A Novel WAC Loss of Function Mutation in an Individual Presenting with Encephalopathy Related to Status Epilepticus during Sleep (ESES)

 , , , , ,
, , , , ,
Abstract
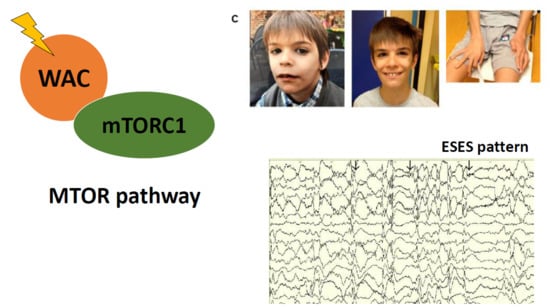
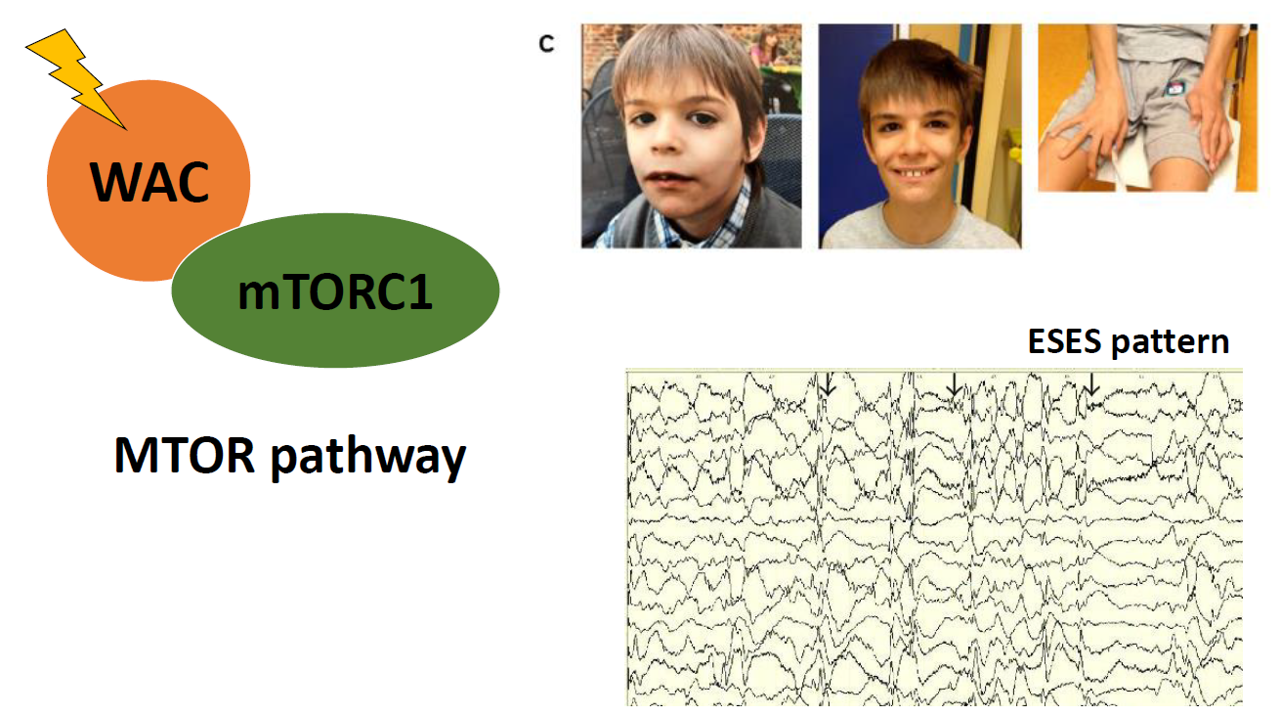
1. Introduction
2. Materials and Methods
2.1. Cohort Description
2.2. Gene Panel Sequencing
2.3. Transcript Analysis
3. Results
3.1. Clinical Assessment
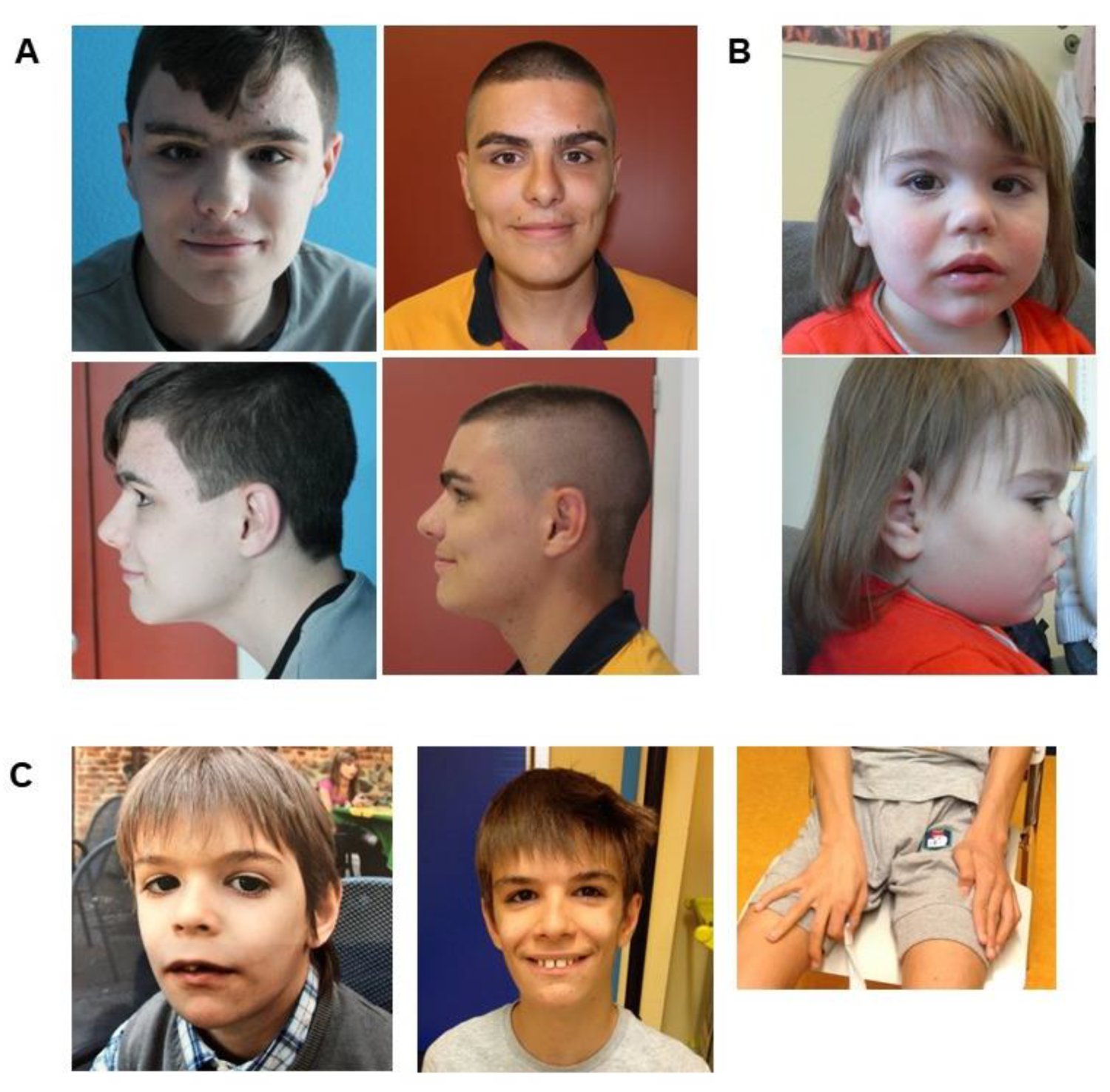
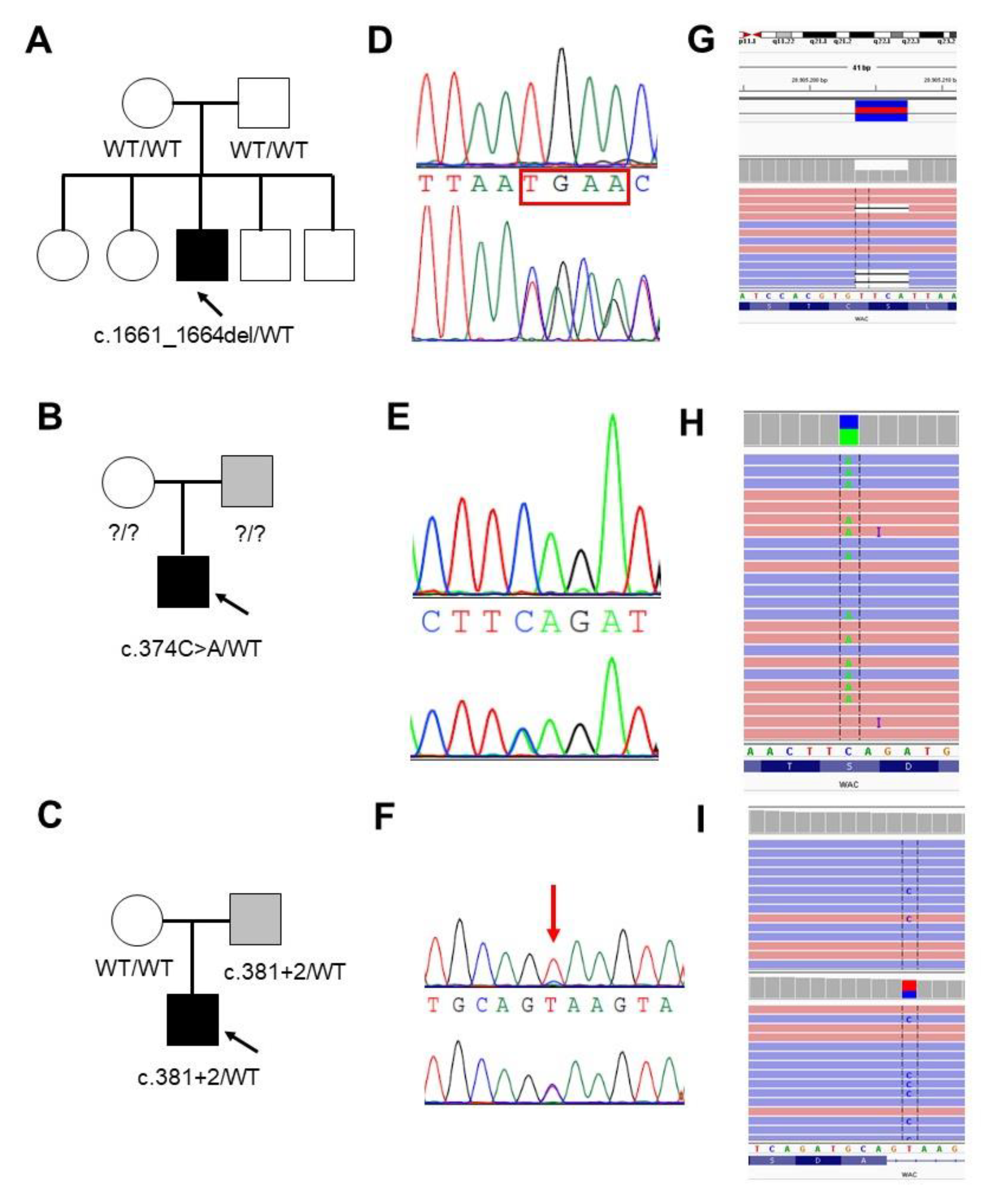
3.1.1. Family 1
3.1.2. Family 2
3.1.3. Family 3
3.2. Molecular Data
3.3. WAC Mutation Spectrum in Public Databases
4. Discussion
4.1. WAC Mutations Spectrum
4.2. Germinal Mosaicism of WAC Mutations
4.3. WAC Phenotype Heterogeneity
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Nagase, T.; Nakayama, M.; Nakajima, D.; Kikuno, R.; Ohara, O. Prediction of the Coding Sequences of Unidentified Human Genes. XX. The Complete Sequences of 100 New cDNA Clones from Brain Which Code for Large Proteins in vitro. DNA Res. 2001, 8, 85–95. [Google Scholar] [CrossRef] [PubMed]
- Zhang, F.; Yu, X. WAC, a functional partner of RNF20/40, regulates histone H2B ubiquitination and gene transcription. Mol. Cell 2011, 41, 384–397. [Google Scholar] [CrossRef] [PubMed]
- Abbas, T.; Dutta, A. p21 in cancer: Intricate networks and multiple activities. Nat. Rev. Cancer 2009, 9, 400–414. [Google Scholar] [CrossRef] [PubMed]
- Totsukawa, G.; Kaneko, Y.; Uchiyama, K.; Toh, H.; Tamura, K.; Kondo, H. VCIP135 deubiquitinase and its binding protein, WAC, in p97ATPase-mediated membrane fusion. EMBO J. 2011, 30, 3581–3593. [Google Scholar] [CrossRef]
- David-Morrison, G.; Xu, Z.; Rui, Y.-N.; Charng, W.-L.; Jaiswal, M.; Yamamoto, S.; Xiong, B.; Zhang, K.; Sandoval, H.; Duraine, L.; et al. WAC Regulates mTOR Activity by Acting as an Adaptor for the TTT and Pontin/Reptin Complexes. Dev. Cell 2016, 36, 139–151. [Google Scholar] [CrossRef]
