Combining Experimental Evolution and Genomics to Understand How Seed Beetles Adapt to a Marginal Host Plant

 , and
, and
Abstract
1. Introduction
2. Materials and Methods
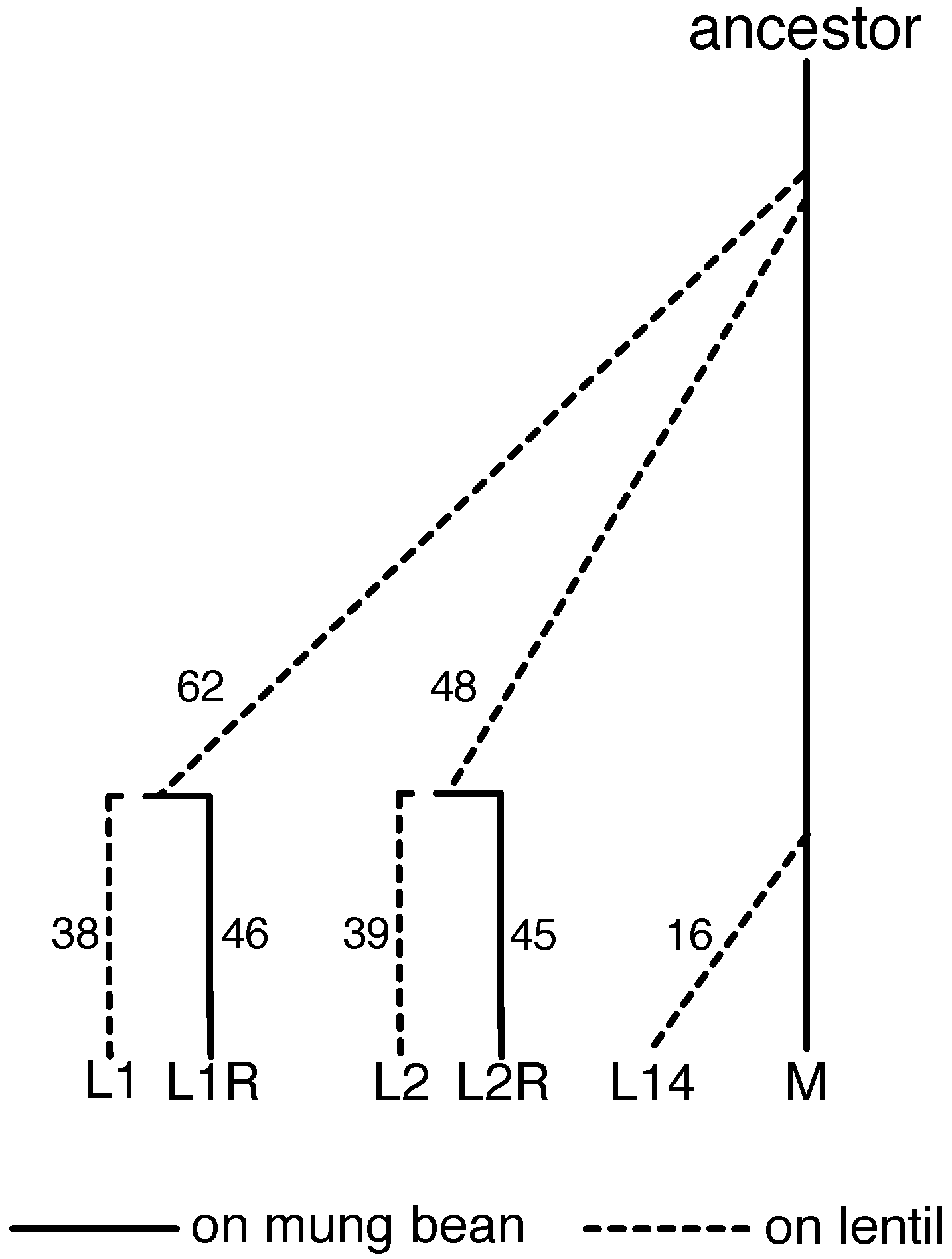
2.1. Evolve-and-Resequence Experiments
2.2. Trait Mapping Experiment
2.3. Gene Expression Experiment
2.4. DNA Sequence Alignment and Variant Calling
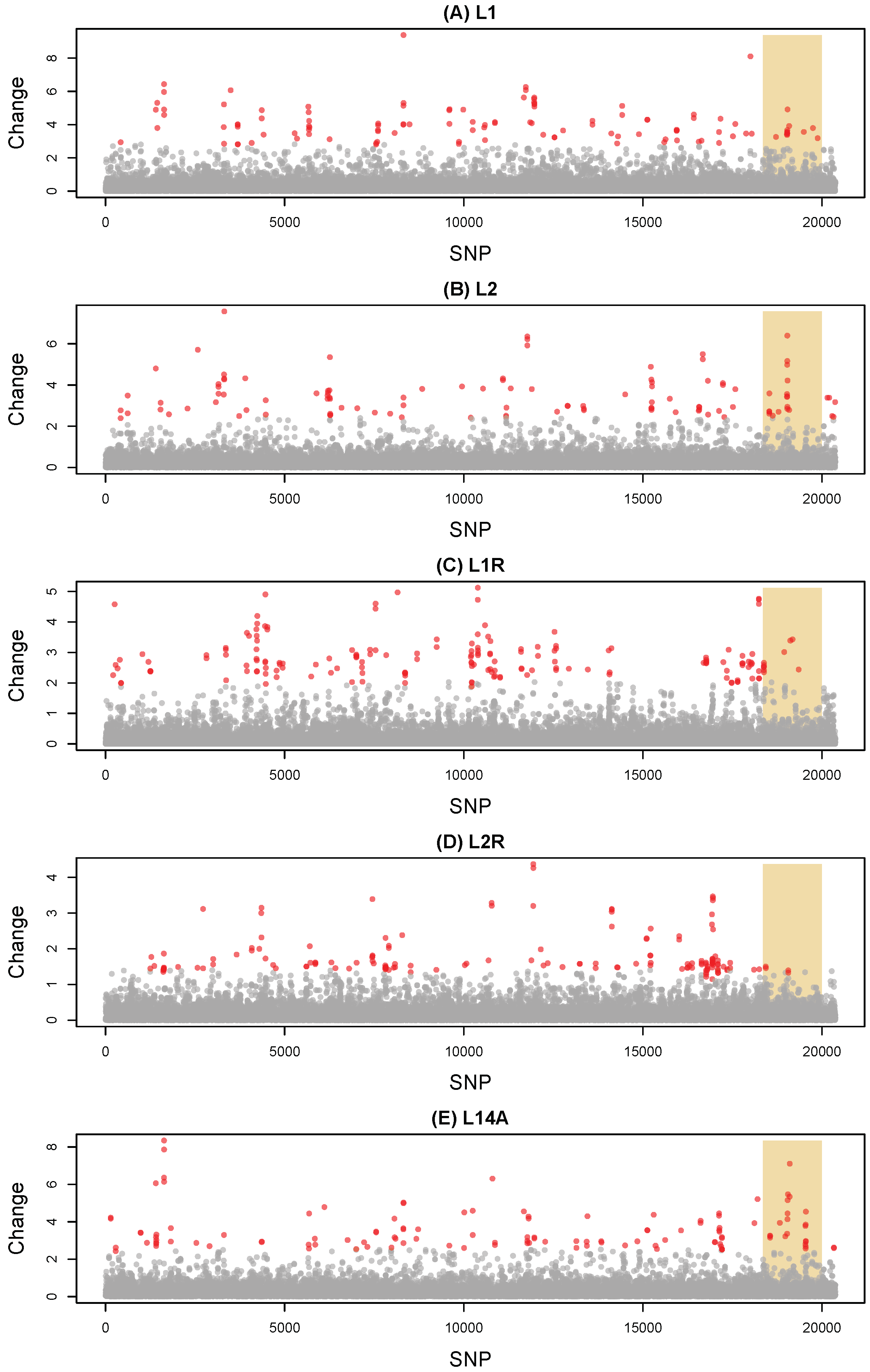
2.5. Measuring Evolutionary Change during the Evolve-and-Resequence Experiments
2.6. Multilocus Genome-Wide Association Mapping
2.7. Gene Expression
2.8. Comparisons across Data Sets
3. Results
3.1. Evolutionary Change
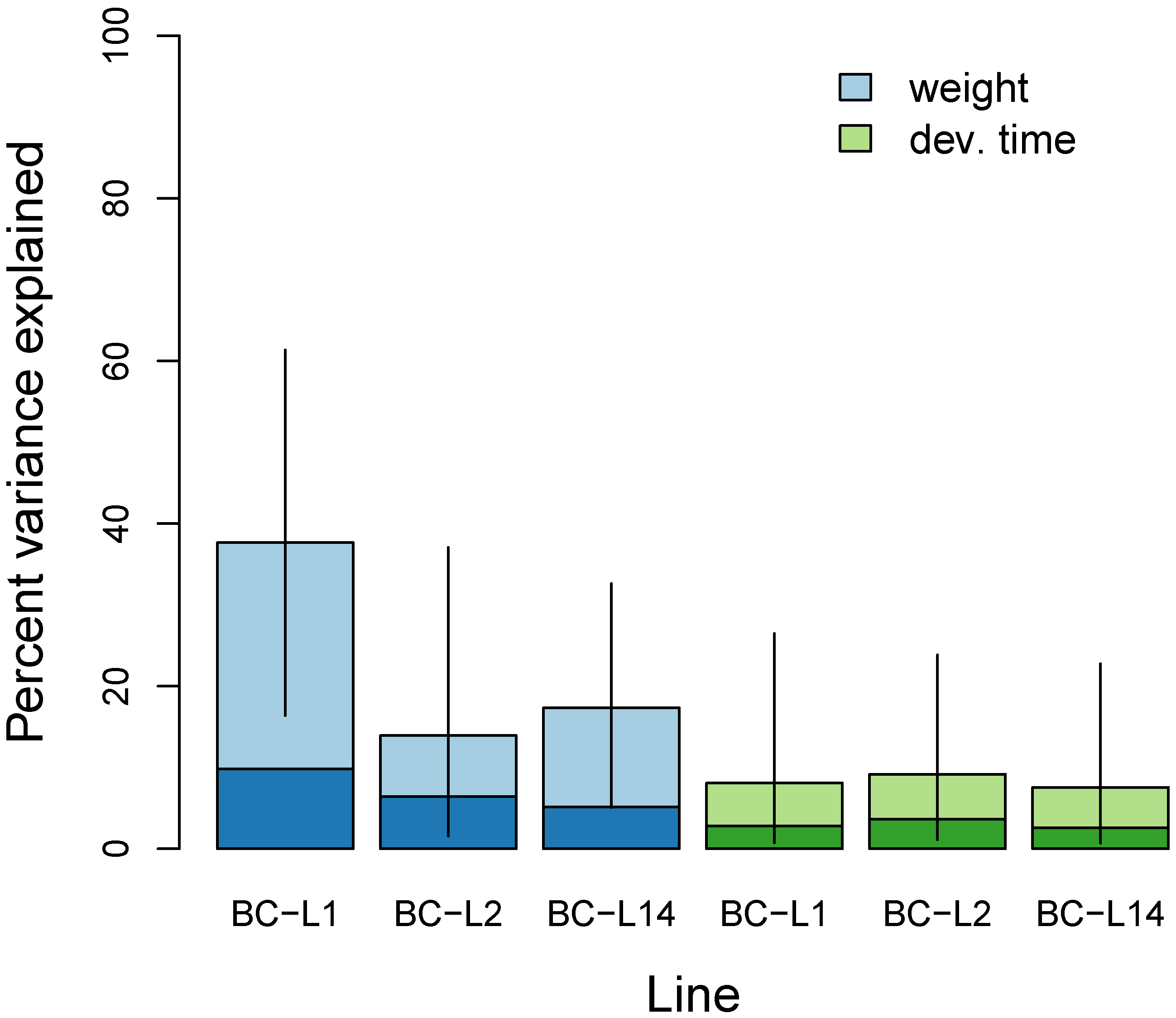
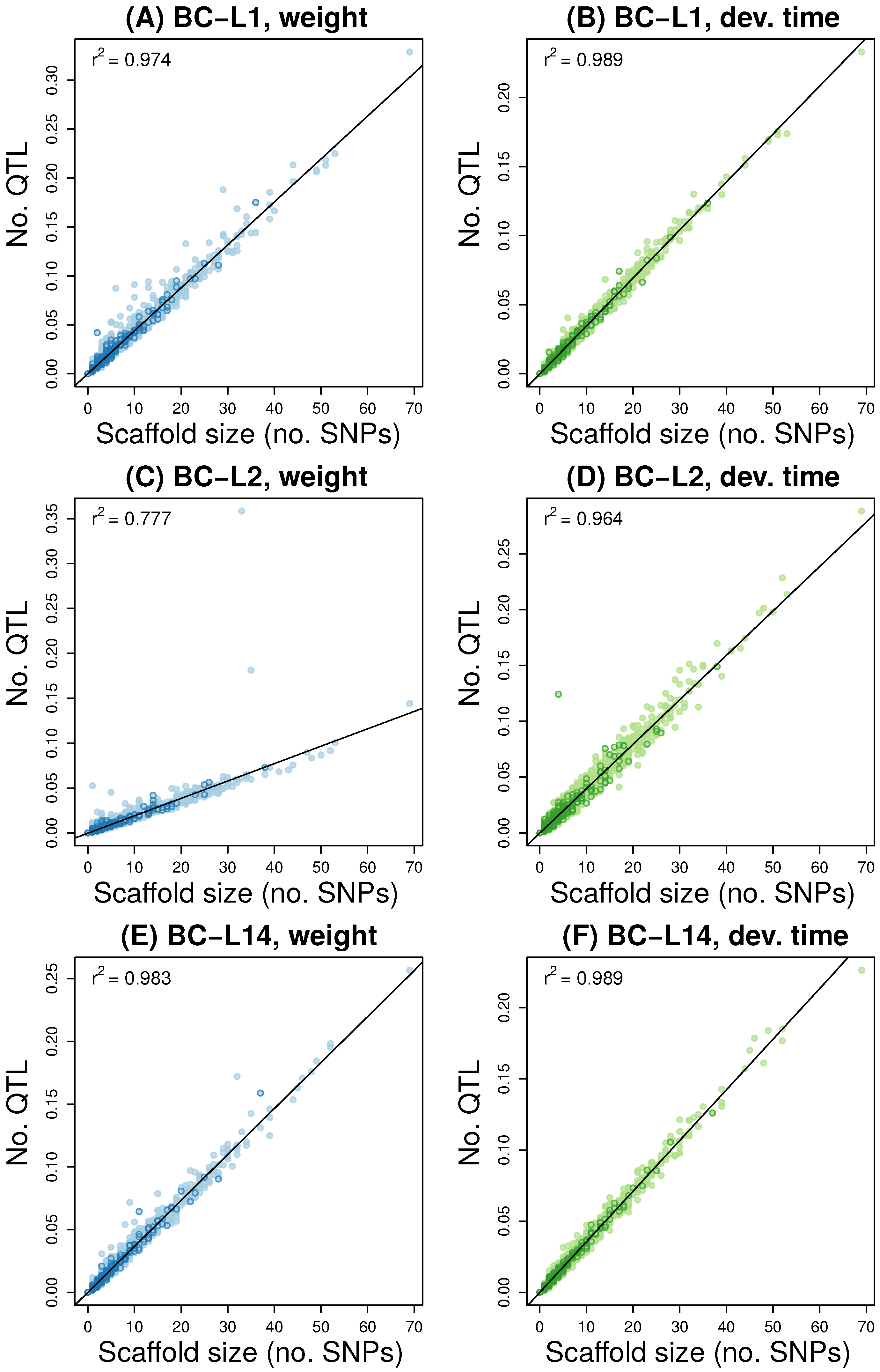
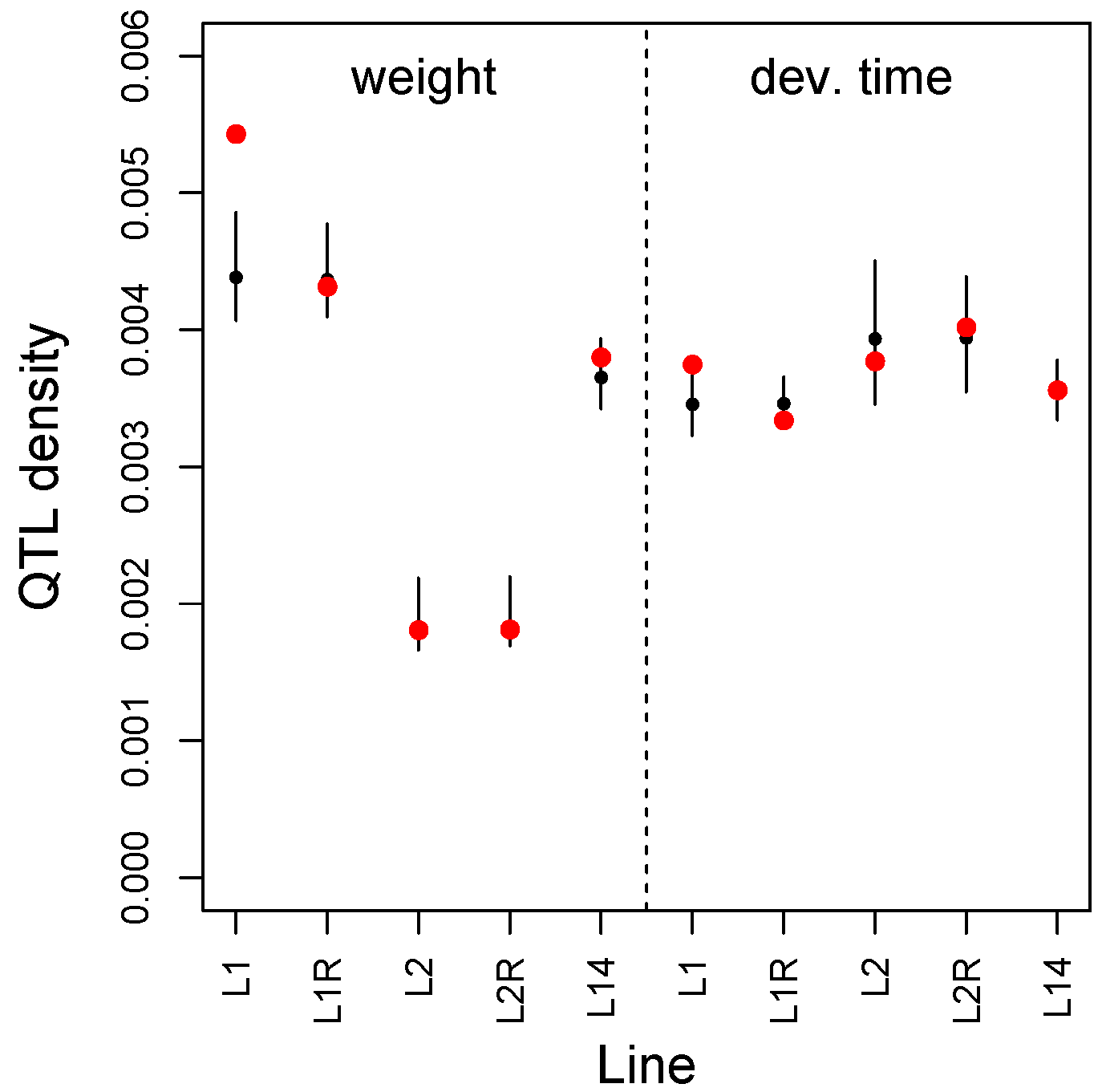
3.2. Multilocus Genome-Wide Association Mapping
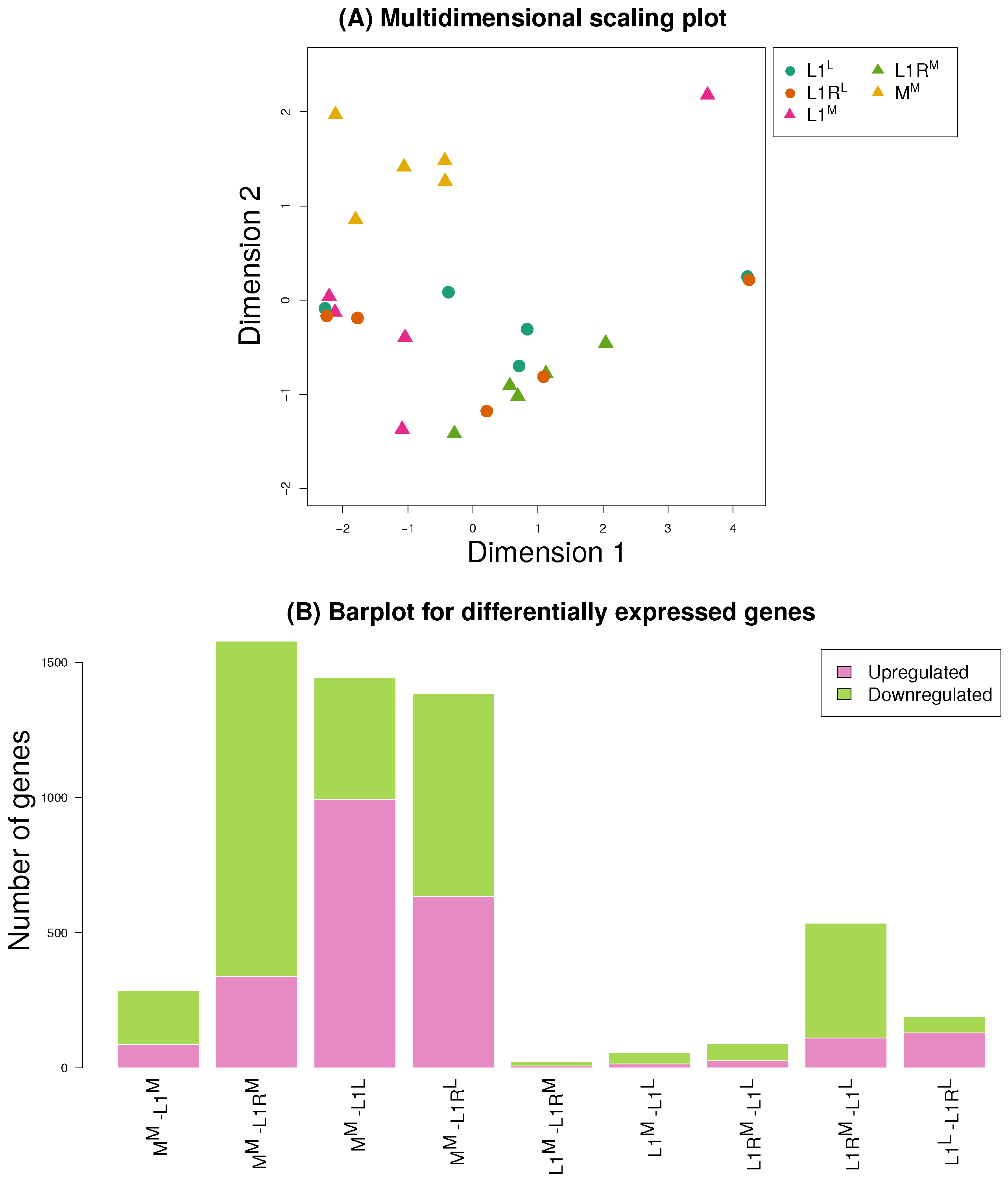
3.3. Gene Expression
3.4. Comparisons Across Data Sets
4. Discussion
4.1. Genetics of Performance Traits
4.2. Parallelism in Change Versus Traits
4.3. Genomics of Host Use and Adaptation
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Martin, A.; Orgogozo, V. The loci of repeated evolution: A catalog of genetic hotspots of phenotypic variation. Evolution 2013, 67, 1235–1250. [Google Scholar] [CrossRef] [PubMed]
- Jones, F.C.; Grabherr, M.G.; Chan, Y.F.; Russell, P.; Mauceli, E.; Johnson, J.; Swofford, R.; Pirun, M.; Zody, M.C.; White, S.; et al. The genomic basis of adaptive evolution in threespine sticklebacks. Nature 2012, 484, 55–61. [Google Scholar] [CrossRef] [PubMed]
- Xie, K.T.; Wang, G.; Thompson, A.C.; Wucherpfennig, J.I.; Reimchen, T.E.; MacColl, A.D.; Schluter, D.; Bell, M.A.; Vasquez, K.M.; Kingsley, D.M. DNA fragility in the parallel evolution of pelvic reduction in stickleback fish. Science 2019, 363, 81–84. [Google Scholar] [CrossRef] [PubMed]
- Nachman, M.W.; Hoekstra, H.E.; D’Agostino, S.L. The genetic basis of adaptive melanism in pocket mice. Proc. Natl. Acad. Sci. USA 2003, 100, 5268–5273. [Google Scholar] [CrossRef]
- Linnen, C.R.; Poh, Y.P.; Peterson, B.K.; Barrett, R.D.; Larson, J.G.; Jensen, J.D.; Hoekstra, H.E. Adaptive evolution of multiple traits through multiple mutations at a single gene. Science 2013, 339, 1312–1316. [Google Scholar] [CrossRef]
- Reed, R.D.; Papa, R.; Martin, A.; Hines, H.M.; Counterman, B.A.; Pardo-Diaz, C.; Jiggins, C.D.; Chamberlain, N.L.; Kronforst, M.R.; Chen, R.; et al. Optix drives the repeated convergent evolution of butterfly wing pattern mimicry. Science 2011, 333, 1137–1141. [Google Scholar] [CrossRef]
- Van Belleghem, S.M.; Rastas, P.; Papanicolaou, A.; Martin, S.H.; Arias, C.F.; Supple, M.A.; Hanly, J.J.; Mallet, J.; Lewis, J.J.; Hines, H.M.; et al. Complex modular architecture around a simple toolkit of wing pattern genes. Nat. Ecol. Evol. 2017, 1, 1–12. [Google Scholar] [CrossRef]
- Concha, C.; Wallbank, R.W.; Hanly, J.J.; Fenner, J.; Livraghi, L.; Rivera, E.S.; Paulo, D.F.; Arias, C.; Vargas, M.; Sanjeev, M.; et al. Interplay between developmental flexibility and determinism in the evolution of mimetic Heliconius wing patterns. Curr. Biol. 2019, 29, 3996–4009. [Google Scholar] [CrossRef]
- Williams, K.D.; Busto, M.; Suster, M.L.; So, A.K.C.; Ben-Shahar, Y.; Leevers, S.J.; Sokolowski, M.B. Natural variation in Drosophila melanogaster diapause due to the insulin-regulated PI3-kinase. Proc. Natl. Acad. Sci. USA 2006, 103, 15911–15915. [Google Scholar] [CrossRef]
- Tauber, E.; Zordan, M.; Sandrelli, F.; Pegoraro, M.; Osterwalder, N.; Breda, C.; Daga, A.; Selmin, A.; Monger, K.; Benna, C.; et al. Natural selection favors a newly derived timeless allele in Drosophila melanogaster. Science 2007, 316, 1895–1898. [Google Scholar] [CrossRef]
