Transcriptional Regulation of CD40 Expression by 4 Ribosomal Proteins via a Functional SNP on a Disease-Associated CD40 Locus

 ,
,
Abstract
1. Introduction
2. Materials and Methods
2.1. Cells and Culture
2.2. Isolation of FLS
2.3. Primers and Antibodies
2.4. Western Blot
2.5. AIDP-Wb
2.6. RNA Isolation
2.7. RNAi Knockdown
2.8. ChIP Assay
2.9. Luciferase Report Assay
2.10. Flow Cytometry
2.11. Scratch-Wound Assay
2.12. Statistical Analysis
3. Results
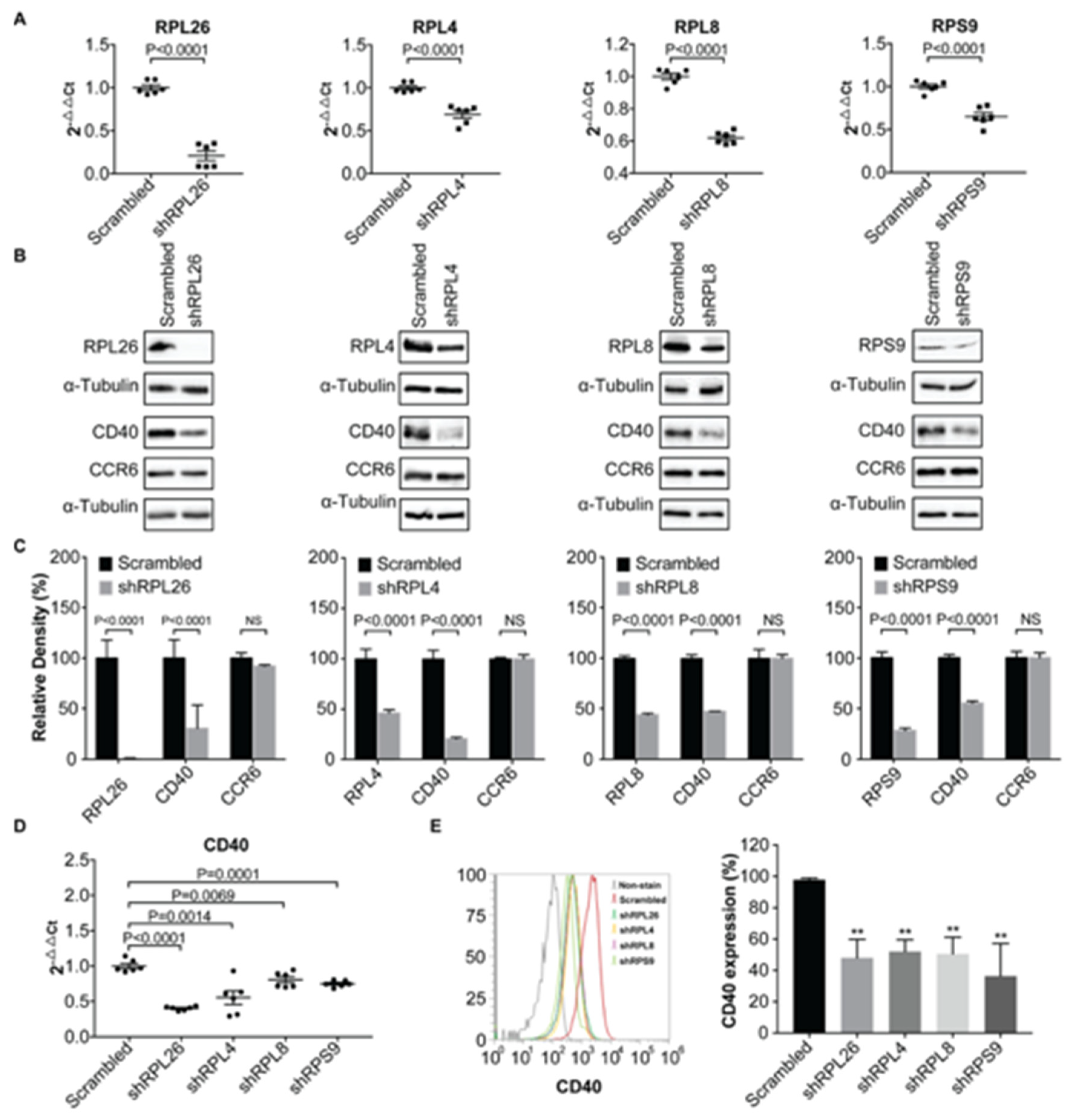
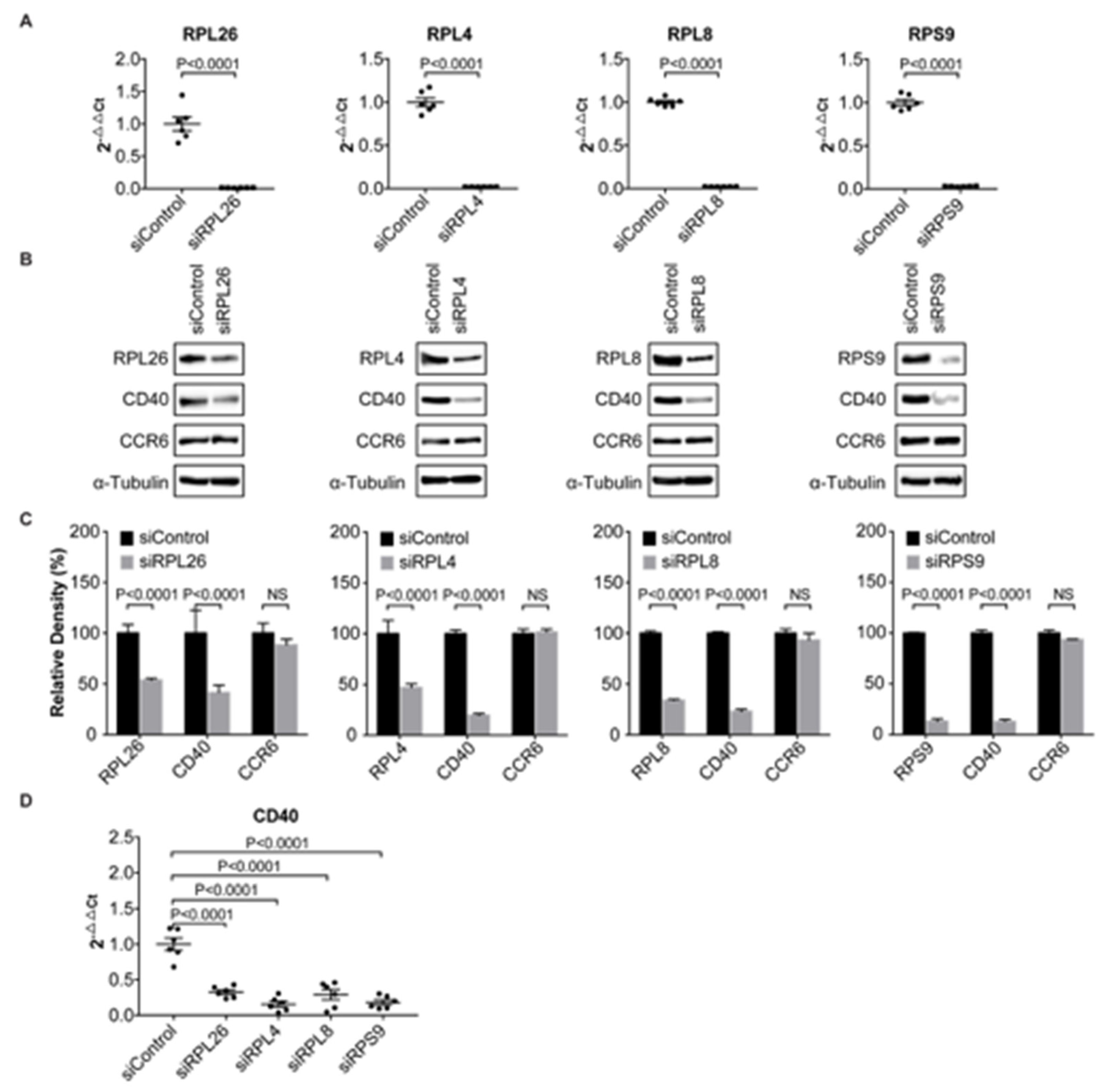
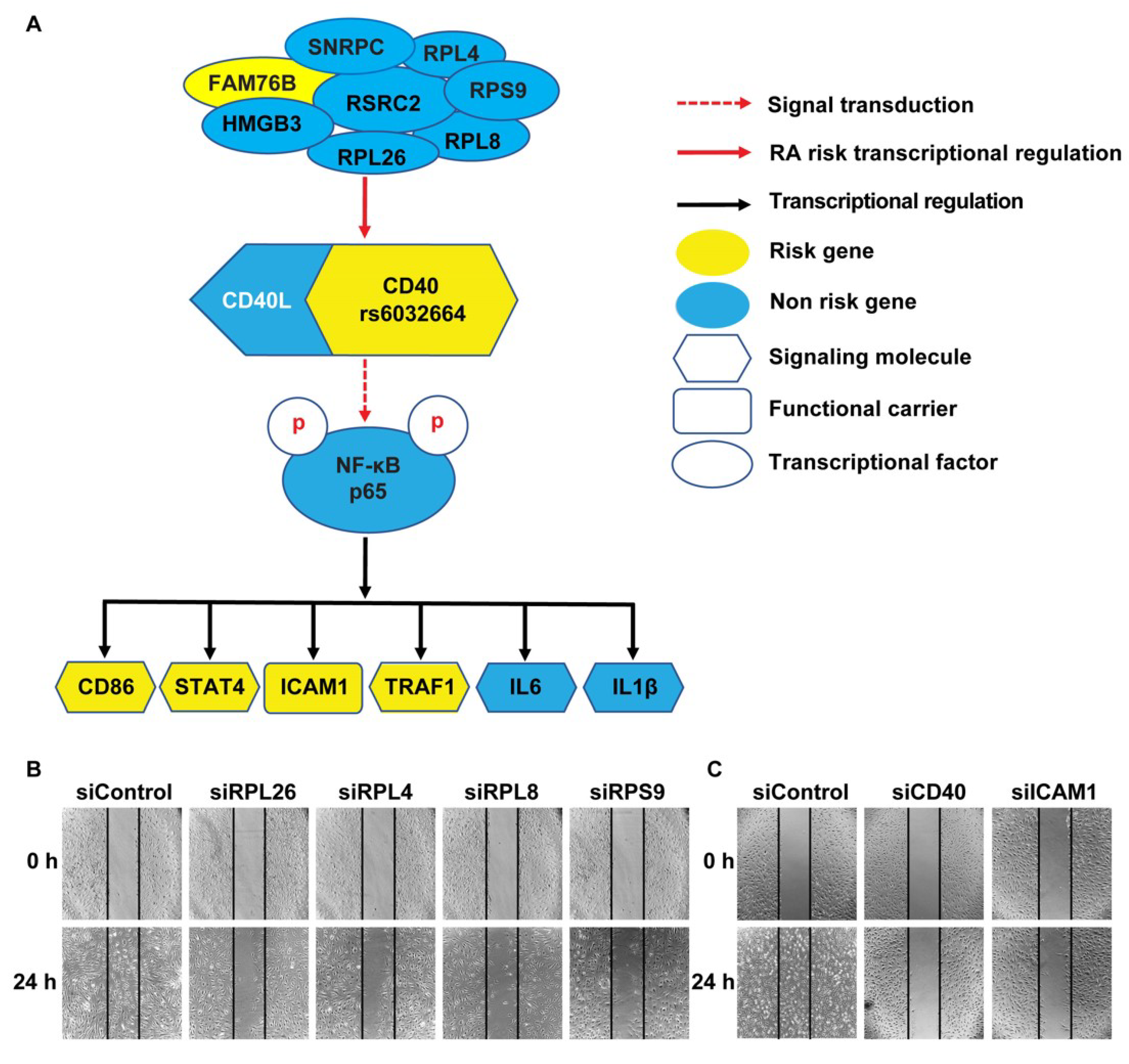
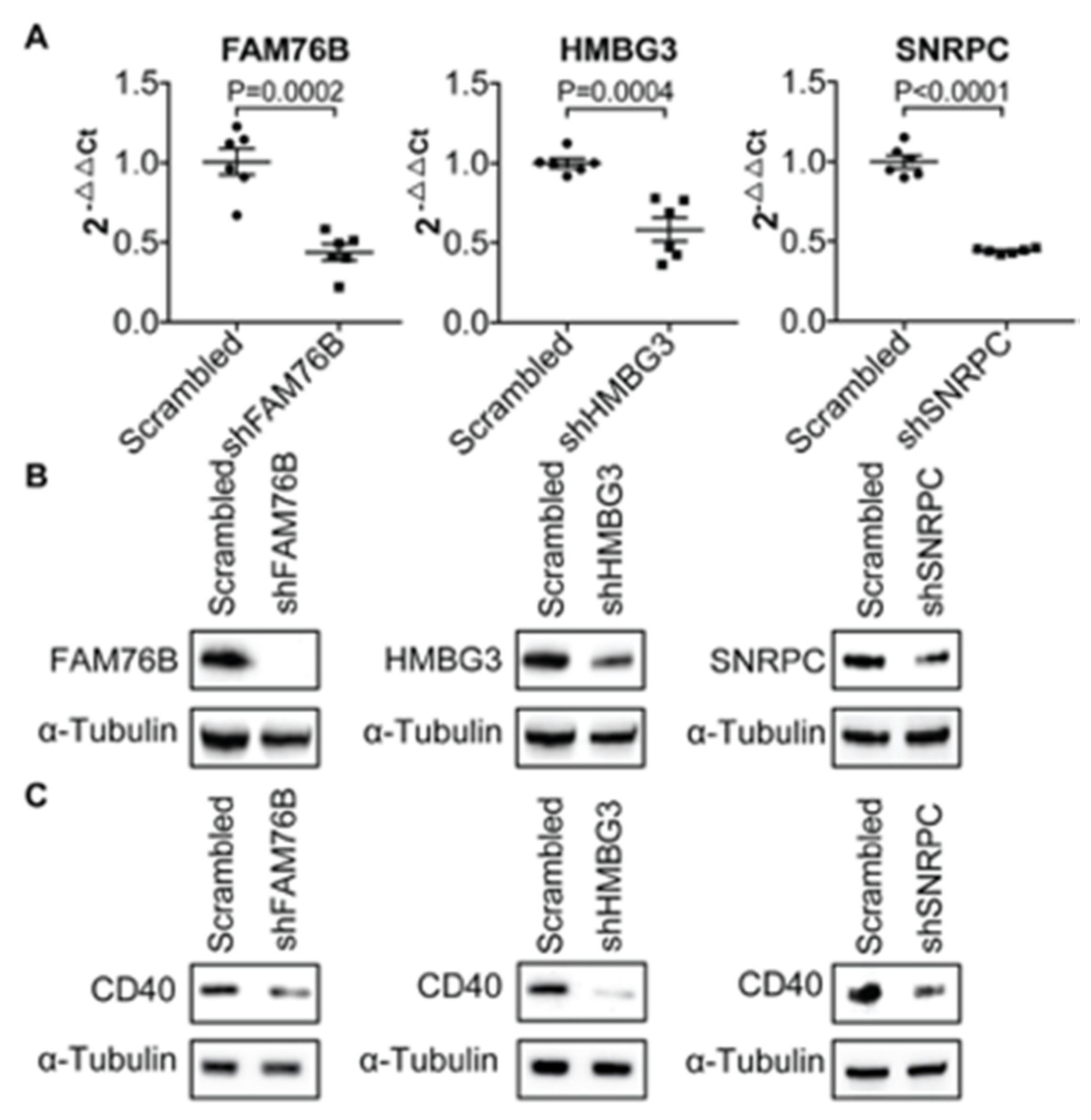
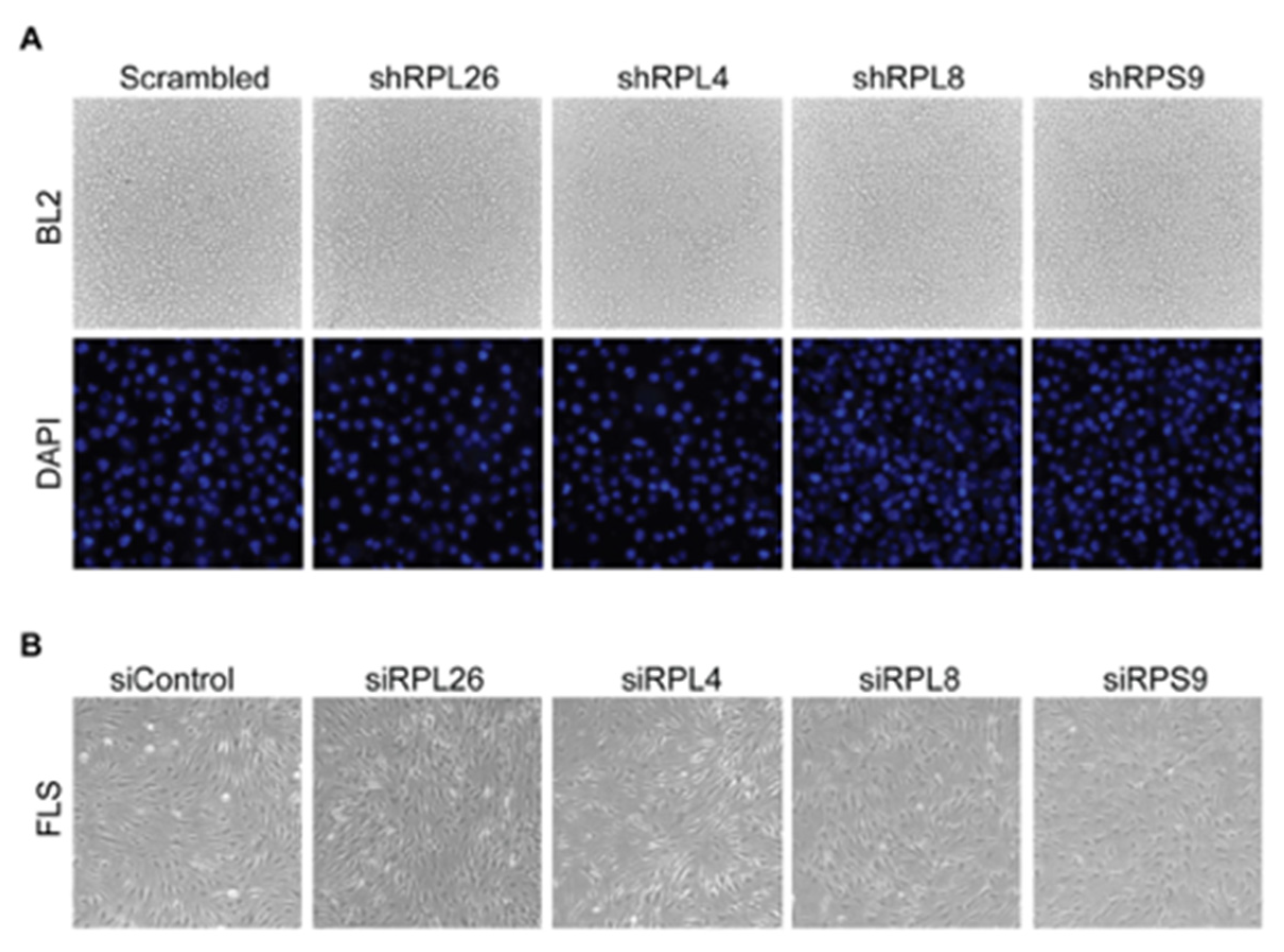
3.1. Transcriptional Regulation of CD40 Expression by a Protein Complex Containing RPL26, RPL4, RPL8, RPS9, in Human B Cells and FLS
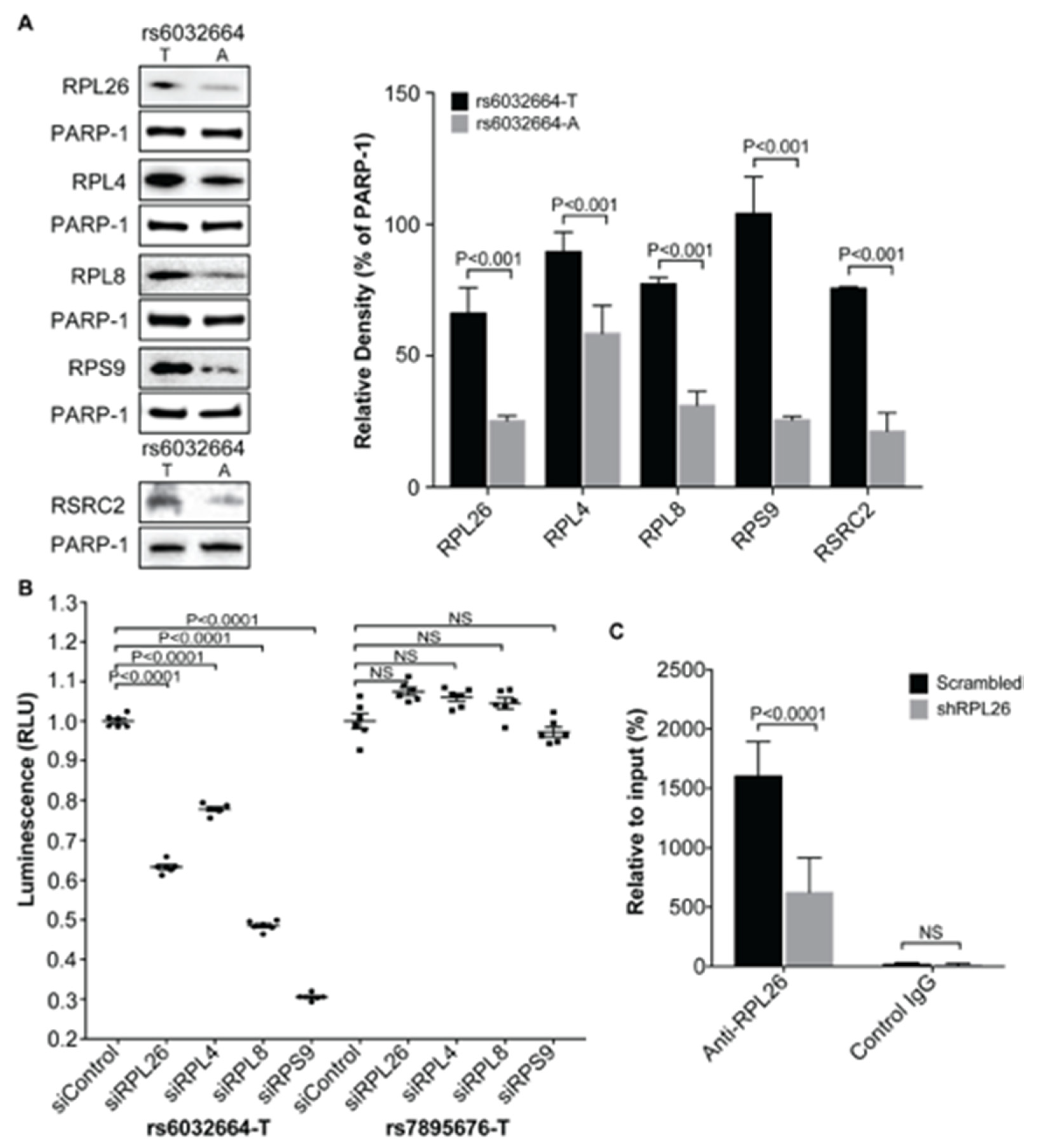
3.2. Demonstration of the Specific Binding of Ribosomal Protein RPL26, RPL4, RPL8 and RPS9 to fSNP rs6032664
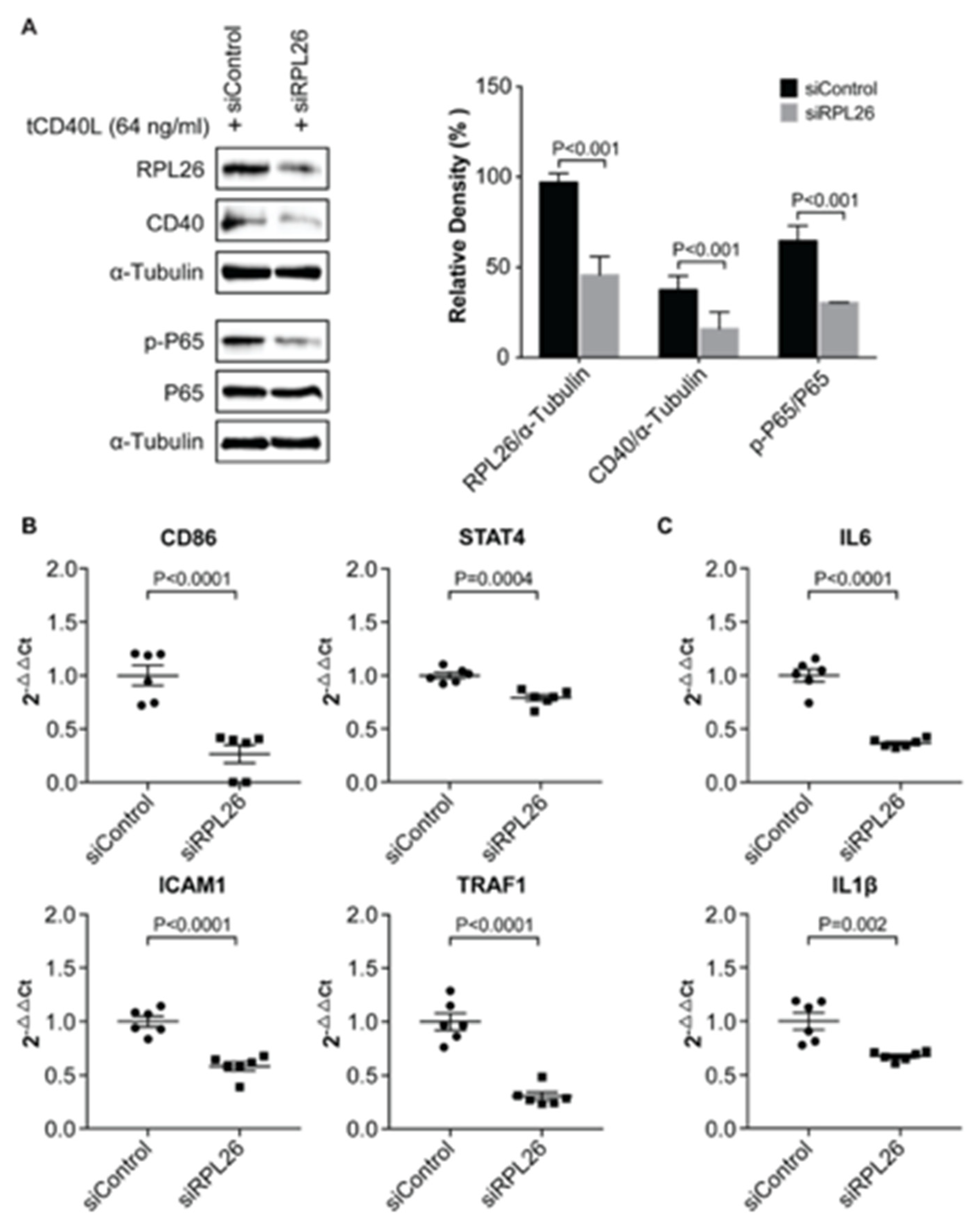
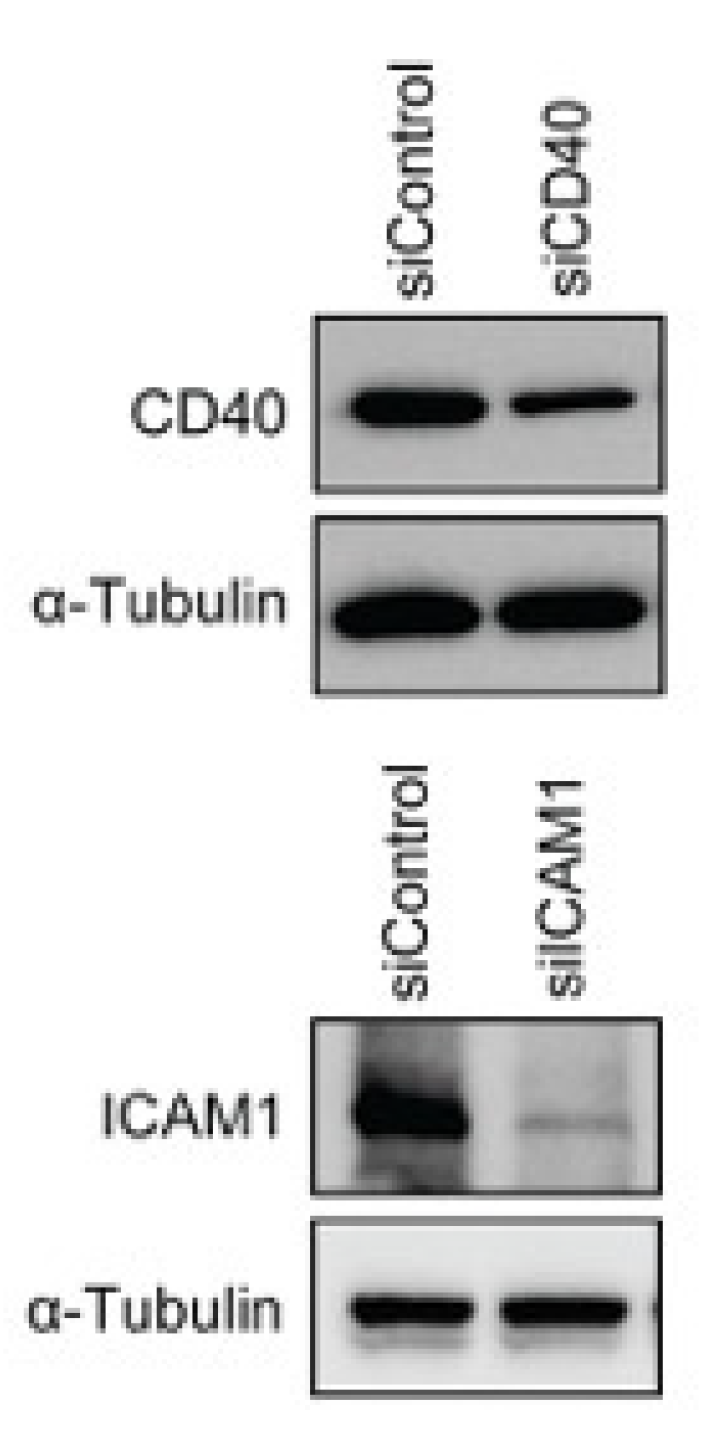
3.3. A Disease-Associated TRN Linked by CD40-Induced NF-κB Signaling Pathway
4. Discussion
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Data Availability
Abbreviations
| FREP-MS | Flanking Restriction Enhanced DNA pulldown-Mass Spectrometry |
| TRN | Transcriptional Regulation Network |
| AIDP-Wb | Allele Imbalanced DNA Pulldown-Western blot |
Appendix A




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Name | Sequence (5’→3’) |
|---|---|
| RPL4-F | GGC TAC AAG AAG ACC AAG GAA |
| RPL4-R | CTC ATT CGC TGA GAG GCA TAG |
| RPL26-F | CCC ATC CGA AAG GAT GAT GAA |
| RPL26-R | CTG CAC CCG TTC AAT GTA GAT A |
| RPL8-F | CAC AACCCT GAG ACC AAG AA |
| RPL8-R | CTC TGT TGG CTG AGG AGA TTA C |
| RPS9-F | GTG AAC ATC CCG TCC TTC AT |
| RPS9-R | CCC TTC TTG GCA TTC TTC CT |
| SNRPC-F | TCA CCC ATG ACT CTC CAT CT |
| SNRPC-R | CCT GCT CTT CCA TCC ATT TCT |
| FAM76B-F | GAC TGT GGA AAC AGA GCC ATA |
