Metabolome and Transcriptome Analysis of Hexaploid Solidago canadensis Roots Reveals its Invasive Capacity Related to Polyploidy


Abstract

1. Introduction
2. Materials and Methods
2.1. Sampling
2.2. Root Metabolite Extraction
2.3. Ultraperformance LC Quadrupole Time-of-Flight Tandem Mass Spectrometry (UPLC-Q-TOF-MS) Metabolite Profiling and Identification
2.4. Differentially Accumulated Metabolite Analysis
2.5. KEGG Pathway Analysis for Differentially Accumulated Metabolites
2.6. cDNA Library Construction and Sequence Assembly
2.7. Functional Annotation and Transcription Factor Gene Identification
2.8. Calculation of Gene Expression Level and Analysis of Differently Expressed Genes
2.9. Association Analysis of the Metabolome and Transcriptome Data Sets
2.10. Small RNA Library Construction and Sequencing
2.11. Alignment and Identification of Known and Novel miRNA Predication
2.12. miRNA Expression Analysis and Target Gene Prediction
2.13. RT-qPCR of mRNAs and Stem-Loop RT-PCR of miRNAs
3. Results
3.1. Putative Metabolite Identification in Roots of Two Cytotypes of S. canadensis
3.2. Differentially Accumulated Metabolites between Two Cytotypes of S. canadensis
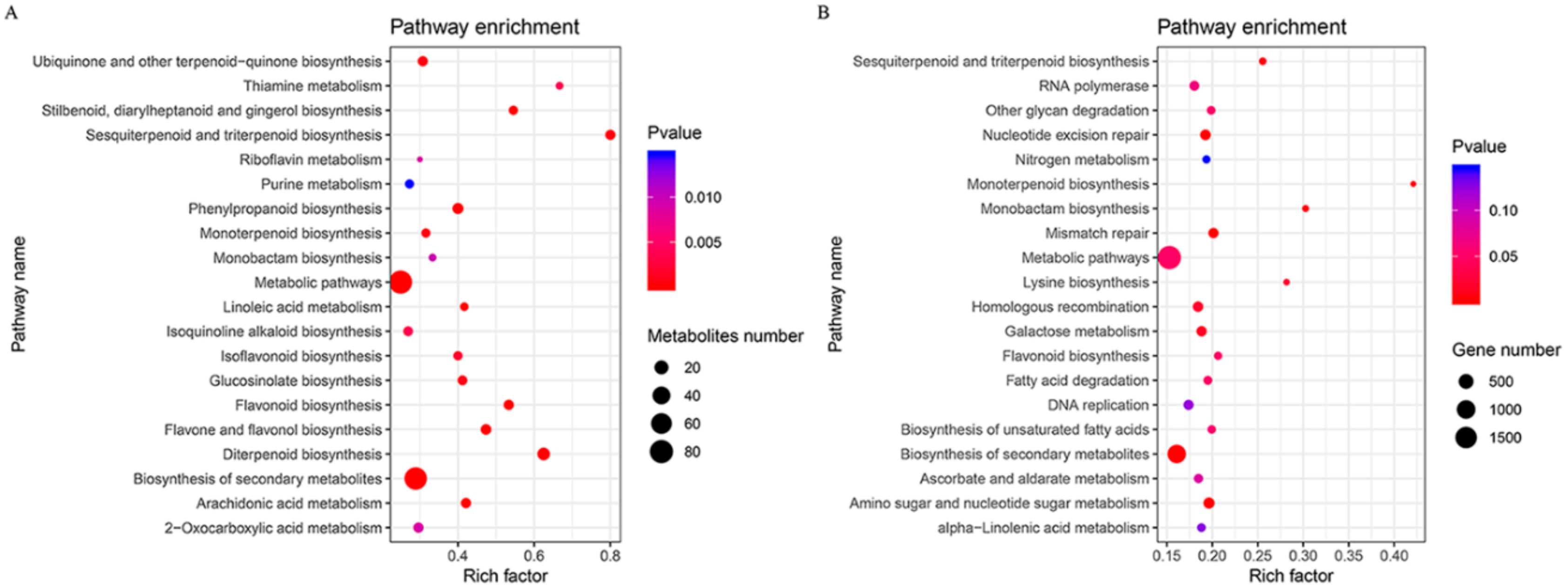
3.3. KEGG Enrichment Analysis of Differentially Accumulated Metabolites
3.4. Overview of Transcript Construction and Annotation for Two Cytotypes of S. canadensis
3.5. Comparison of Transcriptomes between Two Cytotypes of S. canadensis
3.6. Allelopathic Compound Synthesis-Related DEGs
3.7. The Regulation of Allelopathic Compound Synthesis-Related Genes between Two Cytotypes
3.8. Correlation Analysis between the Differentially Accumulated Allelopathic Metabolites and Related DEGs
3.9. Small RNA Expression and Target Gene Prediction
3.10. Visualization of Differentially Expressed miRNA and Target Gene Interaction Network
3.11. qRT-PCR and Stem-Loop RT-PCR Confirmed Differentially Expressed mRNAs and miRNAs
4. Discussion
4.1. Allelopathic Compound Synthesis Ability Was Enhanced in Hexaploid S. canadensis
4.2. Polyploidy Affects the Expression of Allelopathic Metabolite Synthesis Related Genes in Hexaploid S. canadensis
4.3. The Expression of Genes Involved in Regulation May Be Changed in Hexaploid S. canadensis
4.4. The Post-Transcriptional Regulation May Affect the Gene Expression of Hexaploid S. canadensis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Guido, A.; Pillar, V.D. Invasive plant removal: Assessing community impact and recovery from invasion. J. Appl. Ecol. 2017, 54, 1230–1237. [Google Scholar] [CrossRef]
- Powell, K.I.; Chase, J.M.; Knight, T.M. Invasive plants have scale-dependent effects on diversity by altering species-area relationships. Science 2013, 339, 316. [Google Scholar] [CrossRef] [PubMed]
- Rosche, C.; Hensen, I.; Mráz, P.; Durka, W.; Hartmann, M.; Lachmuth, S. Invasion success in polyploids: The role of inbreeding in the contrasting colonization abilities of diploid versus tetraploid populations of Centaurea stoebe s.I. J. Ecol. 2017, 105, 425–435. [Google Scholar] [CrossRef]