- Bosse, M.; Spurgin, L.G.; Laine, V.N.; Cole, E.F.; Firth, J.A.; Gienapp, P.; Gosler, A.G.; McMahon, K.; Poissant, J.; Verhagen, I.; et al. Recent natural selection causes adaptive evolution of an avian polygenic trait. Science 2017, 358, 365–368. [Google Scholar] [CrossRef] [PubMed]
- Lucas, L.K.; Nice, C.C.; Gompert, Z. Genetic constraints on wing pattern variation in Lycaeides butterflies: A case study on mapping complex, multifaceted traits in structured populations. Mol. Ecol. Resour. 2018, 18, 892–907. [Google Scholar] [CrossRef] [PubMed]
- Gompert, Z.; Brady, M.; Chalyavi, F.; Saley, T.C.; Philbin, C.S.; Tucker, M.J.; Forister, M.L.; Lucas, L.K. Genomic evidence of genetic variation with pleiotropic effects on caterpillar fitness and plant traits in a model legume. Mol. Ecol. 2019, 28, 2967–2985. [Google Scholar] [CrossRef] [PubMed]
- Schluter, D. Adaptive radiation along genetic lines of least resistance. Evolution 1996, 50, 1766–1774. [Google Scholar] [CrossRef] [PubMed]
- Hughes, K.A.; Leips, J. Pleiotropy, constraint, and modularity in the evolution of life histories: Insights from genomic analyses. Ann. N. Y. Acad. Sci. 2017, 1389, 76. [Google Scholar] [CrossRef] [PubMed]
- Bell, G. Fluctuating selection: The perpetual renewal of adaptation in variable environments. Philos. Trans. R. Soc. B Biol. Sci. 2010, 365, 87–97. [Google Scholar] [CrossRef]
- Rockman, M.V. The QTN program and the alleles that matter for evolution: All that’s gold does not glitter. Evolution 2012, 66, 1–17. [Google Scholar] [CrossRef]
- Lee, Y.W.; Gould, B.A.; Stinchcombe, J.R. Identifying the genes underlying quantitative traits: A rationale for the QTN programme. AoB Plants 2014, 6. [Google Scholar] [CrossRef]
- Tiffin, P.; Ross-Ibarra, J. Advances and limits of using population genetics to understand local adaptation. Trends Ecol. Evol. 2014, 29, 673–680. [Google Scholar] [CrossRef]
- Linnen, C.R. Predicting evolutionary predictability. Mol. Ecol. 2018, 27, 2647–2650. [Google Scholar] [CrossRef]
- Colosimo, P.F.; Hosemann, K.E.; Balabhadra, S.; Villarreal, G.; Dickson, M.; Grimwood, J.; Schmutz, J.; Myers, R.M.; Schluter, D.; Kingsley, D.M. Widespread parallel evolution in sticklebacks by repeated fixation of Ectodysplasin alleles. Science 2005, 307, 1928–1933. [Google Scholar] [CrossRef] [PubMed]
- Baxter, S.W.; Nadeau, N.J.; Maroja, L.S.; Wilkinson, P.; Counterman, B.A.; Dawson, A.; Beltran, M.; Perez-Espona, S.; Chamberlain, N.; Ferguson, L.; et al. Genomic hotspots for adaptation: The population genetics of Müllerian mimicry in the Heliconius melpomene clade. PLoS Genet. 2010, 6, e1000794. [Google Scholar] [CrossRef] [PubMed]
- Stern, D.L. The genetic causes of convergent evolution. Nat. Rev. Genet. 2013, 14, 751–764. [Google Scholar] [CrossRef]
- Graves, J., Jr.; Hertweck, K.; Phillips, M.; Han, M.; Cabral, L.; Barter, T.; Greer, L.; Burke, M.; Mueller, L.; Rose, M. Genomics of parallel experimental evolution in Drosophila. Mol. Biol. Evol. 2017, 34, 831–842. [Google Scholar] [CrossRef] [PubMed]
- Turner, C.B.; Marshall, C.W.; Cooper, V.S. Parallel genetic adaptation across environments differing in mode of growth or resource availability. Evol. Lett. 2018, 2, 355–367. [Google Scholar] [CrossRef] [PubMed]
- Miller, S.E.; Roesti, M.; Schluter, D. A single interacting species leads to widespread parallel evolution of the stickleback genome. Curr. Biol. 2019, 29, 530–537. [Google Scholar] [CrossRef] [PubMed]
- Soria-Carrasco, V.; Gompert, Z.; Comeault, A.A.; Farkas, T.E.; Parchman, T.L.; Johnston, J.S.; Buerkle, C.A.; Feder, J.L.; Bast, J.; Schwander, T.; et al. Stick insect genomes reveal natural selection’s role in parallel speciation. Science 2014, 344, 738–742. [Google Scholar] [CrossRef]
- Fan, S.; Hansen, M.E.; Lo, Y.; Tishkoff, S.A. Going global by adapting local: A review of recent human adaptation. Science 2016, 354, 54–59. [Google Scholar] [CrossRef]