- Joachim, J.; Jefferies, H.B.J.; Razi, M.; Frith, D.; Snijders, A.P.; Chakravarty, P.; Judith, D.; Tooze, S.A. Activation of ULK Kinase and Autophagy by GABARAP Trafficking from the Centrosome Is Regulated by WAC and GM130. Mol. Cell 2015, 60, 899–913. [Google Scholar] [CrossRef]
- Lugtenberg, D.; Reijnders, M.R.F.; Fenckova, M.; Bijlsma, E.K.; Bernier, R.; van Bon, B.W.M.; Smeets, E.; Vulto-van Silfhout, A.T.; Bosch, D.; Eichler, E.E.; et al. De novo loss-of-function mutations in WAC cause a recognizable intellectual disability syndrome and learning deficits in Drosophila. Eur. J. Hum. Genet. EJHG 2016, 24, 1145–1153. [Google Scholar] [CrossRef]
- DeSanto, C.; D’Aco, K.; Araujo, G.C.; Shannon, N.; Study, D.D.D.; Vernon, H.; Rahrig, A.; Monaghan, K.G.; Niu, Z.; Vitazka, P.; et al. WAC loss-of-function mutations cause a recognisable syndrome characterised by dysmorphic features, developmental delay and hypotonia and recapitulate 10p11.23 microdeletion syndrome. J. Med. Genet. 2015, 52, 754–761. [Google Scholar] [CrossRef]
- Zhang, Y.J.; Yao, P.L.; Zhou, Y.F.; Qiu, T.; Wang, J.; Wang, X.H.; Zhou, S.Z.; Wu, B.B.; Wang, Y. WAC gene pathogenic variation cause DeSanto-Shinawi syndrome with electrical status epilepticus during sleep. Zhonghua Er Ke Za Zhi Chin. J. Pediatr. 2019, 57, 802–804. [Google Scholar]
- Uehara, T.; Ishige, T.; Hattori, S.; Yoshihashi, H.; Funato, M.; Yamaguchi, Y.; Takenouchi, T.; Kosaki, K. Three patients with DeSanto-Shinawi syndrome: Further phenotypic delineation. Am. J. Med. Genet. A. 2018, 176, 1335–1340. [Google Scholar] [CrossRef]
- Vanegas, S.; Ramirez-Montaño, D.; Candelo, E.; Shinawi, M.; Pachajoa, H. DeSanto-Shinawi Syndrome: First Case in South America. Mol. Syndromol. 2018, 9, 154–158. [Google Scholar] [CrossRef] [PubMed]
- Mondini, S.; Mapelli, D.; Vestri, A. Esame Neuropsicologico Breve 2; Raffaello Cortina Editore: Milano, Italy, 2011. [Google Scholar]