| FAM76B-R | CCC ACT ATC TGC TGA CTG ATT T |
| HMGB3-F | CGA TGT CCG GGA AAG AGA AA |
| HMGB3-R | CTC CCT TAG CTG GTC CAT AAT C |
| P53-F | CCC CTC CTG GCC CCT GTC ATC TTC |
| P53-R | GCA GCG CCT CAC AAC CTC CGT CAT |
| P73-F | CAG ACA GCA CCT ACT TCG AC |
| P73-R | CTG CTC ATC TGG TCC ATG G |
| ICAM1-F | AGCGGCTGACGTGTGCAGTAAT |
| ICAM1-R | TCTGAGACCTCTGGCTTCGTCA |
| STAT4-F | CAGTGAAAGCCATCTCGGAGGA |
| STAT4-R | TGTAGTCTCGCAGGATGTCAGC |
| TRAF1-F | CGATGGCACTTTCCTGTGGAAG |
| TRAF1-R | TACAGCCGCAGGCACAACTTGT |
| CD86-F | CCATCAGCTTGTCTGTTTCATTCC |
| CD86-R | GCTGTAATCCAAGGAATGTGGTC |
| IL6-F | GCAGAAAACAACCTGAACCTT |
| IL6-R | ACCTCAAACTCCAAAAGACCA |
| IL1β-F | ACAGATGAAGTGCTCCTTCCA |
| IL1β-R | GTCGGAGATTCGTAGCTGGAT |
| rs6032664-ChIP-F1 | CGATTACTCACTGGGCTATGG |
| rs6032664-ChIP-R1 | CCAGCTAATTCACTGGGCAG |
| rs6032664-ChIP-F2 | GAAGGTCCCTATAGGCAAGG |
| rs6032664-ChIP-R2 | ACGTAAGAACCAGAGAGGGC |
| rs6032664-AIDP-F1 | Bio-CAGAATTGTATCTCTTGATCCTGATTCTCAA |
| rs6032664-AIDP-R1 | TTGAGAATCAGGATCAAGAGATACAATTCTG |
| rs6032664-AIDP-F2 | Bio-CAGAATTGTATCTCTAGATCCTGATTCTCAA |
| rs6032664-AIDP-R2 | TTGAGAATCAGGATCTAGAGATACAATTCTG |
| Name | Clone ID | Target Sequence |
|---|---|---|
| RPL26 | TRCN0000117402 | CCAGGTTTACAGGAAGAAATA |
| RPL4 | TRCN0000117637 | CCCTGGAATTACTCTGCTTAA |
| RPL8 | TRCN0000117468 | CCGAATTGACAAACCCATCTT |
| RPS9 | TRCN0000074794 | GCTGAAGCTGATCGGCGAGTA |
| SNRPC | TRCN0000072288 | GAGGCCTTATTGTATCGGTTT |
| FAM76B | TRCN0000128244 | GCTTTGTAAATTCCAAGGCTA |
| HMGB3 | TRCN0000018520 | GCAAAGCTGAAGGAGAAGTAT |
References
- Smolen, J.S.; Aletaha, D.; Barton, A.; Burmester, G.R.; Emery, P.; Firestein, G.S.; Kavanaugh, A.; McInnes, I.B.; Solomon, D.H.; Strand, V.; et al. Rheumatoid arthritis. Nat. Rev. Dis. Primers 2018, 4, 18001. [Google Scholar] [CrossRef] [PubMed]