- Chaturvedi, S.; Lucas, L.K.; Nice, C.C.; Fordyce, J.A.; Forister, M.L.; Gompert, Z. The predictability of genomic changes underlying a recent host shift in Melissa blue butterflies. Mol. Ecol. 2018, 27, 2651–2666. [Google Scholar] [CrossRef]
- Blount, Z.D.; Borland, C.Z.; Lenski, R.E. Historical contingency and the evolution of a key innovation in an experimental population of Escherichia coli. Proc. Natl. Acad. Sci. USA 2008, 105, 7899–7906. [Google Scholar] [CrossRef]
- Bedhomme, S.; Lafforgue, G.; Elena, S.F. Genotypic but not phenotypic historical contingency revealed by viral experimental evolution. BMC Evol. Biol. 2013, 13, 46. [Google Scholar] [CrossRef]
- Weiss, K.M. Tilting at quixotic trait loci (QTL): An evolutionary perspective on genetic causation. Genetics 2008, 179, 1741–1756. [Google Scholar] [CrossRef][Green Version]
- Burke, M.K.; Dunham, J.P.; Shahrestani, P.; Thornton, K.R.; Rose, M.R.; Long, A.D. Genome-wide analysis of a long-term evolution experiment with Drosophila. Nature 2010, 467, 587–590. [Google Scholar] [CrossRef] [PubMed]
- Bigham, A.; Bauchet, M.; Pinto, D.; Mao, X.; Akey, J.M.; Mei, R.; Scherer, S.W.; Julian, C.G.; Wilson, M.J.; López Herráez, D.; et al. Identifying signatures of natural selection in Tibetan and Andean populations using dense genome scan data. PLoS Genet. 2010, 6, e1001116. [Google Scholar] [CrossRef] [PubMed]
- Elyashiv, E.; Sattath, S.; Hu, T.T.; Strutsovsky, A.; McVicker, G.; Andolfatto, P.; Coop, G.; Sella, G. A genomic map of the effects of linked selection in Drosophila. PLoS Genet. 2016, 12, e1006130. [Google Scholar] [CrossRef] [PubMed]
- Westram, A.M.; Rafajlović, M.; Chaube, P.; Faria, R.; Larsson, T.; Panova, M.; Ravinet, M.; Blomberg, A.; Mehlig, B.; Johannesson, K.; et al. Clines on the seashore: The genomic architecture underlying rapid divergence in the face of gene flow. Evol. Lett. 2018, 2, 297–309. [Google Scholar] [CrossRef] [PubMed]
- Bailey, S.F.; Bataillon, T. Can the experimental evolution programme help us elucidate the genetic basis of adaptation in nature? Mol. Ecol. 2016, 25, 203–218. [Google Scholar] [CrossRef]
- Rêgo, A.; Messina, F.J.; Gompert, Z. Dynamics of genomic change during evolutionary rescue in the seed beetle Callosobruchus maculatus. Mol. Ecol. 2019, 28, 2136–2154. [Google Scholar] [CrossRef]
- Long, A.; Liti, G.; Luptak, A.; Tenaillon, O. Elucidating the molecular architecture of adaptation via evolve and resequence experiments. Nat. Rev. Genet. 2015, 16, 567. [Google Scholar] [CrossRef]
- Gompert, Z.; Messina, F.J. Genomic evidence that resource-based trade-offs limit host-range expansion in a seed beetle. Evolution 2016, 70, 1249–1264. [Google Scholar] [CrossRef]
- Kelly, J.K.; Hughes, K.A. Pervasive linked selection and intermediate-frequency alleles are implicated in an evolve-and-resequencing experiment of Drosophila simulans. Genetics 2019, 211, 943–961. [Google Scholar] [CrossRef] [PubMed]
- Turner, T.L.; Stewart, A.D.; Fields, A.T.; Rice, W.R.; Tarone, A.M. Population-based resequencing of experimentally evolved populations reveals the genetic basis of body size variation in Drosophila melanogaster. PLoS Genet. 2011, 7, e1001336. [Google Scholar] [CrossRef] [PubMed]
- Turner, T.L.; Miller, P.M. Investigating natural variation in Drosophila courtship song by the evolve and resequence approach. Genetics 2012, 191, 633–642. [Google Scholar] [CrossRef] [PubMed]
- Parchman, T.L.; Gompert, Z.; Mudge, J.; Schilkey, F.D.; Benkman, C.W.; Buerkle, A.C. Genome-wide association genetics of an adaptive trait in lodgepole pine. Mol. Ecol. 2012, 21, 2991–3005. [Google Scholar] [CrossRef] [PubMed]
- Weber, A.L.; Khan, G.F.; Magwire, M.M.; Tabor, C.L.; Mackay, T.F.C.; Anholt, R.R.H. Genome-Wide Association Analysis of Oxidative Stress Resistance in Drosophila melanogaster. PLoS ONE 2012, 7, e34745. [Google Scholar] [CrossRef] [PubMed]
