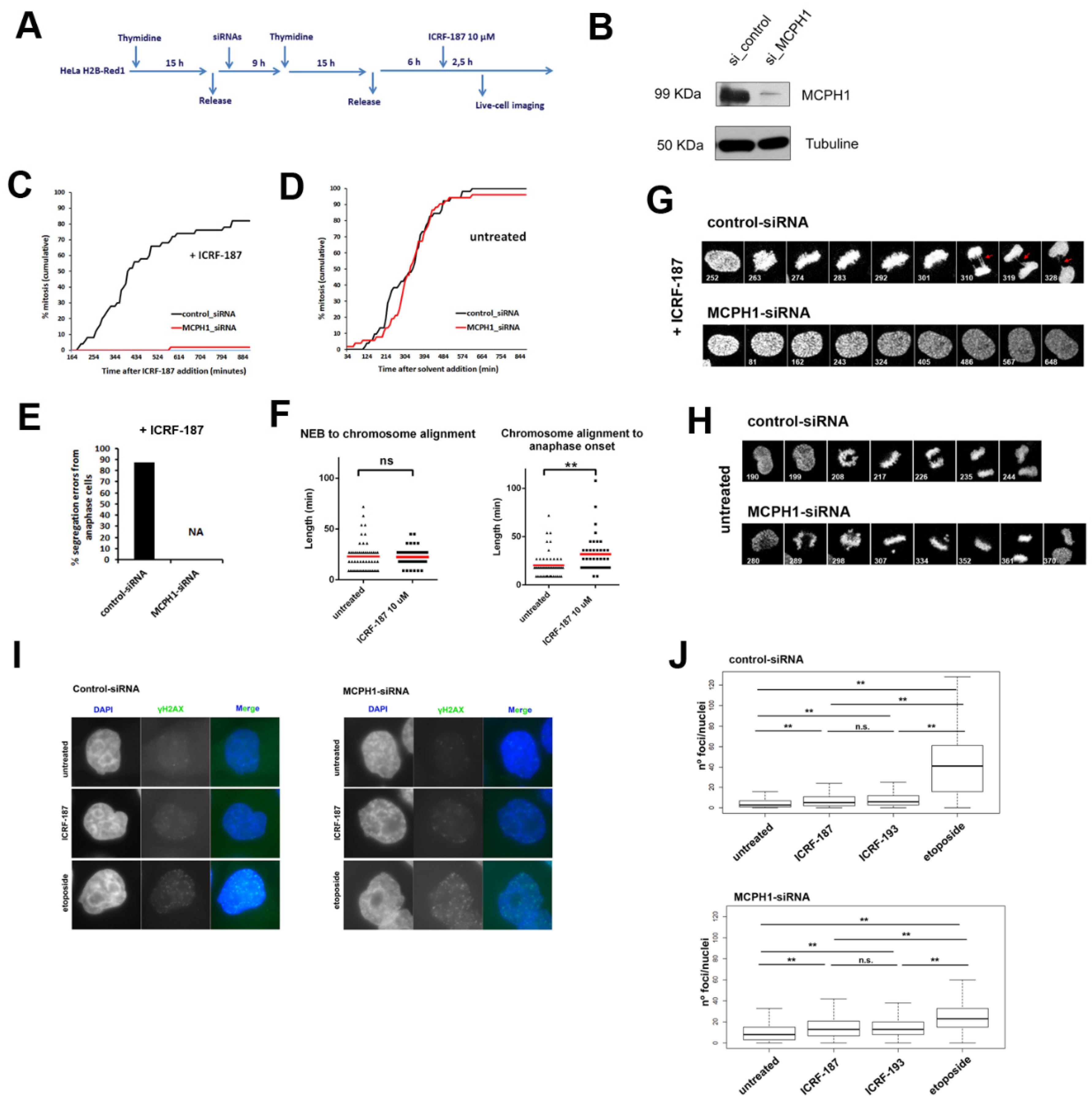
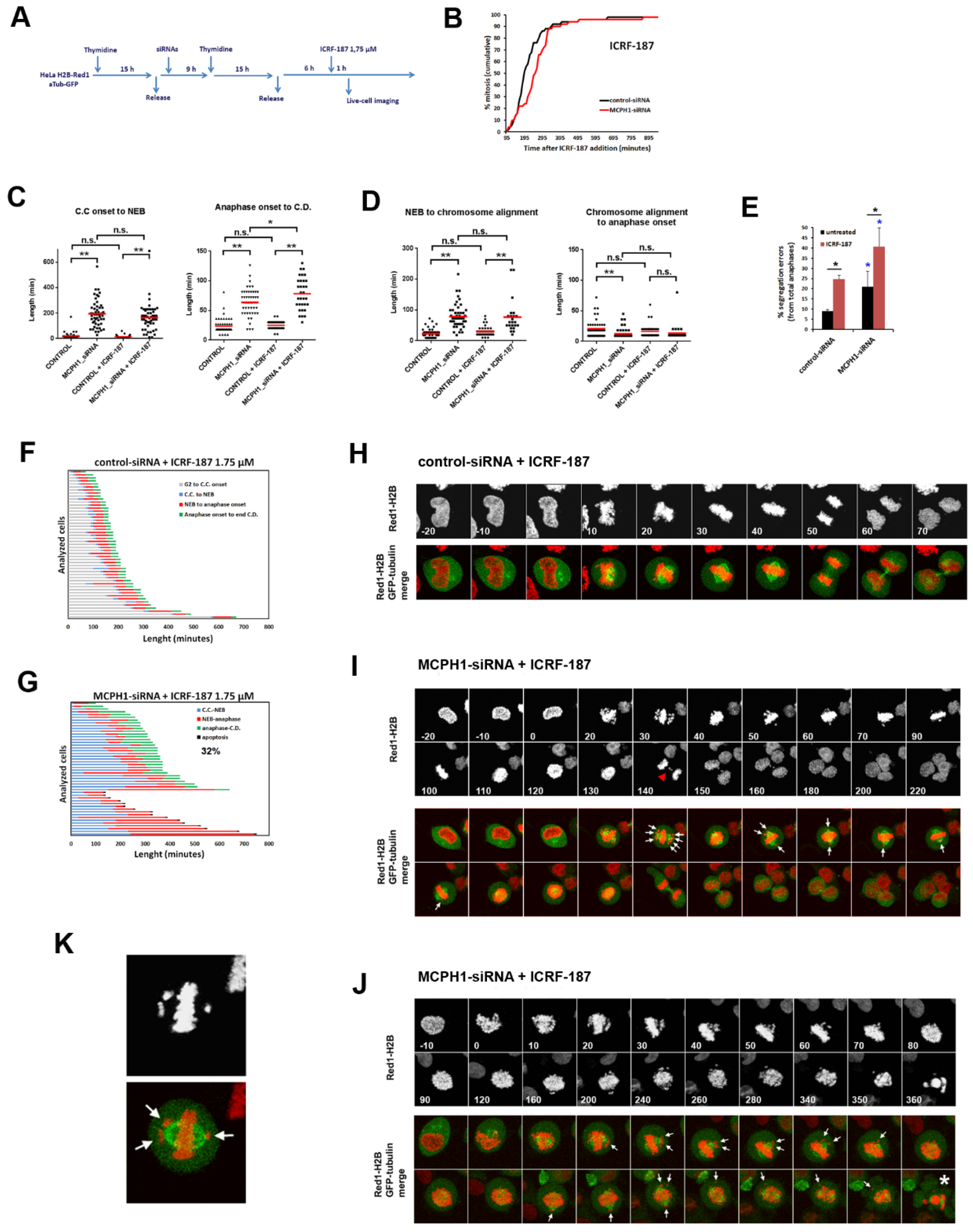
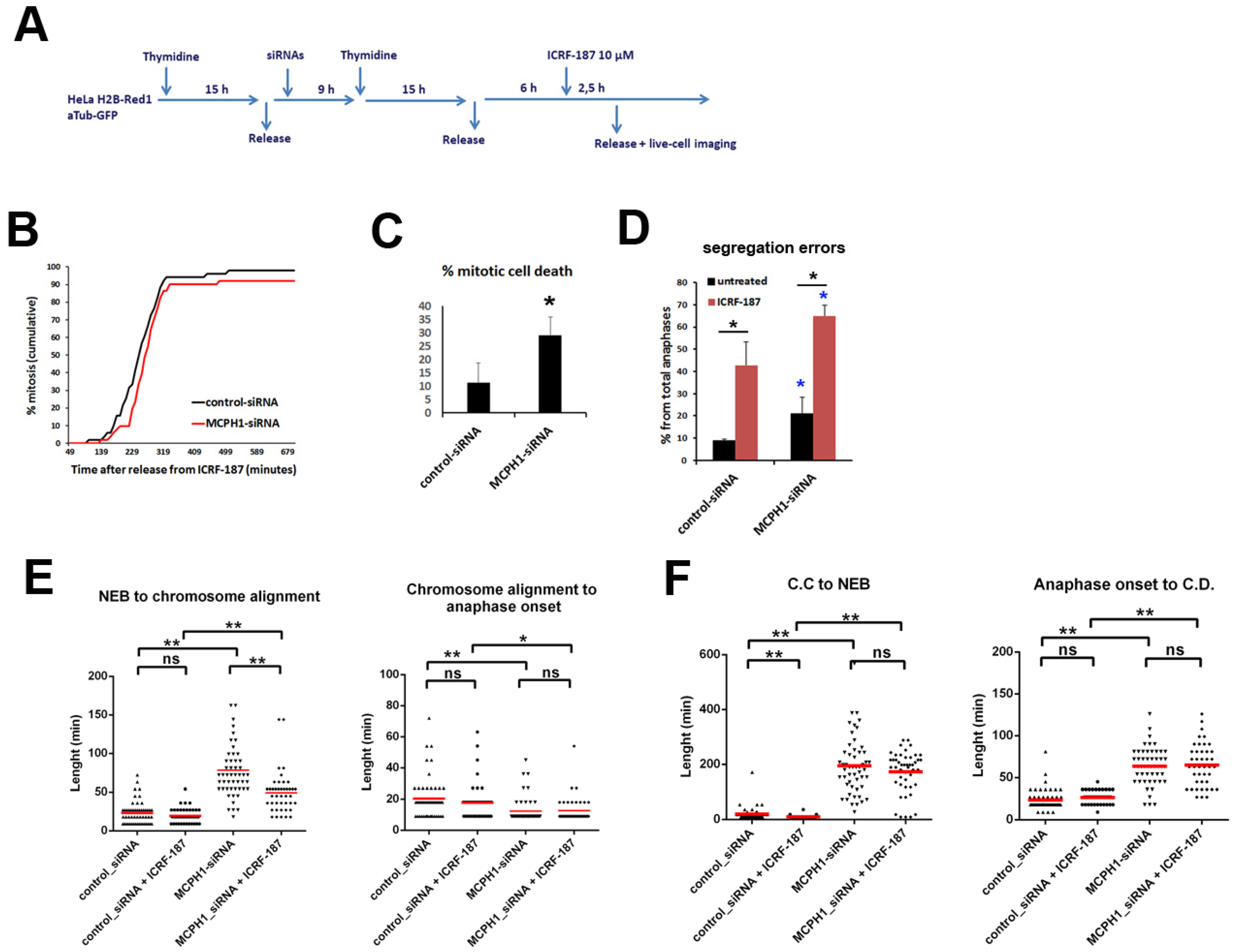
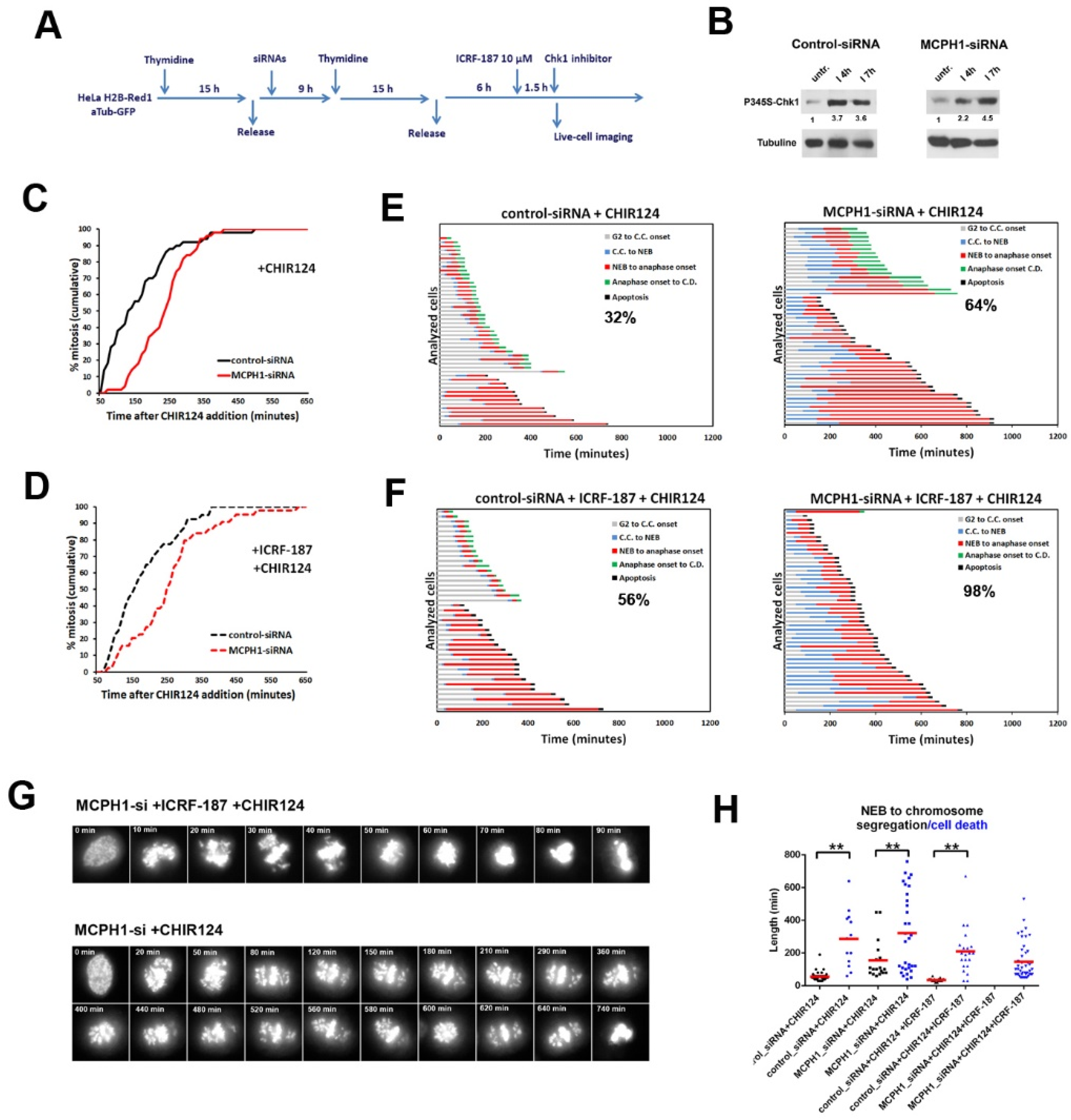
3. Results and Discussion
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Wang, J.C. Cellular roles of DNA topoisomerases: A molecular perspective. Nat. Rev. Mol. Cell Biol. 2002, 3, 430–440. [Google Scholar] [CrossRef]
- Clarke, D.J.; Vas, A.C.; Andrews, C.A.; Díaz-Martínez, L.A.; Giménez-Abián, J.F. Topoisomerase II checkpoints: Universal mechanisms that regulate mitosis. Cell Cycle 2006, 5, 1925–1928. [Google Scholar] [CrossRef]
- Giménez-Abián, J.F.; Clarke, D.J.; Giménez-Martín, G.; Weingartner, M.; Giménez-Abián, M.I.; Carballo, J.A.; Díaz de la Espina, S.M.; Bögre, L.; De la Torre, C. DNA catenations that link sister chromatids until the onset of anaphase are maintained by a checkpoint mechanism. Eur. J. Cell Biol. 2002, 81, 9–16. [Google Scholar] [CrossRef]
- Adachi, Y.; Luke, M.; Laemmli, U.K. Chromosome assembly in vitro: Topoisomerase II is required for condensation. Cell 1991, 64, 137–148. [Google Scholar] [CrossRef]
- Clarke, D.J.; Johnson, R.T.; Downes, C.S. Topoisomerase II inhibition prevents anaphase chromatid segregation in mammalian cells independently of the generation of DNA strand breaks. J. Cell Sci. 1993, 569, 563–569. [Google Scholar]
- Dong, K.C.; Berger, J.M. Structural basis for gate-DNA recognition and bending by type IIA topoisomerases. Nature 2007, 450, 1201–1205. [Google Scholar] [CrossRef]
- Wang, J.C. Unlocking and opening a DNA gate. Proc. Natl. Acad. Sci. USA 2007, 104, 4773–4774. [Google Scholar] [CrossRef]