- Raychaudhuri, S.; Remmers, E.F.; Lee, A.T.; Hackett, R.; Guiducci, C.; Burtt, N.P.; Gianniny, L.; Korman, B.D.; Padyukov, L.; Kurreeman, F.A.; et al. Common variants at CD40 and other loci confer risk of rheumatoid arthritis. Nat. Genet. 2008, 40, 1216–1223. [Google Scholar] [CrossRef] [PubMed]
- Stahl, E.A.; Raychaudhuri, S.; Remmers, E.F.; Xie, G.; Eyre, S.; Thomson, B.P.; Li, Y.; Kurreeman, F.A.; Zhernakova, A.; Hinks, A.; et al. Genome-wide association study meta-analysis identifies seven new rheumatoid arthritis risk loci. Nat. Genet. 2010, 42, 508–514. [Google Scholar] [CrossRef] [PubMed]
- Okada, Y.; Wu, D.; Trynka, G.; Raj, T.; Terao, C.; Ikari, K.; Kochi, Y.; Ohmura, K.; Suzuki, A.; Yoshida, S.; et al. Genetics of rheumatoid arthritis contributes to biology and drug discovery. Nature 2014, 506, 376–381. [Google Scholar] [CrossRef]
- Cheng, T.; Wang, M.; Chen, L.; Guo, Y.; Chen, Z.; Wu, J. Increased expression of CD40/TRAF1 and activation of nuclear factor-kappakappaB-dependent proinflammatory gene expression in collagen-induced arthritis. Scand. J. Rheumatol. 2018, 47, 455–460. [Google Scholar] [CrossRef]
- Schonbeck, U.; Libby, P. The CD40/CD154 receptor/ligand dyad. Cell Mol. Life Sci. 2001, 58, 4–43. [Google Scholar]
- Chatzigeorgiou, A.; Lyberi, M.; Chatzilymperis, G.; Nezos, A.; Kamper, E. CD40/CD40L signaling and its implication in health and disease. Biofactors 2009, 35, 474–483. [Google Scholar] [CrossRef]
- Luo, W.; Weisel, F.; Shlomchik, M.J. B Cell Receptor and CD40 Signaling Are Rewired for Synergistic Induction of the c-Myc Transcription Factor in Germinal Center B Cells. Immunity 2018, 48, 313–326.e315. [Google Scholar] [CrossRef]
- Li, G.; Martinez-Bonet, M.; Wu, D.; Yang, Y.; Cui, J.; Nguyen, H.N.; Cunin, P.; Levescot, A.; Bai, M.; Westra, H.J.; et al. High-throughput identification of noncoding functional SNPs via type IIS enzyme restriction. Nat. Genet. 2018, 50, 1180–1188. [Google Scholar] [CrossRef]
- Peters, A.L.; Stunz, L.L.; Bishop, G.A. CD40 and autoimmunity: The dark side of a great activator. Semin. Immunol. 2009, 21, 293–300. [Google Scholar] [CrossRef]