- Weinig, C.; Ungerer, M.C.; Dorn, L.A.; Kane, N.C.; Toyonaga, Y.; Halldorsdottir, S.S.; Mackay, T.F.C.; Purugganan, M.D.; Schmitt, J. Novel loci control variation in reproductive timing in Arabidopsis thaliana in natural environments. Genetics 2002, 162, 1875–1884. [Google Scholar]
- Rogers, S.M.; Bernatchez, L. Integrating QTL mapping and genome scans towards the characterization of candidate loci under parallel selection in the lake whitefish (Coregonus clupeaformis). Mol. Ecol. 2005, 14, 351–361. [Google Scholar] [CrossRef]
- Gompert, Z.; Lucas, L.K.; Nice, C.C.; Buerkle, C.A. Genome divergence and the genetic architecture of barriers to gene flow between Lycaeides idas and L. melissa. Evolution 2013, 67, 2498–2514. [Google Scholar] [CrossRef]
- Nosil, P.; Villoutreix, R.; de Carvalho, C.F.; Farkas, T.E.; Soria-Carrasco, V.; Feder, J.L.; Crespi, B.J.; Gompert, Z. Natural selection and the predictability of evolution in Timema stick insects. Science 2018, 359, 765–770. [Google Scholar] [CrossRef]
- Price, N.; Moyers, B.T.; Lopez, L.; Lasky, J.R.; Monroe, J.G.; Mullen, J.L.; Oakley, C.G.; Lin, J.; Ågren, J.; Schrider, D.R.; et al. Combining population genomics and fitness QTLs to identify the genetics of local adaptation in Arabidopsis thaliana. Proc. Natl. Acad. Sci. USA 2018, 115, 5028–5033. [Google Scholar] [CrossRef]
- Rajpurohit, S.; Gefen, E.; Bergland, A.O.; Petrov, D.A.; Gibbs, A.G.; Schmidt, P.S. Spatiotemporal dynamics and genome-wide association analysis of desiccation tolerance in Drosophila melanogaster. Mol. Ecol. 2018, 27, 3525–3540. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Mazo-Vargas, A.; Reed, R.D. Single master regulatory gene coordinates the evolution and development of butterfly color and iridescence. Proc. Natl. Acad. Sci. USA 2017, 114, 10707–10712. [Google Scholar] [CrossRef] [PubMed]
- Nallu, S.; Hill, J.A.; Don, K.; Sahagun, C.; Zhang, W.; Meslin, C.; Snell-Rood, E.; Clark, N.L.; Morehouse, N.I.; Bergelson, J.; et al. The molecular genetic basis of herbivory between butterflies and their host plants. Nat. Ecol. Evol. 2018, 2, 1418. [Google Scholar] [CrossRef] [PubMed]
- Ragland, G.J.; Almskaar, K.; Vertacnik, K.L.; Gough, H.M.; Feder, J.L.; Hahn, D.A.; Schwarz, D. Differences in performance and transcriptome-wide gene expression associated with Rhagoletis (Diptera: Tephritidae) larvae feeding in alternate host fruit environments. Mol. Ecol. 2015, 24, 2759–2776. [Google Scholar] [CrossRef] [PubMed]
- Kenkel, C.D.; Matz, M.V. Gene expression plasticity as a mechanism of coral adaptation to a variable environment. Nat. Ecol. Evol. 2016, 1, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Zhu, F.; Moural, T.W.; Nelson, D.R.; Palli, S.R. A specialist herbivore pest adaptation to xenobiotics through up-regulation of multiple Cytochrome P450s. Sci. Rep. 2016, 6, 20421. [Google Scholar] [CrossRef]
- Barrett, R.D.H.; Laurent, S.; Mallarino, R.; Pfeifer, S.P.; Xu, C.C.Y.; Foll, M.; Wakamatsu, K.; Duke-Cohan, J.S.; Jensen, J.D.; Hoekstra, H.E. Linking a mutation to survival in wild mice. Science 2019, 363, 499–504. [Google Scholar] [CrossRef]
- Wasserman, S.S.; Futuyma, D.J. Evolution of host plant utilization in laboratory populations of the southern cowpea weevil, Callosobruchus maculatus Fabricius (Coleoptera: Bruchidae). Evolution 1981, 35, 605–617. [Google Scholar] [CrossRef]
- Messina, F.J. Predictable modification of body size and competitive ability following a host shift by a seed beetle. Evolution 2004, 58, 2788–2797. [Google Scholar] [CrossRef]
- Fricke, C.; Arnqvist, G. Rapid adaptation to a novel host in a seed beetle (Callosobruchus maculatus): The role of sexual selection. Evolution 2007, 61, 440–454. [Google Scholar] [CrossRef]
