Tensor-Decomposition-Based Unsupervised Feature Extraction Applied to Prostate Cancer Multiomics Data



Abstract
1. Introduction
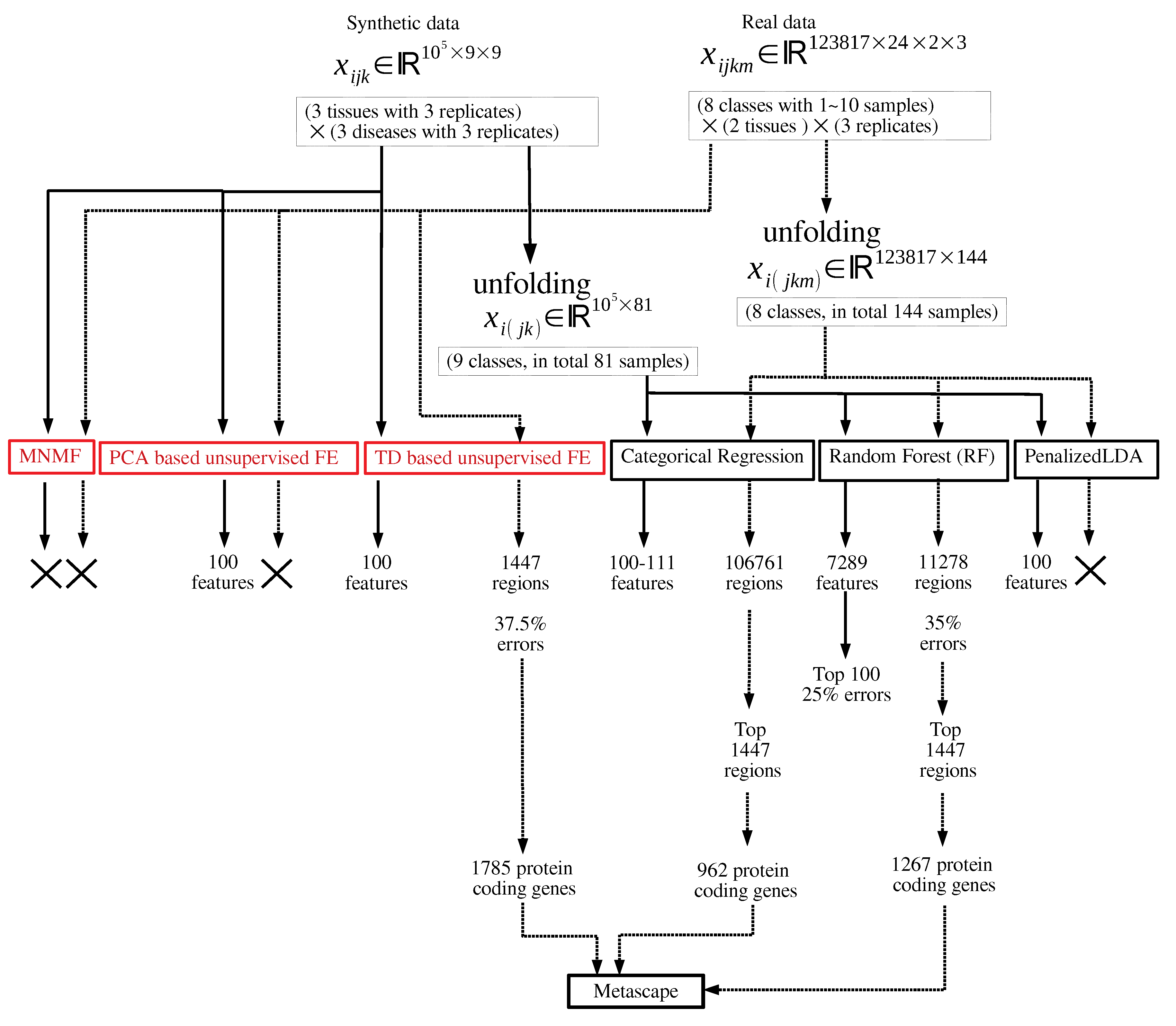
2. Materials and Methods
2.1. Synthetic Data
2.2. Real Dataset
2.3. Categorical Regression
2.4. RF
2.5. PenalizeLDA
2.6. TD-Based Unsupervised FE
2.7. MNMF
2.8. PCA Based Unsupervised FE
3. Results
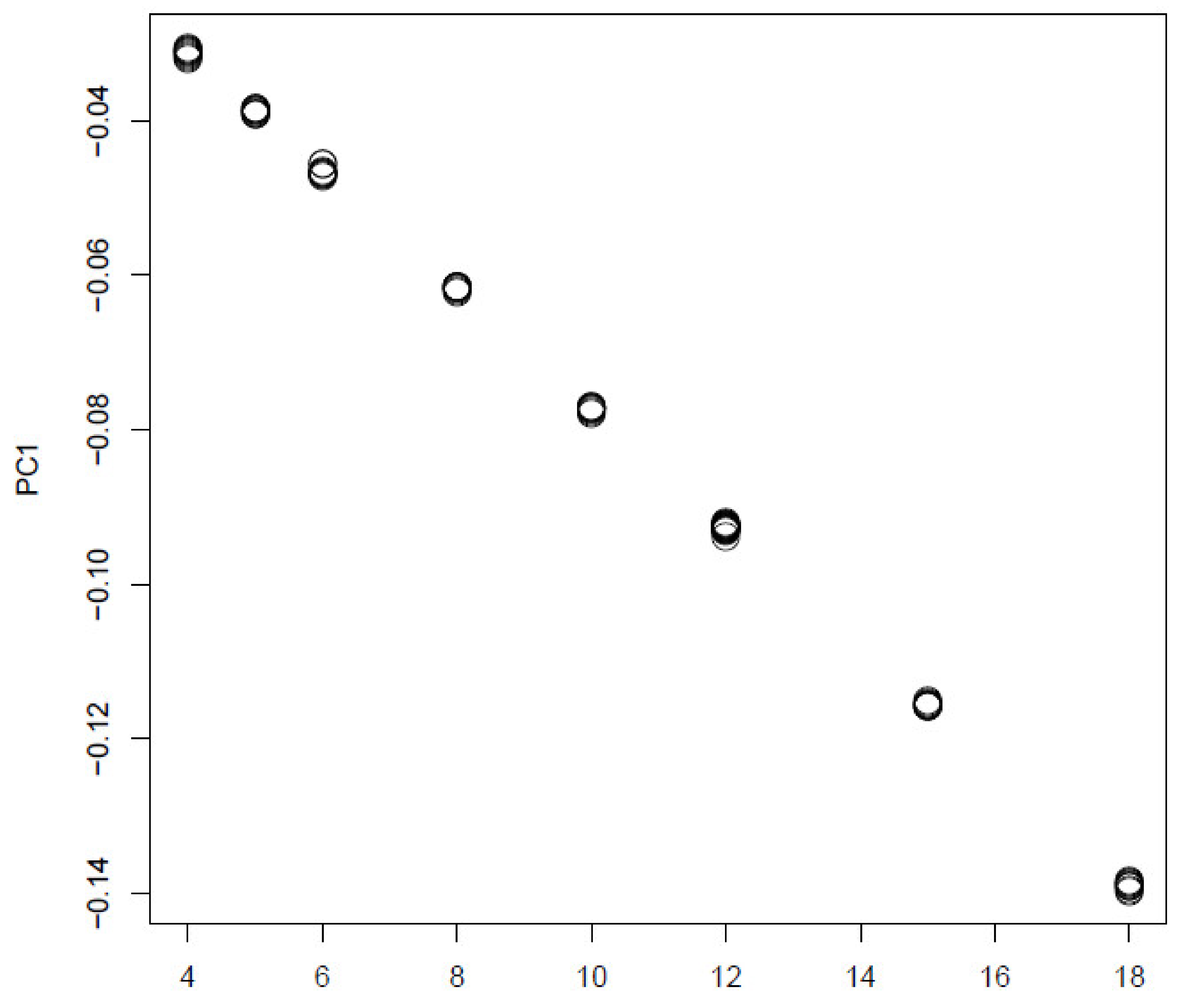
3.1. Synthetic Data
3.2. Real Data
4. Discussion
4.1. Synthetic Data
4.2. Real Data
4.3. Discussions Not to Specific to Either Synthetic or Real Data
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Richter, A.N.; Khoshgoftaar, T.M. Efficient learning from big data for cancer risk modeling: A case study with melanoma. Comput. Biol. Med. 2019, 110, 29–39. [Google Scholar] [CrossRef] [PubMed]
- Awan, M.G.; Eslami, T.; Saeed, F. GPU-DAEMON: GPU algorithm design, data management & optimization template for array based big omics data. Comput. Biol. Med. 2018, 101, 163–173. [Google Scholar] [PubMed]
- Nashaat, M.; Ghosh, A.; Miller, J.; Quader, S.; Marston, C.; Puget, J.F. Hybridization of active learning and data programming for labeling large industrial datasets. In Proceedings of the 2018 IEEE International Conference on Big Data (Big Data), Seattle, WA, USA, 10–13 December 2018; pp. 46–55. [Google Scholar]
- Shah, R.; Zhang, S.; Lin, Y.; Wu, P. xSVM: Scalable Distributed Kernel Support Vector Machine Training. In Proceedings of the 2019 IEEE International Conference on Big Data (Big Data), Los Angeles, CA, USA, 9–12 December 2019; pp. 155–164. [Google Scholar]
- Bekkerman, R.; Bilenko, M.; Langford, J. Scaling up Machine Learning: Parallel and Distributed Approaches; Cambridge University Press: Cambridge, UK, 2011. [Google Scholar]
- Chatterjee, A.; Gupta, U.; Chinnakotla, M.K.; Srikanth, R.; Galley, M.; Agrawal, P. Understanding emotions in text using deep learning and big data. Comput. Hum. Behav. 2019, 93, 309–317. [Google Scholar] [CrossRef]
- Ngiam, K.Y.; Khor, W. Big data and machine learning algorithms for health-care delivery. Lancet Oncol. 2019, 20, e262–e273. [Google Scholar] [CrossRef]
- Santosh, T.; Ramesh, D.; Reddy, D. LSTM based prediction of malaria abundances using big data. Comput. Biol. Med. 2020, 124, 103859. [Google Scholar] [CrossRef] [PubMed]