- Rosche, C.; Durka, W.; Hensen, I.; Mráz, P.; Hartmann, M.; Müller-Schärer, H.; Lachmuth, S. The population genetics of the fundamental cytotype-shift in invasive Centaurea stoebe s.l.: Genetic diversity, genetic differentiation and small-scale genetic structure differ between cytotypes but not between ranges. Biol. Invasions 2016, 18, 1895–1910. [Google Scholar] [CrossRef]
- Münzbergová, Z.; Haisel, D. Effects of polyploidization on the contents of photosynthetic pigments are largely population-specific. Photosynth. Res. 2018, 140, 289–299. [Google Scholar] [CrossRef] [PubMed]
- Pavlikova, Z.; Hola, D.; Vlasakova, B.; Prochazka, T.; Munzbergova, Z. Physiological and fitness differences between cytotypes vary with stress in a grassland perennial herb. PLoS One 2017, 12, e0188795. [Google Scholar] [CrossRef] [PubMed]
- Te Beest, M.; Roux, J.J.L.; Richardson, D.M.; Brysting, A.K.; Suda, J.; Kubešová, M.; Pyšek, P. The more the better? The role of polyploidy in facilitating plant invasions. Ann. Bot. 2012, 109, 19–45. [Google Scholar] [CrossRef]
- Rosche, C.; Hensen, I.; Lachmuth, S. Local pre-adaptation to disturbance and inbreeding-environment interactions affect colonisation abilities of diploid and tetraploid Centaurea stoebe. Plant Biol. 2018, 20, 75–84. [Google Scholar] [CrossRef]
- Xie, L.J.; Zeng, R.S.; Bi, H.H.; Song, Y.Y.; Wang, R.L.; Su, Y.J.; Chen, M.; Chen, S.; Liu, Y.H. Allelochemical mediated invasion of exotic plants in China. Allelopathy J. 2010, 25, 31–50. [Google Scholar]
- He, W.M.; Feng, Y.L.; Ridenour, W.M.; Thelen, G.C.; Pollock, J.L.; Diaconu, A.; Callaway, R.M. Novel weapons and invasion: Biogeographic differences in the competitive effects of Centaurea maculosa and its root exudate (±)-catechin. Oecologia 2009, 159, 803–815. [Google Scholar] [CrossRef]
- Grosso, V.; Farina, A.; Giorgi, D.; Nardi, L.; Diretto, G.; Lucretti, S. A high-throughput flow cytometry system for early screening of in vitro made polyploids in Dendrobium hybrids. Plant Cell Tiss. Org. 2018, 132, 57–70. [Google Scholar] [CrossRef]
- Lavania, U.C.; Sarita, S.; Seshu, L.; Surochita, B.; Nandeesh Kumar, M.; Yasuhiko, M. Autopolyploidy differentially influences body size in plants, but facilitates enhanced accumulation of secondary metabolites, causing increased cytosine methylation. Plant J. 2012, 71, 539–549. [Google Scholar] [CrossRef] [PubMed]
- Melville, M.R.; Morton, J.K. A biosystematic study of the Solidago canadensis (Compositae) complex. I. The Ontario populations. Can. J. Bot. 1982, 60, 976–997. [Google Scholar] [CrossRef]
- Wang, C.Y.; Jiang, K.; Zhou, J.W.; Wu, B.D. Solidago canadensis invasion affects soil N-fixing bacterial communities in heterogeneous landscapes in urban ecosystems in East China. Sci. Total Environ. 2018, 631, 702–713. [Google Scholar] [CrossRef] [PubMed]
- Yang, R.Y.; Mei, L.X.; Tang, J.J.; Chen, X.; An, M.; Wu, H.; Pratley, J. Allelopathic effects of invasive Solidago canadensis L. on germination and growth of native Chinese plant species. Allelopathy J. 2007, 19, 241–248. [Google Scholar]