- Eady, P.E.; Hamilton, L.; Lyons, R.E. Copulation, genital damage and early death in Callosobruchus maculatus. Proc. R. Soc. B Biol. Sci. 2007, 274, 247–252. [Google Scholar] [CrossRef] [PubMed]
- Berger, D.; Grieshop, K.; Lind, M.I.; Goenaga, J.; Maklakov, A.A.; Arnqvist, G. Intralocus sexual conflict and environmental stress. Evolution 2014, 68, 2184–2196. [Google Scholar] [CrossRef] [PubMed]
- Berger, D.; Martinossi-Allibert, I.; Grieshop, K.; Lind, M.I.; Maklakov, A.A.; Arnqvist, G. Intralocus sexual conflict and the tragedy of the commons in seed beetles. Am. Nat. 2016, 188, E98–E112. [Google Scholar] [CrossRef] [PubMed]
- Dougherty, L.R.; van Lieshout, E.; McNamara, K.B.; Moschilla, J.A.; Arnqvist, G.; Simmons, L.W. Sexual conflict and correlated evolution between male persistence and female resistance traits in the seed beetle Callosobruchus maculatus. Proc. R. Soc. B Biol. Sci. 2017, 284, 20170132. [Google Scholar] [CrossRef] [PubMed]
- Grieshop, K.; Arnqvist, G. Sex-specific dominance reversal of genetic variation for fitness. PLoS Biol. 2018, 16, e2006810. [Google Scholar] [CrossRef]
- Sayadi, A.; Immonen, E.; Bayram, H.; Arnqvist, G. The de novo transcriptome and its functional annotation in the seed beetle Callosobruchus maculatus. PLoS ONE 2016, 11, e0158565. [Google Scholar] [CrossRef]
- Sayadi, A.; Barrio, A.M.; Immonen, E.; Dainat, J.; Berger, D.; Tellgren-Roth, C.; Nystedt, B.; Arnqvist, G. The genomic footprint of sexual conflict. Nat. Ecol. Evol. 2019, 3, 1725–1730. [Google Scholar] [CrossRef]
- Tuda, M.; Kagoshima, K.; Toquenaga, Y.; Arnqvist, G. Global genetic differentiation in a cosmopolitan pest of stored beans: Effects of geography, host-plant usage and anthropogenic factors. PLoS ONE 2014, 9, e106268. [Google Scholar] [CrossRef]
- Tuda, M.; Rönn, J.; Buranapanichpan, S.; Wasano, N.; Arnqvist, G. Evolutionary diversification of the bean beetle genus Callosobruchus (Coleoptera: Bruchidae): Traits associated with stored-product pest status. Mol. Ecol. 2006, 15, 3541–3551. [Google Scholar] [CrossRef]
- Messina, F.J.; Jones, J.C.; Mendenhall, M.; Muller, A. Genetic modification of host acceptance by a seed beetle, Callosobruchus maculatus (Coleoptera: Bruchidae). Ann. Entomol. Soc. Am. 2009, 102, 181–188. [Google Scholar] [CrossRef]
- Messina, F.J.; Mendenhall, M.; Jones, J.C. An experimentally induced host shift in a seed beetle. Entomol. Exp. Appl. 2009, 132, 39–49. [Google Scholar] [CrossRef]
- Messina, F.J.; Lish, A.M.; Gompert, Z. Variable responses to novel hosts by populations of the seed beetle Callosobruchus maculatus (Coleoptera: Chrysomelidae: Bruchinae). Environ. Entomol. 2018, 47, 1194–1202. [Google Scholar] [CrossRef] [PubMed]
- Credland, P.F. Effects of host change on the fecundity and development of an unusual strain of Callosobruchus maculatus (F.) (Coleoptera: Bruchidae). J. Stored Prod. Res. 1987, 23, 91–98. [Google Scholar] [CrossRef]
- Credland, P.F. Biotype variation and host change in bruchids: Causes and effects in the evolution of bruchid pests. In Bruchids and Legumes: Economics, Ecology and Coevolution; Fujii, K., Gatehouse, A.M.R., Johnson, C.D., Mitchel, R., Yoshida, T., Eds.; Springer: Dordrecht, The Netherlands, 1990; pp. 271–287. [Google Scholar]
- Zhu-Salzman, K.; Zeng, R.S. Molecular mechanisms of insect adaptation to plant defense: Lessons learned from a Bruchid beetle. Insect Sci. 2008, 15, 477–481. [Google Scholar] [CrossRef]
- Birnbaum, S.S.; Abbot, P. Gene expression and diet breadth in plant-feeding insects: Summarizing trends. Trends Ecol. Evol. 2019. [Google Scholar] [CrossRef] [PubMed]
- Messina, F.J. Life-history variation in a seed beetle: Adult egg-laying vs. larval competitive ability. Oecologia 1991, 85, 447–455. [Google Scholar] [CrossRef]