- Ge, J.; Li, X.; Jiang, H.; Liu, H.; Zhang, T.; Wang, M.; Zhao, T. Picasso: A Sparse Learning Library for High Dimensional Data Analysis in R and Python. J. Mach. Learn. Res. 2019, 20, 1–5. [Google Scholar]
- Wen, F.; Chu, L.; Ying, R.; Liu, P. Fast and Positive Definite Estimation of Large Covariance Matrix for High-Dimensional Data Analysis. IEEE Trans. Big Data 2019. [Google Scholar] [CrossRef]
- Yang, S.; Wen, J.; Zhan, X.; Kifer, D. ET-lasso: A new efficient tuning of lasso-type regularization for high-dimensional data. In Proceedings of the 25th ACM SIGKDD International Conference on Knowledge Discovery & Data Mining, Anchorage, AK, USA, 4–8 August 2019; pp. 607–616. [Google Scholar]
- Mei, B.; Wang, Z. An efficient method to handle the ‘large p, small n’ problem for genomewide association studies using Haseman-Elston regression. J. Genet. 2016, 95, 847–852. [Google Scholar] [CrossRef]
- Johnstone, I.M.; Titterington, D.M. Statistical challenges of high-dimensional data. Philos. Trans. R. Soc. A 2009, 367, 4237–4253. [Google Scholar] [CrossRef]
- Zhang, M.; Zhang, D.; Wells, M.T. Variable selection for large p small n regression models with incomplete data: Mapping QTL with epistases. BMC Bioinform. 2008, 9. [Google Scholar] [CrossRef]
- Huynh, P.H.; Nguyen, V.H.; Do, T.N. Improvements in the Large p, Small n Classification Issue. Comput. Sci. 2020, 1. [Google Scholar] [CrossRef]
- Hood, L.; Rowen, L. The human genome project: Big science transforms biology and medicine. Genome Med. 2013, 5, 79. [Google Scholar] [CrossRef] [PubMed]
- Taguchi, Y.H. Unsupervised Feature Extraction Applied to Bioinformatics; Springer International Publishing: Cham, Switzerland, 2020. [Google Scholar] [CrossRef]
- Witten, D.M.; Tibshirani, R. Penalized classification using Fisher’s linear discriminant. J. R. Stat. Soc. Ser. B 2011, 73, 753–772. [Google Scholar] [CrossRef] [PubMed]
- Baldwin, E.; Han, J.; Luo, W.; Zhou, J.; An, L.; Liu, J.; Zhang, H.H.; Li, H. On fusion methods for knowledge discovery from multi-omics datasets. Comput. Struct. Biotechnol. J. 2020, 18, 509–517. [Google Scholar] [CrossRef]
- Subramanian, I.; Verma, S.; Kumar, S.; Jere, A.; Anamika, K. Multi-omics Data Integration, Interpretation, and Its Application. Bioinform. Biol. Insights 2020, 14, 1177932219899051. [Google Scholar] [CrossRef]