- Abhilasha, D.; Quintana, N.; Vivanco, J.; Joshi, J. Do allelopathic compounds in invasive Solidago canadensis s.l. restrain the native European flora? J. Ecol. 2008, 96, 993–1001. [Google Scholar] [CrossRef]
- Gruľová, D.; Baranová, B.; Ivanova, V.; Martino, L.D.; Mancini, E.; Feo, V.D. Composition and bio activity of essential oils of Solidago spp. and their impact on radish and garden cress. Allelopathy J. 2016, 39, 129–142. [Google Scholar]
- Zhang, Z.; Fu, T.; Liu, Z.; Wang, X.; Xun, H.; Li, G.; Ding, B.; Dong, Y.; Lin, X.; Sanguinet, K.A.; et al. Extensive changes in gene expression and alternative splicing due to homoeologous exchange in rice segmental allopolyploids. Theor. Appl. Genet. 2019, 132, 2295–2308. [Google Scholar] [CrossRef]
- Kajal, M.; Singh, K. Small RNA profiling for identification of miRNAs involved in regulation of saponins biosynthesis in Chlorophytum borivilianum. BMC Plant Biol. 2017, 17, 265. [Google Scholar] [CrossRef]
- Dunn, W.B.; David, B.; Paul, B.; Eva, Z.; Sue, F.M.; Nadine, A.; Marie, B.; Knowles, J.D.; Antony, H.; Haselden, J.N. Procedures for large-scale metabolic profiling of serum and plasma using gas chromatography and liquid chromatography coupled to mass spectrometry. Nat. Protoc. 2011, 6, 1060–1083. [Google Scholar] [CrossRef]
- Haiyan, L.; Yuanyuan, D.; Jing, Y.; Xiuming, L.; Yanfang, W.; Na, Y.; Lili, G.; Nan, W.; Jinyu, W.; Xiaokun, L. De novo transcriptome of safflower and the identification of putative genes for oleosin and the biosynthesis of flavonoids. PLoS One 2012, 7, e30987. [Google Scholar]
- Mizrachi, E.; Hefer, C.A.; Ranik, M.; Joubert, F.; Myburg, A.A. De novo assembled expressed gene catalog of a fast-growing Eucalyptus tree produced by Illumina mRNA-Seq. BMC Genomics. 2010, 11, 681. [Google Scholar] [CrossRef] [PubMed]
- De Lima, J.C.; Loss-Morais, G.; Margis, R. MicroRNAs play critical roles during plant development and in response to abiotic stresses. Genet. Mol. Biol. 2012, 35, 1069–1077. [Google Scholar]
- Wen, B.; Mei, Z.; Zeng, C.; Liu, S. metaX: A flexible and comprehensive software for processing metabolomics data. BMC Bioinf. 2017, 18, 183. [Google Scholar] [CrossRef]
- Szymanska, E.; Saccenti, E.; Smilde, A.K.; Westerhuis, J.A. Double-check: Validation of diagnostic statistics for PLS-DA models in metabolomics studies. Metabolomics 2012, 8, S3–S16. [Google Scholar] [CrossRef]
- Grabherr, M.G.; Haas, B.J.; Yassour, M.; Levin, J.Z.; Thompson, D.A.; Amit, I.; Adiconis, X.; Fan, L.; Raychowdhury, R.; Zeng, Q.D.; et al. Full-length transcriptome assembly from RNA-Seq data without a reference genome. Nat. Biotechnol. 2011, 29, 644–652. [Google Scholar] [CrossRef]
- Pertea, G.; Huang, X.Q.; Liang, F.; Antonescu, V.; Sultana, R.; Karamycheva, S.; Lee, Y.; White, J.; Cheung, F.; Parvizi, B.; et al. TIGR Gene Indices clustering tools (TGICL): A software system for fast clustering of large EST datasets. Bioinformatics 2003, 19, 651–652. [Google Scholar] [CrossRef]
- Kanehisa, M.; Sato, Y.; Furumichi, M.; Morishima, J.; Tanabe, M. New approach for understanding genome variations in KEGG. Nucleic Acids Res. 2019, 47, D590–D595. [Google Scholar] [CrossRef]