- Nitiss, J. Targeting DNA topoisomerase II in cancer chemotherapy. Nat. Rev. Cancer 2009, 9, 338–350. [Google Scholar] [CrossRef]
- Damelin, M.; Bestor, T.H. The decatenation checkpoint. Br. J. Cancer 2007, 96, 201–205. [Google Scholar] [CrossRef]
- Downes, C.S.; Clarke, D.J.; Mullinger, A.M.; Giménez-Abián, J.F.; Creighton, A.M.; Johson, R.T. A topoisomerase II-dependent G2 cycle checkpoint in mammalian cells. Nature 1994, 372, 467–470. [Google Scholar] [CrossRef]
- Deming, P.B.; Cistulli, C.A.; Zhao, H.; Graves, P.R.; Piwnica-Worms, H.; Paules, R.S.; Downes, C.S.; Kaufmann, W.K. The human decatenation checkpoint. Proc. Natl. Acad. Sci. USA 2001, 98, 12044–12049. [Google Scholar] [CrossRef] [PubMed]
- Luo, K.; Yuan, J.; Chen, J.; Lou, Z. . Topoisomerase IIalpha controls the decatenation checkpoint. Nat. Cell Biol. 2009, 19, 389–399. [Google Scholar]
- Arroyo, M.; Kuriyama, R.; Guerrero, I.; Keifenheim, D.; Cañuelo, A.; Calahorra, J.; Sánchez, A.; Clarke, D.J.; Marchal, J.A. MCPH1 is essential for cellular adaptation to the G2-phase decatenation checkpoint. FASEB J. 2019, 33, 8363–8374. [Google Scholar] [CrossRef] [PubMed]
- Skoufias, D.A.; Lacroix, F.B.; Andreassen, P.R.; Wilson, L.; Margolis, R.L. Inhibition of DNA decatenation, but not DNA damage, arrests cells at metaphase. Mol. Cell 2004, 15, 977–990. [Google Scholar] [CrossRef]
- Brownlow, N.; Pike, T.; Zicha, D.; Collinson, L.; Parker, P.J. Mitotic catenation is monitored and resolved by a PKC-regulated pathway. Nat. Commun. 2014, 5, 5685. [Google Scholar] [CrossRef]