- Aspromonte, M.C.; Bellini, M.; Gasparini, A.; Carraro, M.; Bettella, E.; Polli, R.; Cesca, F.; Bigoni, S.; Boni, S.; Carlet, O.; et al. Characterization of intellectual disability and autism comorbidity through gene panel sequencing. Hum. Mutat. 2019, 40, 1346–1363. [Google Scholar] [CrossRef] [PubMed]
- Desmet, F.-O.; Hamroun, D.; Lalande, M.; Collod-Béroud, G.; Claustres, M.; Béroud, C. Human Splicing Finder: An online bioinformatics tool to predict splicing signals. Nucleic Acids Res. 2009, 37, e67. [Google Scholar] [CrossRef] [PubMed]
- Fokkema, I.F.A.C.; Taschner, P.E.M.; Schaafsma, G.C.P.; Celli, J.; Laros, J.F.J.; den Dunnen, J.T. LOVD v. 2.0: The next generation in gene variant databases. Hum. Mutat. 2011, 32, 557–563. [Google Scholar] [CrossRef]
- Gataullina, S.; Bienvenu, T.; Nabbout, R.; Huberfeld, G.; Dulac, O. Gene mutations in paediatric epilepsies cause NMDA-pathy, and phasic and tonic GABA-pathy. Dev. Med. Child Neurol. 2019, 61, 891–898. [Google Scholar] [CrossRef]
- Kessi, M.; Peng, J.; Yang, L.; Xiong, J.; Duan, H.; Pang, N.; Yin, F. Genetic etiologies of the electrical status epilepticus during slow wave sleep: Systematic review. BMC Genet. 2018, 19, 40. [Google Scholar] [CrossRef]
- Robinson, J.T.; Thorvaldsdóttir, H.; Winckler, W.; Guttman, M.; Lander, E.S.; Getz, G.; Mesirov, J.P. Integrative genomics viewer. Nat. Biotechnol. 2011, 29, 24–26. [Google Scholar] [CrossRef]
- Wang, T.; Guo, H.; Xiong, B.; Stessman, H.A.F.; Wu, H.; Coe, B.P.; Turner, T.N.; Liu, Y.; Zhao, W.; Hoekzema, K.; et al. De novo genic mutations among a Chinese autism spectrum disorder cohort. Nat. Commun. 2016, 7, 13316. [Google Scholar] [CrossRef]
- Deciphering Developmental Disorders Study Prevalence and architecture of de novo mutations in developmental disorders. Nature 2017, 542, 433–438. [CrossRef]