- Ashburner, M.; Ball, C.A.; Blake, J.A.; Botstein, D.; Cherry, J.M. Gene ontology: Tool for the unification of biology. Nat. Genet. 2000, 25, 25–29. [Google Scholar] [CrossRef]
- Conesa, A.; Gotz, S.; Garcia-Gomez, J.M.; Terol, J.; Talon, M.; Robles, M. Blast2GO: A universal tool for annotation, visualization and analysis in functional genomics research. Bioinformatics 2005, 21, 3674–3676. [Google Scholar] [CrossRef]
- Usadel, B.; Nagel, A.; Thimm, O.; Redestig, H.; Blaesing, O.E.; Palacios-Rojas, N.; Selbig, J.; Hannemann, J.; Piques, M.C.; Steinhauser, D.; et al. Extension of the visualization tool MapMan to allow statistical analysis of arrays, display of coresponding genes, and comparison with known responses. Plant physiol. 2005, 138, 1195–1204. [Google Scholar] [CrossRef] [PubMed]
- Rice, P.; Longden, I.; Bleasby, A. EMBOSS: The European molecular biology open software suite. Trends Genet. 2000, 16, 276–277. [Google Scholar] [CrossRef]
- Mistry, J.; Finn, R.D.; Eddy, S.R.; Bateman, A.; Punta, M. Challenges in homology search: HMMER3 and convergent evolution of coiled-coil regions. Nucleic Acids Res. 2013, 41, e121. [Google Scholar] [CrossRef] [PubMed]
- Langmead, B.; Salzberg, S.L. Fast gapped-read alignment with Bowtie 2. Nat. Methods. 2012, 9, 357–359. [Google Scholar] [CrossRef] [PubMed]
- Li, B.; Dewey, C.N. RSEM: Accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinformatics 2011, 12, 323. [Google Scholar] [CrossRef]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef]
- Nawrocki, E.P.; Eddy, S.R. Infernal 1.1: 100-fold faster RNA homology searches. Bioinformatics. 2013, 29, 2933–2935. [Google Scholar] [CrossRef]
- Breakfield, N.W.; Corcoran, D.L.; Petricka, J.J.; Shen, J.; Saeseaw, J.; Rubiosomoza, I.; Weigel, D.; Ohler, U.; Benfey, P.N. High-resolution experimental and computational profiling of tissue-specific known and novel miRNAs in Arabidopsis. Genome Res. 2012, 22, 163–176. [Google Scholar] [CrossRef]
- Wang, L.K.; Feng, Z.X.; Wang, X.; Wang, X.W.; Zhang, X.G. DEGseq: An R package for identifying differentially expressed genes from RNA-seq data. Bioinformatics 2010, 26, 136–138. [Google Scholar] [CrossRef]
- Wu, H.J.; Ma, Y.K.; Chen, T.; Wang, M.; Wang, X.J. PsRobot: A web-based plant small RNA meta-analysis toolbox. Nucleic Acids Res. 2012, 40, W22–W28. [Google Scholar] [CrossRef]
- Fahlgren, N.; Carrington, J.C. miRNA target prediction in plants. Methods Mol. Biol. 2010, 592, 51–57. [Google Scholar] [PubMed]
- Chen, C.; Ridzon, D.A.; Broomer, A.J.; Zhou, Z.; Lee, D.H.; Nguyen, J.T.; Barbisin, M.; Xu, N.L.; Mahuvakar, V.R.; Andersen, M.R. Real-time quantification of microRNAs by stem-loop RT-PCR. Nucleic Acids Res. 2005, 33, e179. [Google Scholar] [CrossRef] [PubMed]
- Riddle, N.C.; Jiang, H.M.; An, L.L.; Doerge, R.W.; Birchler, J.A. Gene expression analysis at the intersection of ploidy and hybridity in maize. Theor. Appl. Genet. 2010, 120, 341–353. [Google Scholar] [CrossRef] [PubMed]