- Nakagawa, T.; Hayashita, Y.; Maeno, K.; Masuda, A.; Sugito, N.; Osada, H.; Yanagisawa, K.; Ebi, H.; Shimokata, K.; Takahashi, T. Identification of decatenation G2 checkpoint impairment independently of DNA damage G2 checkpoint in human lung cancer cell lines. Cancer Res. 2004, 64, 4826–4832. [Google Scholar] [CrossRef]
- Franchitto, A.; Oshima, J.; Pichierri, P. The G2-phase decatenation checkpoint is defective in Werner syndrome cells. Cancer Res. 2003, 63, 3289–3295. [Google Scholar]
- Jain, C.K.; Roychoudhury, S.; Majumder, H.K. Selective killing of G2 decatenation checkpoint defective colon cancer cells by catalytic topoisomerase II inhibitor. Biochim. Biophys. Acta. 2015, 1853, 1195–1204. [Google Scholar] [CrossRef]
- Brooks, K.; Chia, K.M.; Spoerri, L.; Mukhopadhyay, P.; Wigan, M.; Stark, M.; Pavey, S.; Gabrielli, B. Defective decatenation checkpoint function is a common feature of melanoma. J. Investig. Dermatol. 2014, 134, 150–158. [Google Scholar] [CrossRef]
- Deiss, K.; Lockwood, N.; Howell, M.; Segeren, H.A.; Saunders, R.E.; Chakravarty, P.; Soliman, T.N.; Martini, S.; Rocha, N.; Semple, R.; et al. A genome-wide RNAi screen identifies the SMC5/6 complex as a non-redundant regulator of a Topo2a-dependent G2 arrest. Nucleic Acids Res. 2018, 1–16. [Google Scholar] [CrossRef]
- Woods, C.G.; Bond, J.; Enard, W. Autosomal recessive primary microcephaly (MCPH): A review of clinical, molecular, and evolutionary findings. Am. J. Hum. Genet. 2005, 76, 717–728. [Google Scholar] [CrossRef] [PubMed]
- Woods, C.G. Human microcephaly. Curr. Opin. Neurobiol. 2004, 14, 112–117. [Google Scholar] [CrossRef] [PubMed]