- Criswell, L.A. Gene discovery in rheumatoid arthritis highlights the CD40/NF-kappaB signaling pathway in disease pathogenesis. Immunol. Rev. 2010, 233, 55–61. [Google Scholar] [CrossRef] [PubMed]
- Yazdany, J.; Davis, J. The role of CD40 ligand in systemic lupus erythematosus. Lupus 2004, 13, 377–380. [Google Scholar] [CrossRef] [PubMed]
- Zheng, X.; Suzuki, M.; Zhang, X.; Ichim, T.E.; Zhu, F.; Ling, H.; Shunnar, A.; Wang, M.H.; Garcia, B.; Inman, R.D.; et al. RNAi-mediated CD40-CD154 interruption promotes tolerance in autoimmune arthritis. Arthritis Res. 2010, 12, R13. [Google Scholar] [CrossRef] [PubMed]
- Vaitaitis, G.M.; Olmstead, M.H.; Waid, D.M.; Carter, J.R.; Wagner, D.H., Jr. A CD40-targeted peptide controls and reverses type 1 diabetes in NOD mice. Diabetologia 2014, 57, 2366–2373. [Google Scholar] [CrossRef] [PubMed]
- Grammer, A.C.; Slota, R.; Fischer, R.; Gur, H.; Girschick, H.; Yarboro, C.; Illei, G.G.; Lipsky, P.E. Abnormal germinal center reactions in systemic lupus erythematosus demonstrated by blockade of CD154-CD40 interactions. J. Clin. Investig. 2003, 112, 1506–1520. [Google Scholar] [CrossRef] [PubMed]
- Kalunian, K.C.; Davis, J.C., Jr.; Merrill, J.T.; Totoritis, M.C.; Wofsy, D.; Group, I.-L.S. Treatment of systemic lupus erythematosus by inhibition of T cell costimulation with anti-CD154: A randomized, double-blind, placebo-controlled trial. Arthritis Rheum. 2002, 46, 3251–3258. [Google Scholar] [CrossRef] [PubMed]
- Davis, J.C., Jr.; Totoritis, M.C.; Rosenberg, J.; Sklenar, T.A.; Wofsy, D. Phase I clinical trial of a monoclonal antibody against CD40-ligand (IDEC-131) in patients with systemic lupus erythematosus. J. Rheumatol. 2001, 28, 95–101. [Google Scholar]
- Huang, W.; Sinha, J.; Newman, J.; Reddy, B.; Budhai, L.; Furie, R.; Vaishnaw, A.; Davidson, A. The effect of anti-CD40 ligand antibody on B cells in human systemic lupus erythematosus. Arthritis Rheum. 2002, 46, 1554–1562. [Google Scholar] [CrossRef]