- Fernandez, C.; Monnier, Y.; Santonja, M.; Gallet, C.; Weston, L.A.; Prevosto, B.; Saunier, A.; Baldy, V.; Bousquet-Melou, A. The impact of competition and allelopathy on the trade-off between plant defense and growth in two contrasting tree species. Front. Plant Sci. 2016, 7, 594. [Google Scholar] [CrossRef]
- Fasano, C.; Diretto, G.; Aversano, R.; D’Agostino, N.; Di Matteo, A.; Frusciante, L.; Giuliano, G.; Carputo, D. Transcriptome and metabolome of synthetic Solanum autotetraploids reveal key genomic stress events following polyploidization. New Phytol. 2016, 210, 1382–1394. [Google Scholar] [CrossRef]
- Pradhan, S.K.; Gupta, R.C.; Goel, R.K. Differential content of secondary metabolites in diploid and tetraploid cytotypes of Siegesbeckia orientalis L. Nat. Prod. Res. 2018, 32, 2476–2482. [Google Scholar] [CrossRef]
- Xu, C.G.; Tang, T.X.; Chen, R.; Liang, C.H.; Liu, X.Y.; Wu, C.L.; Yang, Y.S.; Yang, D.P.; Wu, H. A comparative study of bioactive secondary metabolite production in diploid and tetraploid Echinacea purpurea (L.) Moench. Plant Cell Tiss. Org. 2014, 116, 323–332. [Google Scholar] [CrossRef]
- Yuan, Y.; Wang, B.; Zhang, S.; Tang, J.; Tu, C.; Hu, S.; Yong, J.W.H.; Chen, X. Enhanced allelopathy and competitive ability of invasive plant Solidago canadensis in its introduced range. J. Plant Ecol. 2013, 6, 253–263. [Google Scholar] [CrossRef]
- Zhang, S.; Zhu, W.; Wang, B.; Tang, J.; Chen, X. Secondary metabolites from the invasive Solidago canadensis L. accumulation in soil and contribution to inhibition of soil pathogen Pythium ultimum. Appl. Soil Ecol. 2011, 48, 280–286. [Google Scholar] [CrossRef]
- El Ayeb-Zakhama, A.; Sakka-Rouis, L.; Bergaoui, A.; Flamini, G.; Ben Jannet, H.; Harzallah-Skhiri, F. Chemical composition and allelopathic potential of essential oils from Tipuana tipu (Benth.) Kuntze cultivated in Tunisia. Chem. Biodivers. 2016, 13, 309–318. [Google Scholar] [CrossRef]
- Weston, L.A.; Mathesius, U. Flavonoids: Their structure, biosynthesis and role in the rhizosphere, including allelopathy. J. Chem. Ecol. 2013, 39, 283–297. [Google Scholar] [CrossRef] [PubMed]
- Mancini, E.; Arnold, N.A.; De Martino, L.; De Feo, V.; Formisano, C.; Rigano, D.; Senatore, F. Chemical composition and phytotoxic effects of essential oils of Salvia hierosolymitana Boiss. and Salvia multicaulis Vahl. var. simplicifolia Boiss. growing wild in Lebanon. Molecules 2009, 14, 4725–4736. [Google Scholar] [CrossRef] [PubMed]
- Jackson, S.; Chen, Z.J. Genomic and expression plasticity of polyploidy. Curr. Opin. Plant Biol. 2010, 13, 153–159. [Google Scholar]
- Bottani, S.; Zabet, N.R.; Wendel, J.F.; Veitia, R.A. Gene expression dominance in allopolyploids: Hypotheses and models. Trends Plant Sci. 2018, 23, 393–402. [Google Scholar] [CrossRef]
- Javadian, N.; Karimzadeh, G.; Sharifi, M.; Moieni, A.; Behmanesh, M. In vitro polyploidy induction: Changes in morphology, podophyllotoxin biosynthesis, and expression of the related genes in Linum album (Linaceae). Planta 2017, 245, 1165–1178. [Google Scholar] [CrossRef]