- Woods, C.G.; Basto, R. Microcephaly. Curr. Biol. 2014, 24, 1109–1111. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Venkatesh, T.; Suresh, P.S. Emerging roles of MCPH1: Expedition from primary microcephaly to cancer. Eur. J. Cell Biol. 2014, 93, 98–105. [Google Scholar] [CrossRef] [PubMed]
- Neitzel, H.; Neumann, L.M.; Schindler, D.; Wirges, A.; Tönnies, H.; Trimborn, M.; Krebsova, A.; Richter, R.; Sperling, K. Premature chromosome condensation in humans associated with microcephaly and mental retardation: A novel autosomal recessive condition. Am. J. Hum. Genet. 2002, 70, 1015–1022. [Google Scholar] [CrossRef]
- Yamashita, D.; Shintomi, K.; Ono, T.; Gavvovidis, I.; Schindler, D.; Neitzel, H.; Trimborn, M.; Hirano, T. MCPH1 regulates chromosome condensation and shaping as a composite modulator of condensin II. J. Cell Biol. 2011, 194, 841–854. [Google Scholar] [CrossRef]
- Trimborn, M.; Schindler, D.; Neitzel, H.; Hirano, T. Misregulated chromosome condensation in MCPH1 primary microcephaly is mediated by condensin II. Cell Cycle 2006, 5, 322–326. [Google Scholar]
- Arroyo, M.; Trimborn, M.; Sánchez, A.; Hirano, T.; Neitzel, H.; Marchal, J.A. Chromosome structure deficiencies in MCPH1 syndrome. Chromosoma 2015, 124, 491–501. [Google Scholar] [CrossRef]
- Arroyo, M.; Kuriyama, R.; Trimborn, M.; Keifenheim, D.; Cañuelo, A.; Sánchez, A.; Clarke, D.J.; Marchal, J.A. MCPH1, mutated in primary microcephaly, is required for efficient chromosome alignment during mitosis. Sci. Rep. 2017, 7. [Google Scholar] [CrossRef]