- Gerritse, K.; Laman, J.D.; Noelle, R.J.; Aruffo, A.; Ledbetter, J.A.; Boersma, W.J.; Claassen, E. CD40-CD40 ligand interactions in experimental allergic encephalomyelitis and multiple sclerosis. Proc. Natl. Acad. Sci. USA 1996, 93, 2499–2504. [Google Scholar] [CrossRef]
- Early, G.S.; Zhao, W.; Burns, C.M. Anti-CD40 ligand antibody treatment prevents the development of lupus-like nephritis in a subset of New Zealand black × New Zealand white mice. Response correlates with the absence of an anti-antibody response. J. Immunol. 1996, 157, 3159–3164. [Google Scholar]
- Kalled, S.L.; Cutler, A.H.; Datta, S.K.; Thomas, D.W. Anti-CD40 ligand antibody treatment of SNF1 mice with established nephritis: Preservation of kidney function. J. Immunol. 1998, 160, 2158–2165. [Google Scholar] [PubMed]
- Wang, X.; Huang, W.; Schiffer, L.E.; Mihara, M.; Akkerman, A.; Hiromatsu, K.; Davidson, A. Effects of anti-CD154 treatment on B cells in murine systemic lupus erythematosus. Arthritis Rheum. 2003, 48, 495–506. [Google Scholar] [CrossRef] [PubMed]
- Li, G.; Diogo, D.; Wu, D.; Spoonamore, J.; Dancik, V.; Franke, L.; Kurreeman, F.; Rossin, E.J.; Duclos, G.; Hartland, C.; et al. Human genetics in rheumatoid arthritis guides a high-throughput drug screen of the CD40 signaling pathway. PLoS Genet. 2013, 9, e1003487. [Google Scholar] [CrossRef] [PubMed]
- Margolles-Clark, E.; Kenyon, N.S.; Ricordi, C.; Buchwald, P. Effective and specific inhibition of the CD40-CD154 costimulatory interaction by a naphthalenesulphonic acid derivative. Chem. Biol. Drug Des. 2010, 76, 305–313. [Google Scholar] [CrossRef] [PubMed]
- Zarzycka, B.; Seijkens, T.; Nabuurs, S.B.; Ritschel, T.; Grommes, J.; Soehnlein, O.; Schrijver, R.; van Tiel, C.M.; Hackeng, T.M.; Weber, C.; et al. Discovery of small molecule CD40-TRAF6 inhibitors. J. Chem. Inf. Model. 2015, 55, 294–307. [Google Scholar] [CrossRef]