- Vaske, C.J.; Benz, S.C.; Sanborn, J.Z.; Earl, D.; Szeto, C.; Zhu, J.; Haussler, D.; Stuart, J.M. Inference of patient-specific pathway activities from multi-dimensional cancer genomics data using PARADIGM. Bioinformatics 2010, 26, i237–i245. [Google Scholar] [CrossRef]
- Shen, R.; Olshen, A.B.; Ladanyi, M. Integrative clustering of multiple genomic data types using a joint latent variable model with application to breast and lung cancer subtype analysis. Bioinformatics 2009, 25, 2906–2912. [Google Scholar] [CrossRef]
- Mo, Q.; Wang, S.; Seshan, V.E.; Olshen, A.B.; Schultz, N.; Sander, C.; Powers, R.S.; Ladanyi, M.; Shen, R. Pattern discovery and cancer gene identification in integrated cancer genomic data. Proc. Natl. Acad. Sci. USA 2013, 110, 4245–4250. [Google Scholar] [CrossRef]
- Wu, D.; Wang, D.; Zhang, M.Q.; Gu, J. Fast dimension reduction and integrative clustering of multi-omics data using low-rank approximation: Application to cancer molecular classification. BMC Genom. 2015, 16. [Google Scholar] [CrossRef]
- Chin, S.F.; Teschendorff, A.E.; Marioni, J.C.; Wang, Y.; Barbosa-Morais, N.L.; Thorne, N.P.; Costa, J.L.; Pinder, S.E.; van de Wiel, M.A.; Green, A.R.; et al. High-resolution aCGH and expression profiling identifies a novel genomic subtype of ER negative breast cancer. Genome Biol. 2007, 8, R215. [Google Scholar] [CrossRef]
- Lock, E.F.; Dunson, D.B. Bayesian consensus clustering. Bioinformatics 2013, 29, 2610–2616. [Google Scholar] [CrossRef] [PubMed]
- Wang, B.; Mezlini, A.M.; Demir, F.; Fiume, M.; Tu, Z.; Brudno, M.; Haibe-Kains, B.; Goldenberg, A. Similarity network fusion for aggregating data types on a genomic scale. Nat. Methods 2014, 11, 333–337. [Google Scholar] [CrossRef] [PubMed]
- Shi, Q.; Zhang, C.; Peng, M.; Yu, X.; Zeng, T.; Liu, J.; Chen, L. Pattern fusion analysis by adaptive alignment of multiple heterogeneous omics data. Bioinformatics 2017, 33, 2706–2714. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, H.; Shrestha, S.; Draghici, S.; Nguyen, T. PINSPlus: A tool for tumor subtype discovery in integrated genomic data. Bioinformatics 2018, 35, 2843–2846. [Google Scholar] [CrossRef]
- Rappoport, N.; Shamir, R. NEMO: Cancer subtyping by integration of partial multi-omic data. Bioinformatics 2019, 35, 3348–3356. [Google Scholar] [CrossRef]
- Rohart, F.; Gautier, B.; Singh, A.; Lê Cao, K.A. mixOmics: An R package for ‘omics feature selection and multiple data integration. PLoS Comput. Biol. 2017, 13, e1005752. [Google Scholar] [CrossRef]