- Kim, J.I.; Zhang, X.; Pascuzzi, P.E.; Liu, C.J.; Chapple, C. Glucosinolate and phenylpropanoid biosynthesis are linked by proteasome-dependent degradation of PAL. New Phytol. 2020, 225, 154–168. [Google Scholar] [CrossRef]
- Tang, Y.H.; Liu, F.; Mao, K.Q.; Xing, H.C.; Chen, J.R.; Guo, Q.Q. Cloning and characterization of the key 4-coumarate CoA ligase genes in Boehmeria nivea. S. Afr. J. Bot. 2018, 116, 123–130. [Google Scholar] [CrossRef]
- Liu, W.; Xiao, Z.D.; Fan, C.; Jiang, N.H.; Meng, X.C.; Xiang, X. Cloning and characterization of a flavonol synthase gene from Litchi chinensis and its variation among Litchi cultivars with different fruit maturation periods. Front. Plant Sci. 2018, 9, 567. [Google Scholar] [CrossRef]
- Sun, Q.; Wang, G.H.; Zhang, X.; Zhang, X.R.; Qiao, P.; Long, L.; Yuan, Y.L.; Cai, Y.F. Genome-wide identification of the TIFY gene family in three cultivated Gossypium species and the expression of JAZ genes. Sci. Rep. 2017, 7, 42418. [Google Scholar] [CrossRef]
- Li, J.P.; Chen, F.J.; Li, Y.Q.; Li, P.C.; Wang, Y.Q.; Mi, G.H.; Yuan, L.X. ZmRAP2.7, an AP2 transcription factor, is involved in maize brace roots development. Front. Plant Sci. 2019, 10, 820. [Google Scholar] [CrossRef]
- Li, B.Z.; Fan, R.N.; Guo, S.Y.; Wang, P.T.; Zhu, X.H.; Fan, Y.T.; Chen, Y.X.; He, K.Y.; Kumar, A.; Shi, J.P.; et al. The Arabidopsis MYB transcription factor, MYB111 modulates salt responses by regulating flavonoid biosynthesis. Environ. Exp. Bot. 2019, 166, 103807. [Google Scholar] [CrossRef]
- Afrin, S.; Huang, J.J.; Luo, Z.Y. JA-mediated transcriptional regulation of secondary metabolism in medicinal plants. Sci. Bull 2015, 60, 1062–1072. [Google Scholar] [CrossRef]
- Li, C.; Zheng, L.L.; Wang, X.N.; Hu, Z.B.; Zheng, Y.; Chen, Q.H.; Hao, X.C.; Xiao, X.; Wang, X.B.; Wang, G.D.; et al. Comprehensive expression analysis of Arabidopsis GA2-oxidase genes and their functional insights. Plant Sci. 2019, 285, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.Y.; Li, N.; Yao, C.P.; Meng, S.S.; Song, C.P. Functional analysis of the ABA-responsive protein family in ABA and stress signal transduction in Arabidopsis. Chinese Sci. Bull. 2013, 58, 3721–3730. [Google Scholar] [CrossRef]
- Sun, L.R.; Wang, Y.B.; He, S.B.; Hao, F.S. Mechanisms for abscisic acid inhibition of primary root growth. Plant Signal. Behav. 2018, 13, e1500069. [Google Scholar] [CrossRef]
- Mou, W.; Li, D.; Luo, Z.; Mao, L.; Ying, T. Transcriptomic analysis reveals possible influences of ABA on secondary metabolism of pigments, flavonoids and antioxidants in tomato fruit during ripening. PLoS ONE 2015, 10, e0129598. [Google Scholar] [CrossRef]
- Han, S.A.; Fang, L.; Ren, X.J.; Wang, W.L.; Jiang, J. MPK6 controls H2O2-induced root elongation by mediating Ca2+ influx across the plasma membrane of root cells in Arabidopsis seedlings. New Phytol. 2015, 205, 695–706. [Google Scholar] [CrossRef]
- Li, K.; Yang, F.B.; Miao, Y.C.; Song, C.P. Abscisic acid signaling is involved in regulating the mitogen-activated protein kinase cascade module, AIK1-MKK5-MPK6. Plant Signal. Behav. 2017, 12, e1321188. [Google Scholar] [CrossRef]