- Homsy, J.; Zaidi, S.; Shen, Y.; Ware, J.S.; Samocha, K.E.; Karczewski, K.J.; DePalma, S.R.; McKean, D.; Wakimoto, H.; Gorham, J.; et al. De novo mutations in congenital heart disease with neurodevelopmental and other congenital anomalies. Science 2015, 350, 1262–1266. [Google Scholar] [CrossRef]
- Karczewski, K.J.; Francioli, L.C.; Tiao, G.; Cummings, B.B.; Alföldi, J.; Wang, Q.; Collins, R.L.; Laricchia, K.M.; Ganna, A.; Birnbaum, D.P.; et al. Variation across 141,456 human exomes and genomes reveals the spectrum of loss-of-function intolerance across human protein-coding genes. bioRxiv 2019, 531210. [Google Scholar]
- De, S. Somatic mosaicism in healthy human tissues. June 2011, 27, 217–223. [Google Scholar] [CrossRef] [PubMed]
- D’Gama, A.M.; Walsh, C.A. Somatic mosaicism and neurodevelopmental disease. Nat. Neurosci. 2018, 21, 1504–1514. [Google Scholar] [CrossRef]
- Freed, D.; Pevsner, J. The Contribution of Mosaic Variants to Autism Spectrum Disorder. PLoS Genet. 2016, 12, e1006245. [Google Scholar] [CrossRef]
- Krupp, D.R.; Barnard, R.A.; Duffourd, Y.; Evans, S.A.; Mulqueen, R.M.; Bernier, R.; Rivière, J.-B.; Fombonne, E.; O’Roak, B.J. Exonic Mosaic Mutations Contribute Risk for Autism Spectrum Disorder. Am. J. Hum. Genet. 2017, 101, 369–390. [Google Scholar] [CrossRef]
- Lim, E.T.; Uddin, M.; Rubeis, S.D.; Chan, Y.; Kamumbu, A.S.; Zhang, X.; D’Gama, A.M.; Kim, S.N.; Hill, R.S.; Goldberg, A.P.; et al. Rates, distribution and implications of postzygotic mosaic mutations in autism spectrum disorder. Nat. Neurosci. 2017, 20, 1217–1224. [Google Scholar] [CrossRef]
- Acuna-Hidalgo, R.; Bo, T.; Kwint, M.P.; van de Vorst, M.; Pinelli, M.; Veltman, J.A.; Hoischen, A.; Vissers, L.E.L.M.; Gilissen, C. Post-zygotic Point Mutations Are an Underrecognized Source of De Novo Genomic Variation. Am. J. Hum. Genet. 2015, 97, 67–74. [Google Scholar] [CrossRef]