- Meng, C.; Helm, D.; Frejno, M.; Kuster, B. moCluster: Identifying Joint Patterns Across Multiple Omics Data Sets. J. Proteome Res. 2015, 15, 755–765. [Google Scholar] [CrossRef]
- Meng, C.; Kuster, B.; Culhane, A.C.; Gholami, A. A multivariate approach to the integration of multi-omics datasets. BMC Bioinform. 2014, 15, 162. [Google Scholar] [CrossRef]
- O’Connell, M.J.; Lock, E.F.R. JIVE for exploration of multi-source molecular data. Bioinformatics 2016, 32, 2877–2879. [Google Scholar] [CrossRef]
- De Tayrac, M.; Le, S.; Aubry, M.; Mosser, J.; Husson, F. Simultaneous analysis of distinct Omics data sets with integration of biological knowledge: Multiple Factor Analysis approach. BMC Genom. 2009, 10, 32. [Google Scholar] [CrossRef]
- Speicher, N.K.; Pfeifer, N. Integrating different data types by regularized unsupervised multiple kernel learning with application to cancer subtype discovery. Bioinformatics 2015, 31, i268–i275. [Google Scholar] [CrossRef] [PubMed]
- Integrated genomic analyses of ovarian carcinoma. Nature 2011, 474, 609–615. [CrossRef] [PubMed]
- Edgar, R.; Domrachev, M.; Lash, A.E. Gene Expression Omnibus: NCBI gene expression and hybridization array data repository. Nucleic Acids Res. 2002, 30, 207–210. [Google Scholar] [CrossRef] [PubMed]
- R Core Team. R: A Language and Environment for Statistical Computing; R Foundation for Statistical Computing: Vienna, Austria, 2019. [Google Scholar]
- Liaw, A.; Wiener, M. Classification and Regression by randomForest. R News 2002, 2, 18–22. [Google Scholar]
- Witten, D. penalizedLDA: Penalized Classification Using Fisher’s Linear Discriminant. 2015. Available online: https://cran.r-project.org/web/packages/penalizedLDA/penalizedLDA.pdf (accessed on 11 December 2020).
- Igolkina, A.A.; Zinkevich, A.; Karandasheva, K.O.; Popov, A.A.; Selifanova, M.V.; Nikolaeva, D.; Tkachev, V.; Penzar, D.; Nikitin, D.M.; Buzdin, A. H3K4me3, H3K9ac, H3K27ac, H3K27me3 and H3K9me3 Histone Tags Suggest Distinct Regulatory Evolution of Open and Condensed Chromatin Landmarks. Cells 2019, 8, 1034. [Google Scholar] [CrossRef]
- Pekowska, A.; Benoukraf, T.; Ferrier, P.; Spicuglia, S. A unique H3K4me2 profile marks tissue-specific gene regulation. Genome Res. 2010, 20, 1493–1502. [Google Scholar] [CrossRef]