- Li, K.; Yang, F.B.; Zhang, G.Z.; Song, S.F.; Li, Y.; Ren, D.T.; Miao, Y.C.; Song, C.P. AIK1, a mitogen-activated protein kinase, modulates abscisic acid responses through the MKK5-MPK6 kinase cascade. Plant Physiol. 2017, 173, 1391–1408. [Google Scholar] [CrossRef]
- Meyers, B.C.; Axtell, M.J. MicroRNAs in plants: Key findings from the early years. Plant Cell 2019, 31, 1206–1207. [Google Scholar] [CrossRef]
- Han, J.Y.; Kim, H.J.; Kwon, Y.S.; Choi, Y.E. The Cyt P450 enzyme CYP716A47 catalyzes the formation of protopanaxadiol from dammarenediol-II during ginsenoside biosynthesis in Panax ginseng. Plant Cell Physiol. 2011, 52, 2062–2073. [Google Scholar] [CrossRef] [PubMed]
- Han, J.Y.; Kim, M.J.; Ban, Y.W.; Hwang, H.S.; Choi, Y.E. The involvement of beta-amyrin 28-oxidase (CYP716A52v2) in oleanane-type ginsenoside biosynthesis in Panax ginseng. Plant Cell Physiol. 2013, 54, 2034–2046. [Google Scholar] [CrossRef] [PubMed]
- Udomsom, N.; Rai, A.; Suzuki, H.; Okuyama, J.; Imai, R.; Mori, T.; Nakabayashi, R.; Saito, K.; Yamazaki, M. Function of AP2/ERF transcription factors involved in the regulation of specialized metabolism in ophiorrhiza pumila revealed by transcriptomics and metabolomics. Front. Plant Sci. 2016, 7, 1861. [Google Scholar] [CrossRef] [PubMed]
- Solofoharivelo, M.C.; Ap, V.D.W.; Stephan, D.; Burger, J.T.; Murray, S.L. MicroRNAs in fruit trees: Discovery, diversity and future research directions. Plant Biol. 2014, 16, 856–865. [Google Scholar] [CrossRef]
- Henriksson, E.; Olsson, A.S.; Johannesson, H.; Johansson, H.; Hanson, J.; Engstrom, P.; Soderman, E. Homeodomain leucine zipper class I genes in Arabidopsis. Expression patterns and phylogenetic relationships. Plant physiol. 2005, 139, 509–518. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Classification | Metabolite Name | log2 Fold Change (Hexaploid/Diploid) | Padj-Value |
|---|---|---|---|
| Terpenoid | Casbene | 5.275391063 | 6.49 × 10−04 |
| Farnesal | 4.154837827 | 1.17 × 10−03 | |
| Germacrene D | 4.08702165 | 4.16 × 10−04 | |
| Baccatin III | 3.978134672 | 6.40 × 10−05 | |
| Nivalenol | 2.742280848 | 1.98 × 10−08 | |
| Cembrene | 2.610800783 | 3.32 × 10−03 | |
| Germacra-1(10),4,11(13)-trien-12-ol | 2.563360372 | 5.93 × 10−05 | |
| trans-Carveol | 2.255799893 | 1.33 × 10−02 | |
| trans-Farnesol | 2.236407545 | 1.28 × 10−11 | |
| trans-Isopiperitenol | 2.224502422 | 1.89 × 10−03 | |
| Myrcene | 2.199510457 | 4.49 × 10−09 | |
| Loganic acid | 2.107521873 | 3.51 × 10−06 | |
| Loganin | 2.097243456 | 6.65 × 10−06 | |
| Geranylgeraniol | 2.00541636 | 4.87 × 10−05 | |
| Deoxyloganin | 1.721160952 | 3.27 × 10−07 | |
| Germacradienol | 1.442421048 | 1.34 × 10−07 | |
| Gibberellin A12 aldehyde | −1.218344661 | 1.23 × 10−10 | |
| Gibberellin A51-catabolite | −1.257611873 | 5.60 × 10−05 | |
| Gibberellin A44 diacid | −1.707646412 | 1.44 × 10−10 | |
| Neoabietal | −1.779434393 | 1.75 × 10−04 | |
| Arabidiol | −1.857123891 | 2.21 × 10−09 | |
| Copal-8-ol diphosphate | −1.924119174 | 1.23 × 10−09 | |
| Aphidicolin | −1.947152337 | 3.92 × 10−16 | |
| Gibberellin A53 | −2.991652225 | 1.12 × 10−08 | |