- Mitchell, R. The traits of a biotype of Callosobruchus maculatus (F.) (Coleoptera: Bruchidae) from South India. J. Stored Prod. Res. 1991, 27, 221–224. [Google Scholar] [CrossRef]
- Messina, F.J.; Durham, S.L. Loss of adaptation following reversion suggests trade-offs in host use by a seed beetle. J. Evol. Biol. 2015, 28, 1882–1891. [Google Scholar] [CrossRef]
- Gompert, Z.; Lucas, L.K.; Buerkle, C.A.; Forister, M.L.; Fordyce, J.A.; Nice, C.C. Admixture and the organization of genetic diversity in a butterfly species complex revealed through common and rare genetic variants. Mol. Ecol. 2014, 23, 4555–4573. [Google Scholar] [CrossRef]
- Li, H.; Durbin, R. Fast and accurate short read alignment with Burrows–Wheeler transform. Bioinformatics 2009, 25, 1754–1760. [Google Scholar] [CrossRef]
- Buerkle, C.A.; Gompert, Z. Population genomics based on low coverage sequencing: How low should we go? Mol. Ecol. 2013, 22, 3028–3035. [Google Scholar] [CrossRef] [PubMed]
- Dickson, S.P.; Wang, K.; Krantz, I.; Hakonarson, H.; Goldstein, D.B. Rare variants create synthetic genome-wide associations. PLoS Biol. 2010, 8, e1000294. [Google Scholar] [CrossRef] [PubMed]
- Gompert, Z.; Lucas, L.K.; Nice, C.C.; Fordyce, J.A.; Forister, M.L.; Buerkle, C.A. Genomic regions with a history of divergent selection affect fitness of hybrids between two butterfly species. Evolution 2012, 66, 2167–2181. [Google Scholar] [CrossRef] [PubMed]
- Gompert, Z.; Jahner, J.P.; Scholl, C.F.; Wilson, J.S.; Lucas, L.K.; Soria-Carrasco, V.; Fordyce, J.A.; Nice, C.C.; Buerkle, C.A.; Forister, M.L. The evolution of novel host use is unlikely to be constrained by trade-offs or a lack of genetic variation. Mol. Ecol. 2015, 24, 2777–2793. [Google Scholar] [CrossRef] [PubMed]
- Gingerich, P.D. Rates of Evolution: A Quantitative Synthesis; Cambridge University Press: Cambridge, UK, 2019. [Google Scholar]
- Harte, D. HiddenMarkov: Hidden Markov Models; R Package Version 1.8-11.; Statistics Research Associates: Wellington, New Zealand, 2017. [Google Scholar]
- Bates, D.; Maechler, M.; Bolker, B.; Walker, S.; Christensen, R.H.B.; Singmann, H.; Dai, B.; Grothendieck, G.; Green, P.; Bolker, M.B. Package ‘lme4’. Convergence 2015, 12, 2. [Google Scholar]
- Crainiceanu, C.M.; Ruppert, D. Likelihood ratio tests in linear mixed models with one variance component. J. R. Stat. Soc. Ser. B Stat. Methodol. 2004, 66, 165–185. [Google Scholar] [CrossRef]
- Greven, S.; Crainiceanu, C.M.; Küchenhoff, H.; Peters, A. Restricted likelihood ratio testing for zero variance components in linear mixed models. J. Comput. Graph. Stat. 2008, 17, 870–891. [Google Scholar] [CrossRef]
- Scheipl, F.; Greven, S.; Küchenhoff, H. Size and power of tests for a zero random effect variance or polynomial regression in additive and linear mixed models. Comput. Stat. Data Anal. 2008, 52, 3283–3299. [Google Scholar] [CrossRef]
- Zhou, X.; Carbonetto, P.; Stephens, M. Polygenic modeling with Bayesian sparse linear mixed models. PLoS Genet. 2013, 9, e1003264. [Google Scholar] [CrossRef]
- Guan, Y.; Stephens, M. Bayesian variable selection regression for genome-wide association studies and other large-scale problems. Ann. Appl. Stat. 2011, 5, 1780–1815. [Google Scholar] [CrossRef]
- Song, L.; Florea, L. Rcorrector: Efficient and accurate error correction for Illumina RNA-seq reads. GigaScience 2015, 4, s13742-015. [Google Scholar] [CrossRef] [PubMed]
- Martin, M. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet J. 2011, 17, 10–12. [Google Scholar] [CrossRef]
- Dobin, A.; Davis, C.A.; Schlesinger, F.; Drenkow, J.; Zaleski, C.; Jha, S.; Batut, P.; Chaisson, M.; Gingeras, T.R. STAR: Ultrafast universal RNA-seq aligner. Bioinformatics 2013, 29, 15–21. [Google Scholar] [CrossRef] [PubMed]