- de Boer, A.; Vermeulen, K.; Egger, J.I.M.; Janzing, J.G.E.; de Leeuw, N.; Veenstra-Knol, H.E.; den Hollander, N.S.; van Bokhoven, H.; Staal, W.; Kleefstra, T. EHMT1 mosaicism in apparently unaffected parents is associated with autism spectrum disorder and neurocognitive dysfunction. Mol. Autism 2018, 9. [Google Scholar] [CrossRef]
- Abdelhedi, F.; Khattabi, L.E.; Essid, N.; Viot, G.; Letessier, D.; Lebbar, A.; Dupont, J.-M. A de novo 10p11.23-p12.1 deletion recapitulates the phenotype observed in WAC mutations and strengthens the role of WAC in intellectual disability and behavior disorders. Am. J. Med. Genet. A 2016, 170, 1912–1917. [Google Scholar] [CrossRef]
- Helbig, K.L.; Farwell Hagman, K.D.; Shinde, D.N.; Mroske, C.; Powis, Z.; Li, S.; Tang, S.; Helbig, I. Diagnostic exome sequencing provides a molecular diagnosis for a significant proportion of patients with epilepsy. Genet. Med. Off. J. Am. Coll. Med. Genet. 2016, 18, 898–905. [Google Scholar] [CrossRef]
- Gruber, V.; Scholl, T.; Samueli, S.; Gröppel, G.; Mühlebner, A.; Hainfellner, J.A.; Feucht, M. Pathophysiology of neurodevelopmental mTOR pathway-associated epileptic conditions: Current status of biomedical research. Clin. Neuropathol. 2019, 38, 210–224. [Google Scholar] [CrossRef] [PubMed]
- McMahon, J.; Huang, X.; Yang, J.; Komatsu, M.; Yue, Z.; Qian, J.; Zhu, X.; Huang, Y. Impaired autophagy in neurons after disinhibition of mammalian target of rapamycin and its contribution to epileptogenesis. J. Neurosci. Off. J. Soc. Neurosci. 2012, 32, 15704–15714. [Google Scholar] [CrossRef] [PubMed]
- Schmeisser, K.; Parker, J.A. Pleiotropic Effects of mTOR and Autophagy during Development and Aging. Front. Cell Dev. Biol. 2019, 7. [Google Scholar] [CrossRef] [PubMed]
- Winden, K.D.; Ebrahimi-Fakhari, D.; Sahin, M. Abnormal mTOR Activation in Autism. Annu. Rev. Neurosci. 2018, 41, 1–23. [Google Scholar] [CrossRef]