- Germann, S.M.; Schramke, V.; Pedersen, R.T.; Gallina, I.; Eckert-Boulet, N.; Oestergaard, V.H.; Lisby, M. TopBP1/Dpb11 binds DNA anaphase bridges to prevent genome instability. J. Cell Biol. 2014, 204, 45–59. [Google Scholar] [CrossRef]
- Haarhuis, J.H.I.; Elbatsh, A.M.O.; Rowland, B.D. Cohesin and its regulation: On the logic of X-shaped chromosomes. Dev. Cell 2014, 31, 7–18. [Google Scholar] [CrossRef] [PubMed]
- Gavvovidis, I.; Rost, I.; Trimborn, M.; Kaiser, F.J.; Purps, J.; Wiek, C.; Hanenberg, H.; Neitzel, H.; Schindler, D. A Novel MCPH1 isoform complements the defective chromosome condensation of human MCPH1-deficient cells. PLoS ONE 2012, 7, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Trimborn, M.; Bell, S.M.; Felix, C.; Rashid, Y.; Jafri, H.; Griffiths, P.D.; Neumann, L.M.; Krebs, A.; Reis, A.; Sperling, K.; et al. Mutations in microcephalin cause aberrant regulation of chromosome condensation. Am. J. Hum. Genet. 2004, 75, 261–266. [Google Scholar] [CrossRef] [PubMed]
- Bower, J.J.; Gamze, F.K.; Zhou, Y.; Simpson, D.A.; Cordeiro-Stone, M.; Kaufmann, W.K. Topoisomerase IIα maintains genomic stability through decatenation G2 checkpoint signaling. Oncogene 2010, 29, 4787–4799. [Google Scholar] [CrossRef]
- Arroyo, M.; Cañuelo, A.; Calahorra, J.; Hastert, F.D.; Sánchez, A.; Clarke, D.J.; Marchal, J.A. Mitotic entry upon Topo II catalytic inhibition is controlled by Chk1 and Plk1. FEBS J. 2020. [Epub ahead of print]. [Google Scholar] [CrossRef]