- Liao, Y.; Smyth, G.K.; Shi, W. featureCounts: An efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics 2014, 30, 923–930. [Google Scholar] [CrossRef]
- Robinson, M.D.; McCarthy, D.J.; Smyth, G.K. edgeR: A Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 2010, 26, 139–140. [Google Scholar] [CrossRef]
- Ritchie, M.E.; Phipson, B.; Wu, D.; Hu, Y.; Law, C.W.; Shi, W.; Smyth, G.K. limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucl. Acids Res. 2015, 43, e47. [Google Scholar] [CrossRef]
- Law, C.W.; Chen, Y.; Shi, W.; Smyth, G.K. voom: Precision weights unlock linear model analysis tools for RNA-seq read counts. Genome Biol. 2014, 15, R29. [Google Scholar] [CrossRef]
- Schuler, M.A. The role of cytochrome P450 monooxygenases in plant-insect interactions. Plant Physiol. 1996, 112, 1411. [Google Scholar] [CrossRef]
- Terra, W.R.; Ferreira, C. Insect digestive enzymes: Properties, compartmentalization and function. Comp. Biochem. Physiol. Part B Comp. Biochem. 1994, 109, 1–62. [Google Scholar] [CrossRef]
- Guo, F.; Lei, J.; Sun, Y.; Chi, Y.H.; Ge, F.; Patil, B.S.; Koiwa, H.; Zeng, R.; Zhu-Salzman, K. Antagonistic regulation, yet synergistic defense: Effect of bergapten and protease inhibitor on development of cowpea bruchid Callosobruchus maculatus. PLoS ONE 2012, 7, e41877. [Google Scholar] [CrossRef]
- Benfey, P.N.; Mitchell-Olds, T. From genotype to phenotype: Systems biology meets natural variation. Science 2008, 320, 495–497. [Google Scholar] [CrossRef] [PubMed]
- Dowell, R.D.; Ryan, O.; Jansen, A.; Cheung, D.; Agarwala, S.; Danford, T.; Bernstein, D.A.; Rolfe, P.A.; Heisler, L.E.; Chin, B.; et al. Genotype to phenotype: A complex problem. Science 2010, 328, 469. [Google Scholar] [CrossRef] [PubMed]
- Messina, F.J.; Jones, J.C. Inheritance of traits mediating a major host shift by a seed beetle, Callosobruchus maculatus (Coleoptera: Chrysomelidae: Bruchinae). Ann. Entomol. Soc. Am. 2011, 104, 808–815. [Google Scholar] [CrossRef]
- Sezer, M.; Butlin, R. The genetic basis of host plant adaptation in the brown planthopper (Nilaparvata lugens). Heredity 1998, 80, 499–508. [Google Scholar] [CrossRef]
- Forister, M.L. Independent inheritance of preference and performance in hybrids between host races of Mitoura butterflies (Lepidoptera: Lycaenidae). Evolution 2005, 59, 1149–1155. [Google Scholar] [CrossRef]
- Carroll, S.P.; Dingle, H.; Famula, T.R.; Fox, C.W. Genetic architecture of adaptive differentiation in evolving host races of the soapberry bug, Jadera haematoloma. In Microevolution Rate, Pattern, Process; Springer: Berlin, Germany, 2001; pp. 257–272. [Google Scholar]
- De Jong, P.W.; Nielsen, J.K. Host plant use of Phyllotreta nemorum: Do coadapted gene complexes play a role? Entomol. Exp. Appl. 2002, 104, 207–215. [Google Scholar] [CrossRef]
- Taverner, A.M.; Yang, L.; Barile, Z.J.; Lin, B.; Peng, J.; Pinharanda, A.P.; Rao, A.S.; Roland, B.P.; Talsma, A.D.; Wei, D.; et al. Adaptive substitutions underlying cardiac glycoside insensitivity in insects exhibit epistasis in vivo. eLife 2019, 8, e48224. [Google Scholar] [CrossRef]
- Burke, M.K.; Liti, G.; Long, A.D. Standing genetic variation drives repeatable experimental evolution in outcrossing populations of Saccharomyces cerevisiae. Mol. Biol. Evol. 2014, 31, 3228–3239. [Google Scholar] [CrossRef]
- Alves, J.M.; Carneiro, M.; Cheng, J.Y.; Lemos de Matos, A.; Rahman, M.M.; Loog, L.; Campos, P.F.; Wales, N.; Eriksson, A.; Manica, A.; et al. Parallel adaptation of rabbit populations to myxoma virus. Science 2019, 363, 1319–1326. [Google Scholar] [CrossRef]
- MacPherson, A.; Nuismer, S. The probability of parallel genetic evolution from standing genetic variation. J. Evol. Biol. 2017, 30, 326–337. [Google Scholar] [CrossRef]