- Mühlebner, A.; Bongaarts, A.; Sarnat, H.B.; Scholl, T.; Aronica, E. New insights into a spectrum of developmental malformations related to mTOR dysregulations: Challenges and perspectives. J. Anat. 2019, 235, 521–542. [Google Scholar] [CrossRef]
- Tassinari, C.A.; Cantalupo, G.; Rios-Pohl, L.; Giustina, E.D.; Rubboli, G. Encephalopathy with status epilepticus during slow sleep: “The Penelope syndrome”. Epilepsia 2009, 50, 4–8. [Google Scholar] [CrossRef]
- Mathieu, M.-L.; de Bellescize, J.; Till, M.; Flurin, V.; Labalme, A.; Chatron, N.; Sanlaville, D.; Chemaly, N.; des Portes, V.; Ostrowsky, K.; et al. Electrical status epilepticus in sleep, a constitutive feature of Christianson syndrome? Eur. J. Paediatr. Neurol. 2018, 22, 1124–1132. [Google Scholar] [CrossRef]
- Nissenkorn, A.; Gak, E.; Vecsler, M.; Reznik, H.; Menascu, S.; Ben Zeev, B. Epilepsy in Rett syndrome---the experience of a National Rett Center. Epilepsia 2010, 51, 1252–1258. [Google Scholar] [CrossRef]
- Pacheva, I.; Panov, G.; Gillberg, C.; Neville, B. A girl with tuberous sclerosis complex presenting with severe epilepsy and electrical status epilepticus during sleep, and with high-functioning autism and mutism. Cogn. Behav. Neurol. Off. J. Soc. Behav. Cogn. Neurol. 2014, 27, 88–95. [Google Scholar] [CrossRef]
- Lee, D.Y. Roles of mTOR signaling in Brain Development. Exp. Neurobiol. 2015, 24, 177–185. [Google Scholar] [CrossRef]
- Chiurazzi, P.; Oostra, B.A. Genetics of mental retardation. Curr. Opin. Pediatr. 2000, 12, 529–535. [Google Scholar] [CrossRef] [PubMed]
- Chiurazzi, P.; Pirozzi, F. Advances in understanding—Genetic basis of intellectual disability. F1000Research 2016, 5. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
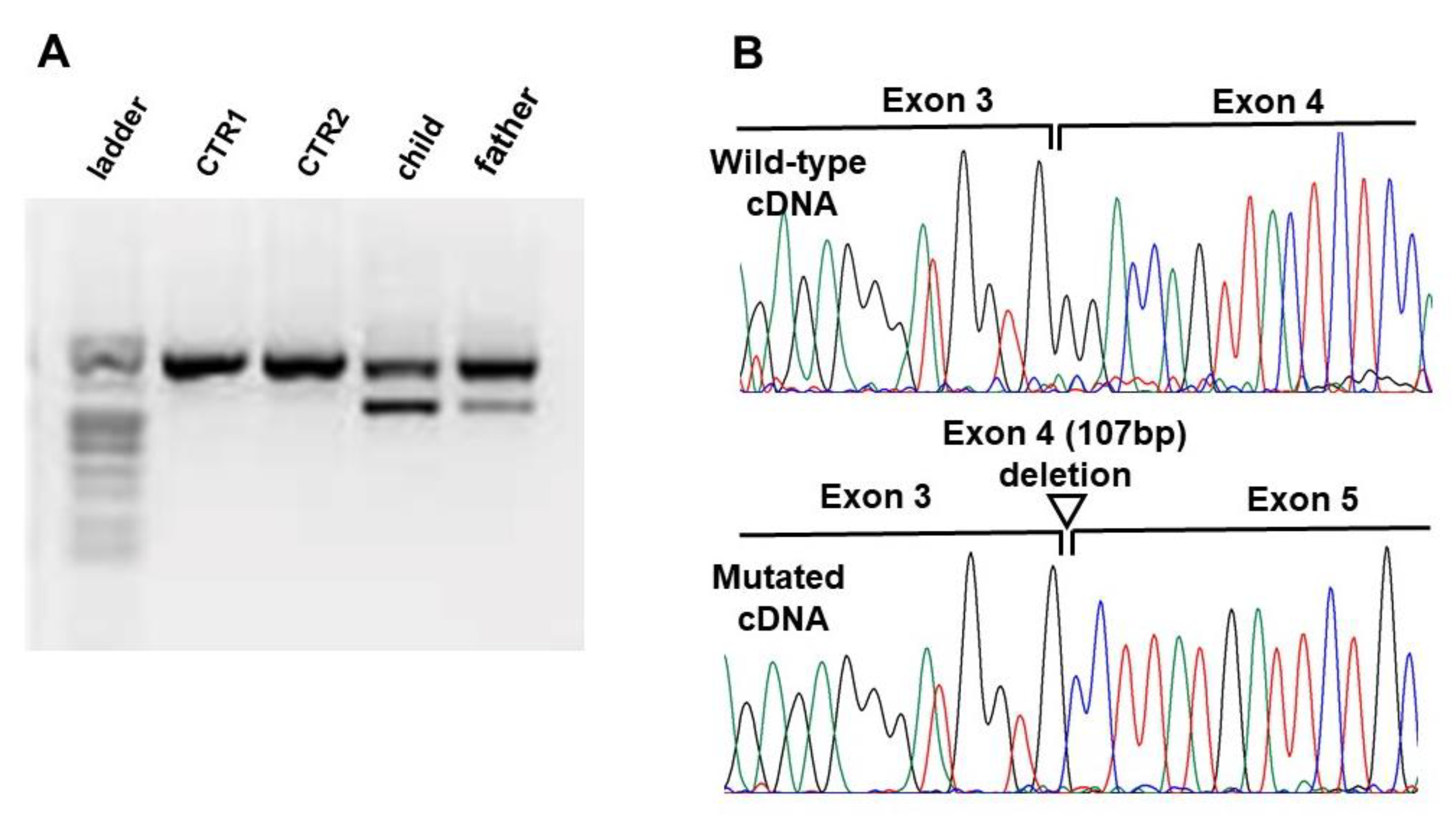{kind=link}
| Case 1 | Case 2 | Case 3 | De Santo 2015 | Lugtenberg 2016 | Uehara 2018 | Vanegas 2018 | Zhang 2019 | |
|---|---|---|---|---|---|---|---|---|
| Sex | M | F | M | 5 F, 1 M | 6 F, 4 M | 3 M | M | F |
| Age | 16 y/o | 7 y/o | 17 y/o | 1.3–11 y/o | 1.5–22 y/o | 3–22 y/o | 4 y/o | 5.8 y/o |