- Fujita, K.; Nonomura, N. Role of Androgen Receptor in Prostate Cancer: A Review. World J. Men’s Health 2019, 37, 288. [Google Scholar] [CrossRef]
- Gerhardt, J.; Montani, M.; Wild, P.; Beer, M.; Huber, F.; Hermanns, T.; Müntener, M.; Kristiansen, G. FOXA1 Promotes Tumor Progression in Prostate Cancer and Represents a Novel Hallmark of Castration-Resistant Prostate Cancer. Am. J. Pathol. 2012, 180, 848–861. [Google Scholar] [CrossRef]
- Navarro, H.I.; Goldstein, A.S. HoxB13 mediates AR-V7 activity in prostate cancer. Proc. Natl. Acad. Sci. USA 2018, 115, 6528–6529. [Google Scholar] [CrossRef]
- Zhou, Y.; Zhou, B.; Pache, L.; Chang, M.; Khodabakhshi, A.H.; Tanaseichuk, O.; Benner, C.; Chanda, S.K. Metascape provides a biologist-oriented resource for the analysis of systems-level datasets. Nat. Commun. 2019, 10. [Google Scholar] [CrossRef]
- Piñero, J.; Ramírez-Anguita, J.M.; Saüch-Pitarch, J.; Ronzano, F.; Centeno, E.; Sanz, F.; Furlong, L.I. The DisGeNET knowledge platform for disease genomics: 2019 update. Nucleic Acids Res. 2019, 48, D845–D855. [Google Scholar] [CrossRef] [PubMed]
- Pan, J.B.; Hu, S.C.; Shi, D.; Cai, M.C.; Li, Y.B.; Zou, Q.; Ji, Z.L. PaGenBase: A Pattern Gene Database for the Global and Dynamic Understanding of Gene Function. PLoS ONE 2013, 8, e80747. [Google Scholar] [CrossRef] [PubMed]
- Horoszewicz, J.S.; Leong, S.S.; Kawinski, E.; Karr, J.P.; Rosenthal, H.; Chu, T.M.; Mirand, E.A.; Murphy, G.P. LNCaP Model of Human Prostatic Carcinoma. Cancer Res. 1983, 43, 1809–1818. [Google Scholar] [PubMed]
- Han, H.; Shim, H.; Shin, D.; Shim, J.E.; Ko, Y.; Shin, J.; Kim, H.; Cho, A.; Kim, E.; Lee, T.; et al. TRRUST: A reference database of human transcriptional regulatory interactions. Sci. Rep. 2015, 5. [Google Scholar] [CrossRef]



















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Omics Data | |||
|---|---|---|---|
| Model Name | Number of Samples | Numbers | Types |
| PARADIGM [21] | 230 patient samples and 10 adjacent normal tissues | two omics data | copy number and mRNA expression |
| iCluster [22] | 37 primary breast cancers and four breast cancer cell lines | two omics data | copy number and mRNA expression |
| 91 lung adenocarcinomas | two omics data | copy number and mRNA expression | |
| iClusterPlus [23] | 729 human cancer cell lines | three omics data | chromosomal copy number, gene expression, and mutation |