| Gibberellin A14 | −3.154745064 | 3.50 × 10−07 | |
| cis-Abienol | −3.453551139 | 2.83 × 10−09 | |
| 5alpha-Acetoxytaxa-4(20),11(12)-dien-10beta-ol | −3.88691206 | 6.02 × 10−11 | |
| 5alpha-Acetoxy-10beta,14beta-dihydroxytaxadiene | −4.134902554 | 7.48 × 10−08 | |
| Phenylpropanoid | 4-Hydroxystyrene | 5.642964883 | 7.17 × 10−12 |
| Coniferyl acetate | 4.620635971 | 8.40 × 10−15 | |
| Chavicol | 3.886054817 | 1.89 × 10−10 | |
| Caffeoyl quinic acid | 3.2138012 | 9.27 × 10−06 | |
| Scopoletin | 2.505276504 | 1.57 × 10−12 | |
| Syringin | 2.135952387 | 6.27 × 10−08 | |
| Ferulaldehyde | 2.097477361 | 8.55 × 10−06 | |
| 4-Coumaroylshikimate | −1.37435324 | 7.53 × 10−08 | |
| Coumaryl acetate | −1.832764994 | 1.92 × 10−11 | |
| Coniferin | −2.093818248 | 9.62 × 10−12 | |
| Flavonoid | Quercetin 3-beta-D-sophoroside | 2.889187046 | 1.49 × 10−07 |
| Glyceollin | 2.745732531 | 3.47 × 10−07 | |
| Laricitrin | 2.477143429 | 7.15 × 10−06 | |
| Quercetin 3-(2G-xylosylrutinoside) | 2.476832075 | 2.18 × 10−03 | |
| Pinobanksin 3-O-acetate | 2.308305371 | 3.46 × 10−13 | |
| Maackiain-3-O-glucosyl-6″-O-malonate | 2.286077494 | 2.65 × 10−04 | |
| 2-Hydroxy-2,3-dihydrogenistein | 2.247817936 | 4.06 × 10−06 | |
| Leucoanthocyanidol | 1.976381763 | 1.20 × 10−04 | |
| Formononetin | 1.870944123 | 1.46 × 10−05 | |
| 8-C-Glucosylnaringenin | 1.53852511 | 1.01 × 10−04 | |
| Glyceocarpin | 1.404825044 | 1.19 × 10−05 | |
| 3′,4′,5-Trihydroxy-3,7-dimethoxyflavone | 1.470845226 | 4.36 × 10−06 | |
| Myricetin | 1.145537801 | 5.03 × 10−05 | |
| 3-O-Methylquercetin | −1.216396205 | 1.37 × 10−03 | |
| Vitexin | −1.3550808 | 1.35 × 10−05 | |
| Rhoifolin | −1.401280595 | 6.82 × 10−07 | |
| Malonyldaidzin | -1.699702739 | 3.71 × 10−11 | |
| Sophoraflavonoloside | -1.720462228 | 5.92 × 10−05 | |
| Hesperetin 7-O-glucoside | -1.779216101 | 1.46 × 10−08 |
| Cytotype | Known miRNA Number | Target Gene Number of Known miRNA | Novel miRNA Number | Target Gene Number of Novel miRNA |
|---|---|---|---|---|
| Diploid | 53 | 930 | 136 | 815 |
| Hexaploid | 54 | 933 | 135 | 820 |
| Total | 54 | 933 | 140 | 827 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wu, M.; Ge, Y.; Xu, C.; Wang, J. Metabolome and Transcriptome Analysis of Hexaploid Solidago canadensis Roots Reveals its Invasive Capacity Related to Polyploidy. Genes 2020, 11, 187. https://doi.org/10.3390/genes11020187
Wu M, Ge Y, Xu C, Wang J. Metabolome and Transcriptome Analysis of Hexaploid Solidago canadensis Roots Reveals its Invasive Capacity Related to Polyploidy. Genes. 2020; 11(2):187. https://doi.org/10.3390/genes11020187
Chicago/Turabian StyleWu, Miao, Yimeng Ge, Chanchan Xu, and Jianbo Wang. 2020. "Metabolome and Transcriptome Analysis of Hexaploid Solidago canadensis Roots Reveals its Invasive Capacity Related to Polyploidy" Genes 11, no. 2: 187. https://doi.org/10.3390/genes11020187
APA StyleWu, M., Ge, Y., Xu, C., & Wang, J. (2020). Metabolome and Transcriptome Analysis of Hexaploid Solidago canadensis Roots Reveals its Invasive Capacity Related to Polyploidy. Genes, 11(2), 187. https://doi.org/10.3390/genes11020187