| Normal perinatal period | + | − | + | 2/6 | 6/10 | 3/3 | + | + |
| Delayed physical Growth | − | + | + | 2/6 | 3/10 | 2/3 | + | − |
| CC size anomaly | − | + (<2SD) | + (<2SD) | N.R. | 2/10 (>2SD) | 1/3 (>2SD) | + | − |
| Development | ||||||||
| Intellectual Disability | Moderate | N.E. | Severe | (IQ > 79) 3/6 | 8/10 (6 Mild, 1 Severe, 1 Moderate) | 3/3 | + | + |
| Language delay | + | + | + | 6/6 | 9/10 | 3/3 | + | + |
| Motor delay | + | + | + | 6/6 | 9/10 | 3/3 | + | + |
| Behavioral problems | ||||||||
| Autistic features | − | + | + | 1/6 | 4/9 | 0/3 | − | N.R. |
| Hyperactivity | − | − | + | 3/6 | 4/10 | 0/3 | + | N.R. |
| Anxiety | − | − | + | 3/6 | 3/10 | 1/3 | + | N.R. |
| Sleep disturbances | − | + | + | 2/6 | 6/10 | 0/3 | − | N.R. |
| Stereotypies | − | + | + | 1/6 | N.R. | 0/3 | − | N.R. |
| Neurological | ||||||||
| Hypotonia | − | + | + | 6/6 | 7/9 | 0/3 | + | + |
| Seizures | − | − | + (focal) | 2/6 | 1/9 | 0/3 | − | + (focal) |
| Epilepsy | − | − | + | 1/6 | 1/9 | 0/3 | − | + |
| Ocular | ||||||||
| Vision | − | − | + | 2/6 | 5/10 | 1/3 | − | |
| Strabismus | − | + | − | 3/6 | 3/10 | 1/3 | − | |
| Dysmorphisms | ||||||||
| Facial shape dysmorphology | − | − | + | N.R. | 5/10 | 2/3 | − | + |
| Prominent forehead | + | − | − | 6/6 | 10/10 | 1/3 | + | N.R. |
| Bulbous nasal tip | + | − | + | 5/6 | N.R. | 3/3 | + | + |
| Long or downslanting palpebral fissures | − | − | + | 1/6 | 5/10 | 3/3 | − | N.R. |
| Synophrys | + | − | − | 3/6 | 2/10 | 1/3 | + | N.R. |
| Deep set eyes | + | − | + | 2/6 | 5/10 | 0/3 | + | N.R. |
| Full lips - thin upper lip | − | − | − | 3/6 | N.R. | 1/3 | − | N.R. |
| Low-set ears | − | − | − | 3/6 | N.R. | 1/3 | + | N.R. |
| Hirsutism/hypertricosis | − | + | − | 2/6 | 1/10 | 1/3 | + | N.R. |
| Digital anomalies | − | + | − | 1/6 | 7/7 | 3/3 | + | N.R. |
| Other | ||||||||