- Endler, J.A. Natural Selection in the Wild; Number 21 in Monographs in Population Biology; Princeton University Press: Princeton, NJ, USA, 1986. [Google Scholar]
- Manolio, T.A.; Collins, F.S.; Cox, N.J.; Goldstein, D.B.; Hindorff, L.A.; Hunter, D.J.; McCarthy, M.I.; Ramos, E.M.; Cardon, L.R.; Chakravarti, A.; et al. Finding the missing heritability of complex diseases. Nature 2009, 461, 747–753. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Benyamin, B.; McEvoy, B.P.; Gordon, S.; Henders, A.K.; Nyholt, D.R.; Madden, P.A.; Heath, A.C.; Martin, N.G.; Montgomery, G.W.; et al. Common SNPs explain a large proportion of the heritability for human height. Nat. Genet. 2010, 42, 565–569. [Google Scholar] [CrossRef] [PubMed]
- Scott, J.G. Cytochromes P450 and insecticide resistance. Insect Biochem. Mol. Biol. 1999, 29, 757–777. [Google Scholar] [CrossRef]
- Li, X.; Schuler, M.A.; Berenbaum, M.R. Jasmonate and salicylate induce expression of herbivore cytochrome P450 genes. Nature 2002, 419, 712–715. [Google Scholar] [CrossRef]
- Etges, W.J. Evolutionary genomics of host plant adaptation: Insights from Drosophila. Curr. Opin. Insect Sci. 2019, 36, 96–102. [Google Scholar] [CrossRef]
- Schoonhoven, L.M.; van Loon, J.J.A.; Dicke, M. Insect-Plant Biology, 2nd ed.; Oxford University Press: Oxford, UK, 2010. [Google Scholar]
- Desroches, P.; Mandon, N.; Baehr, J.; Huignard, J. Mediation of host-plant use by a glucoside in Callosobruchus maculatus F. (Coleoptera: Bruchidae). J. Insect Physiol. 1997, 43, 439–446. [Google Scholar] [CrossRef]
- Nosil, P. Reproductive isolation caused by visual predation on migrants between divergent environments. Proc. R. Soc. Lond. Ser. B Biol. Sci. 2004, 271, 1521–1528. [Google Scholar] [CrossRef]
- Forister, M.L.; Gompert, Z.; Nice, C.C.; Forister, G.W.; Fordyce, J.A. Ant association facilitates the evolution of diet breadth in a lycaenid butterfly. Proc. R. Soc. B Biol. Sci. 2011, 278, 1539–1547. [Google Scholar] [CrossRef]
- Scholl, C.F.; Burls, K.J.; Newton, J.L.; Young, B.; Forister, M.L. Temporal and geographic variation in parasitoid attack with no evidence for ant protection of the Melissa blue butterfly, Lycaeides melissa. Ecol. Entomol. 2014, 39, 168–176. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Comparison | Observed | Expected | P |
|---|---|---|---|
| M × L1 | 0 | 0.85 | 0.42 |
| M × L1R | 3 | 4.93 | 0.14 |
| L1 × L1R | 0 | 0.57 | 0.57 |
| L1 × L1 | 2 | 0.17 | 0.01 |
| L1R × L1R | 6 | 1.67 | 0.01 |
| M × L1 | 7 | 4.51 | 0.08 |
| M × L1R | 6 | 2.24 | 0.12 |
| L1 × L1R | 0 | 0.07 | 0.93 |
| L1 × L1R | 1 | 0.28 | 0.21 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rêgo, A.; Chaturvedi, S.; Springer, A.; Lish, A.M.; Barton, C.L.; Kapheim, K.M.; Messina, F.J.; Gompert, Z. Combining Experimental Evolution and Genomics to Understand How Seed Beetles Adapt to a Marginal Host Plant. Genes 2020, 11, 400. https://doi.org/10.3390/genes11040400
Rêgo A, Chaturvedi S, Springer A, Lish AM, Barton CL, Kapheim KM, Messina FJ, Gompert Z. Combining Experimental Evolution and Genomics to Understand How Seed Beetles Adapt to a Marginal Host Plant. Genes. 2020; 11(4):400. https://doi.org/10.3390/genes11040400
Chicago/Turabian StyleRêgo, Alexandre, Samridhi Chaturvedi, Amy Springer, Alexandra M. Lish, Caroline L. Barton, Karen M. Kapheim, Frank J. Messina, and Zachariah Gompert. 2020. "Combining Experimental Evolution and Genomics to Understand How Seed Beetles Adapt to a Marginal Host Plant" Genes 11, no. 4: 400. https://doi.org/10.3390/genes11040400
APA StyleRêgo, A., Chaturvedi, S., Springer, A., Lish, A. M., Barton, C. L., Kapheim, K. M., Messina, F. J., & Gompert, Z. (2020). Combining Experimental Evolution and Genomics to Understand How Seed Beetles Adapt to a Marginal Host Plant. Genes, 11(4), 400. https://doi.org/10.3390/genes11040400