- Zhou, X.; Liao, W.J.; Liao, J.M.; Liao, P.; Lu, H. Ribosomal proteins: Functions beyond the ribosome. J. Mol. Cell Biol. 2015, 7, 92–104. [Google Scholar] [CrossRef]
- Zhao, Y.; Wu, D.; Jiang, D.; Zhang, X.; Wu, T.; Cui, J.; Qian, M.; Zhao, J.; Oesterreich, S.; Sun, W.; et al. A sequential methodology for the rapid identification and characterization of breast cancer-associated functional SNPs. Nat. Commun. 2020, 11, 3340. [Google Scholar] [CrossRef]
- Alamanos, Y.; Voulgari, P.V.; Drosos, A.A. Incidence and prevalence of rheumatoid arthritis, based on the 1987 American College of Rheumatology criteria: A systematic review. Semin. Arthritis Rheum. 2006, 36, 182–188. [Google Scholar] [CrossRef]
- Kiener, H.P.; Lee, D.M.; Agarwal, S.K.; Brenner, M.B. Cadherin-11 induces rheumatoid arthritis fibroblast-like synoviocytes to form lining layers in vitro. Am. J. Pathol. 2006, 168, 1486–1499. [Google Scholar] [CrossRef]
- Noss, E.H.; Nguyen, H.N.; Chang, S.K.; Watts, G.F.; Brenner, M.B. Genetic polymorphism directs IL-6 expression in fibroblasts but not selected other cell types. Proc. Natl. Acad. Sci. USA 2015, 112, 14948–14953. [Google Scholar] [CrossRef]
- Kotzin, B.L. The role of B cells in the pathogenesis of rheumatoid arthritis. J. Rheumatol. Suppl. 2005, 73, 14–18, 29–30. [Google Scholar] [PubMed]
- Masoumi, M.; Bashiri, H.; Khorramdelazad, H.; Barzaman, K.; Hashemi, N.; Sereshki, H.A.; Sahebkar, A.; Karami, J. Destructive Roles of Fibroblast-like Synoviocytes in Chronic Inflammation and Joint Damage in Rheumatoid Arthritis. Inflammation 2020. [Google Scholar] [CrossRef] [PubMed]
- Scheinman, R. NF-kappaB and Rheumatoid Arthritis: Will Understanding Genetic Risk Lead to a Therapeutic Reward? Immunopathol. Dis. Ther. 2013, 4, 93–110. [Google Scholar] [CrossRef] [PubMed]