| Feeding difficulties | − | − | + | 4/6 | 4/10 | 0/3 | + | N.R. |
| Constipation | − | − | + | 5/6 | N.R. | 1/3 | − | N.R. |
| Frequent infections | − | − | − | N.R. | 6/8 | 0/3 | + | N.R. |
| Hearing impairment | − | − | − | 2/6 | 0/2, N.R./8 | 0/3 | + | N.R. |
| EEG abnormalities | − | − | + | 0/2 | 0/1, N.R./9 | N.R. | N.R. | + |
| MRI abnormalities | − | − | − | 1/6 | 4/9 | N.R. | − | − |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Leonardi, E.; Bellini, M.; Aspromonte, M.C.; Polli, R.; Mercante, A.; Ciaccio, C.; Granocchio, E.; Bettella, E.; Donati, I.; Cainelli, E.; et al. A Novel WAC Loss of Function Mutation in an Individual Presenting with Encephalopathy Related to Status Epilepticus during Sleep (ESES). Genes 2020, 11, 344. https://doi.org/10.3390/genes11030344
Leonardi E, Bellini M, Aspromonte MC, Polli R, Mercante A, Ciaccio C, Granocchio E, Bettella E, Donati I, Cainelli E, et al. A Novel WAC Loss of Function Mutation in an Individual Presenting with Encephalopathy Related to Status Epilepticus during Sleep (ESES). Genes. 2020; 11(3):344. https://doi.org/10.3390/genes11030344
Chicago/Turabian StyleLeonardi, Emanuela, Mariagrazia Bellini, Maria C. Aspromonte, Roberta Polli, Anna Mercante, Claudia Ciaccio, Elisa Granocchio, Elisa Bettella, Ilaria Donati, Elisa Cainelli, and et al. 2020. "A Novel WAC Loss of Function Mutation in an Individual Presenting with Encephalopathy Related to Status Epilepticus during Sleep (ESES)" Genes 11, no. 3: 344. https://doi.org/10.3390/genes11030344
APA StyleLeonardi, E., Bellini, M., Aspromonte, M. C., Polli, R., Mercante, A., Ciaccio, C., Granocchio, E., Bettella, E., Donati, I., Cainelli, E., Boni, S., Sartori, S., Pantaleoni, C., Boniver, C., & Murgia, A. (2020). A Novel WAC Loss of Function Mutation in an Individual Presenting with Encephalopathy Related to Status Epilepticus during Sleep (ESES). Genes, 11(3), 344. https://doi.org/10.3390/genes11030344