- Tse, A.N.; Rendahl, K.G.; Sheikh, T.; Cheema, H.; Aardalen, K.; Embry, M.; Ma, S.; Moler, E.J.; Zhi, J.N.; De Menezes, D.E.L.; et al. CHIR-124, a novel potent inhibitor of Chk1, potentiates the cytotoxicity of topoisomerase I poisons in vitro and in vivo. Clin. Cancer Res. 2007, 13, 591–602. [Google Scholar] [CrossRef]
- Warren, N.J.H.; Eastman, A. Comparison of the different mechanisms of cytotoxicity induced by checkpoint kinase I inhibitors when used as single agents or in combination with DNA damage. Oncogene 2019, 39, 1389–1401. [Google Scholar] [CrossRef]
- Tse, A.N.; Schwartz, G.K. Potentiation of cytotoxicity of topoisomerase I poison by concurrent and sequential treatment with the checkpoint inhibitor UCN-01 involves disparate mechanisms resulting in either p53-independent clonogenic suppression or p53-dependent mitotic catastroph. Cancer Res. 2004, 64, 6635–6644. [Google Scholar] [CrossRef]
- On, K.F.; Chen, Y.; Ma, H.T.; Chow, J.P.H.; Poon, R.Y.C. Determinants of mitotic catastrophe on abrogation of the G2 DNA damage checkpoint by UCN-01. Mol. Cancer Ther. 2011, 10, 784–794. [Google Scholar] [CrossRef]
- Deiss, K.; Lockwood, N.; Howell, M.; Segeren, H.A.; Saunders, R.E.; Chakravarty, P.; Soliman, T.N.; Martini, S.; Rocha, N.; Semple, R.; et al. A genome-wide RNAi screen identifies the SMC5/6 complex as a non-redundant regulator of a Topo2a-dependent G2 arrest. Nucleic Acids Res. 2019, 47, 2906–2921. [Google Scholar] [CrossRef]
- Vogel, C.; Hager, C.; Bastians, H. Mechanisms of mitotic cell death induced by chemotherapy-mediated G2 checkpoint abrogation. Cancer Res. 2007, 67, 339–345. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.F.; Zhang, Y.; Wilde, J.; Hansen, K.C.; Lai, F.; Niswander, L. Microcephaly disease gene Wdr62 regulates mitotic progression of embryonic neural stem cells and brain size. Nat. Commun. 