- Noort, A.R.; Tak, P.P.; Tas, S.W. Non-canonical NF-kappaB signaling in rheumatoid arthritis: Dr Jekyll and Mr Hyde? Arthritis Res. 2015, 17, 15. [Google Scholar] [CrossRef] [PubMed]
- Han, H.; Cho, J.W.; Lee, S.; Yun, A.; Kim, H.; Bae, D.; Yang, S.; Kim, C.Y.; Lee, M.; Kim, E.; et al. TRRUST v2: An expanded reference database of human and mouse transcriptional regulatory interactions. Nucleic Acids Res. 2018, 46, D380–D386. [Google Scholar] [CrossRef] [PubMed]
- Legrand-Poels, S.; Schoonbroodt, S.; Piette, J. Regulation of interleukin-6 gene expression by pro-inflammatory cytokines in a colon cancer cell line. Biochem. J. 2000, 349 Pt 3, 765–773. [Google Scholar] [CrossRef]
- Chandrasekar, B.; Vemula, K.; Surabhi, R.M.; Li-Weber, M.; Owen-Schaub, L.B.; Jensen, L.E.; Mummidi, S. Activation of intrinsic and extrinsic proapoptotic signaling pathways in interleukin-18-mediated human cardiac endothelial cell death. J. Biol. Chem. 2004, 279, 20221–20233. [Google Scholar] [CrossRef]
- Lyck, R.; Enzmann, G. The physiological roles of ICAM-1 and ICAM-2 in neutrophil migration into tissues. Curr. Opin. Hematol. 2015, 22, 53–59. [Google Scholar] [CrossRef]
- Bartok, B.; Hammaker, D.; Firestein, G.S. Phosphoinositide 3-kinase delta regulates migration and invasion of synoviocytes in rheumatoid arthritis. J. Immunol. 2014, 192, 2063–2070. [Google Scholar] [CrossRef]
- Martinotti, S.; Ranzato, E. Scratch Wound Healing Assay. Methods Mol. Biol. 2019, 2109, 225–229. [Google Scholar] [CrossRef]
- Babiano, R.; Gamalinda, M.; Woolford, J.L., Jr.; de la Cruz, J. Saccharomyces cerevisiae ribosomal protein L26 is not essential for ribosome assembly and function. Mol. Cell Biol. 2012, 32, 3228–3241. [Google Scholar] [CrossRef] [PubMed]
- Yildirim, N.; Aktas, M.E.; Ozcan, S.N.; Akbas, E.; Ay, A. Differential transcriptional regulation by alternatively designed mechanisms: A mathematical modeling approach. Silico Biol. 2017, 12, 95–127. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.; Umair, Z.; Kumar, V.; Lee, U.; Choi, S.C.; Kim, J. Ventx1.1 competes with a transcriptional activator Xcad2 to regulate negatively its own expression. BMB Rep. 2019, 52, 403–408. [Google Scholar] [CrossRef] [PubMed]