2014, 5, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Pilaz, L.J.; McMahon, J.J.; Miller, E.E.; Lennox, A.L.; Suzuki, A.; Salmon, E.; Silver, D.L. Prolonged mitosis of neural progenitors alters cell fate in the developing brain. Neuron 2016. [Google Scholar] [CrossRef] [PubMed]




© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Arroyo, M.; Sánchez, A.; Cañuelo, A.; Heredia-Molina, R.F.; Martínez-Molina, E.; Clarke, D.J.; Marchal, J.A. MCPH1 Lack of Function Enhances Mitotic Cell Sensitivity Caused by Catalytic Inhibitors of Topo II. Genes 2020, 11, 406. https://doi.org/10.3390/genes11040406
Arroyo M, Sánchez A, Cañuelo A, Heredia-Molina RF, Martínez-Molina E, Clarke DJ, Marchal JA. MCPH1 Lack of Function Enhances Mitotic Cell Sensitivity Caused by Catalytic Inhibitors of Topo II. Genes. 2020; 11(4):406. https://doi.org/10.3390/genes11040406
Chicago/Turabian StyleArroyo, María, Antonio Sánchez, Ana Cañuelo, Rosalía F. Heredia-Molina, Eduardo Martínez-Molina, Duncan J. Clarke, and Juan Alberto Marchal. 2020. "MCPH1 Lack of Function Enhances Mitotic Cell Sensitivity Caused by Catalytic Inhibitors of Topo II" Genes 11, no. 4: 406. https://doi.org/10.3390/genes11040406
APA StyleArroyo, M., Sánchez, A., Cañuelo, A., Heredia-Molina, R. F., Martínez-Molina, E., Clarke, D. J., & Marchal, J. A. (2020). MCPH1 Lack of Function Enhances Mitotic Cell Sensitivity Caused by Catalytic Inhibitors of Topo II. Genes, 11(4), 406. https://doi.org/10.3390/genes11040406