- Blais, A.; Dynlacht, B.D. Constructing transcriptional regulatory networks. Genes Dev. 2005, 19, 1499–1511. [Google Scholar] [CrossRef] [PubMed]
- Ament, S.A.; Pearl, J.R.; Cantle, J.P.; Bragg, R.M.; Skene, P.J.; Coffey, S.R.; Bergey, D.E.; Wheeler, V.C.; MacDonald, M.E.; Baliga, N.S.; et al. Transcriptional regulatory networks underlying gene expression changes in Huntington’s disease. Mol. Syst. Biol. 2018, 14, e7435. [Google Scholar] [CrossRef] [PubMed]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zou, M.; Zhang, X.; Jiang, D.; Zhao, Y.; Wu, T.; Gong, Q.; Su, H.; Wu, D.; Moreland, L.; Li, G. Transcriptional Regulation of CD40 Expression by 4 Ribosomal Proteins via a Functional SNP on a Disease-Associated CD40 Locus. Genes 2020, 11, 1526. https://doi.org/10.3390/genes11121526
Zou M, Zhang X, Jiang D, Zhao Y, Wu T, Gong Q, Su H, Wu D, Moreland L, Li G. Transcriptional Regulation of CD40 Expression by 4 Ribosomal Proteins via a Functional SNP on a Disease-Associated CD40 Locus. Genes. 2020; 11(12):1526. https://doi.org/10.3390/genes11121526
Chicago/Turabian StyleZou, Meijuan, Xiaoyu Zhang, Danli Jiang, Yihan Zhao, Ting Wu, Qiaoke Gong, Hang Su, Di Wu, Larry Moreland, and Gang Li. 2020. "Transcriptional Regulation of CD40 Expression by 4 Ribosomal Proteins via a Functional SNP on a Disease-Associated CD40 Locus" Genes 11, no. 12: 1526. https://doi.org/10.3390/genes11121526
APA StyleZou, M., Zhang, X., Jiang, D., Zhao, Y., Wu, T., Gong, Q., Su, H., Wu, D., Moreland, L., & Li, G. (2020). Transcriptional Regulation of CD40 Expression by 4 Ribosomal Proteins via a Functional SNP on a Disease-Associated CD40 Locus. Genes, 11(12), 1526. https://doi.org/10.3390/genes11121526