| 189 tumors | four omics data | exome sequence, DNA copy number, promoter methylation, and mRNA expression | |
| LRAcluster [24] | 3319 samples | four omics data | mutation, CNV, DNA methylation, and gene expression |
| PSDF [25] | 106 breast cancer samples | two omics data | Copy number and gene expression |
| BCC [26] | 348 tumor samples | four omics data | RNA expression, methylation, miRNA expression, Reverse phase protein array |
| SNF [27] | 215 GBM data samples | three omics data | DNA methylation, miRNA expression, and gene expression |
| PFA [28] | 415 cell lines | two omics | gene expression and copy number |
| PINSPlus [29] | 12,158 samples | — | — |
| NEMO [30] | 173 samples DNA methylation data from 194 samples, and miRNA expression data from 188 samples | three omics data | — |
| DIABLO [31] | 150 breast cancer samples | three moics | mRNA, miRNA, and protein expression |
| moCluster [32] | 83 samples of colorectal cancer | three omics data | DNA methylation, gene expression, and protein expression |
| MCIA [33] | 266 samples | two omics data | proteomics and transcriptomics |
| JIVE [34] | 348 breast cancer samples | three omics data | gene expression, DNA methylation, and miRNA data |
| MFA [35] | 43 glioma samples | two omics data | CGH-array and transcriptome |
| rMKL-LPP [36] | glioblastoma multiforme (GBM) with 213 samples, breast invasive carcinoma (BIC) with 105 samples, kidney renal clear cell carcinoma (KRCCC) with 122 samples, lung squamous cell carcinoma (LSCC) with 106 samples and colon adenocarcinoma (COAD) with 92 samples | three omics data | gene expression, DNA methylation and miRNA expression |
| iNMF [37] | 592 samples | three omics data | gene expression, DNA methylation, miRNA expression |
| Number of | |||
|---|---|---|---|
| Omics | Multiomics | Tissues | Biological Replicates |
| AR | 2 | 2 | 3 |
| FOXA1 | 4 | 2 | 3 |
| HOXB13 | 4 | 2 | 3 |
| H3K27AC | 10 | 2 | 3 |
| H3K27me3 | 1 | 2 | 3 |
| H3K4me3 | 1 | 2 | 3 |
| K4me2 | 1 | 2 | 3 |
| ATAC | 1 | 2 | 3 |
| total | 24 | — | — |
| p | 0.1 | 0.05 | 0.01 | 0.001 | ||||
|---|---|---|---|---|---|---|---|---|
| not selected | 99,889 | 0 | 99,895 | 0 | 99,899 | 0 | 99,900 | 0 |
| selected | 11 | 100 | 5 | 100 | 1 | 100 | 0 | 100 |
| p | 0.1 | 0.05 | 0.01 | 0.001 | ||||
|---|---|---|---|---|---|---|---|---|
| not selected | 99,900 | 0 | 99,900 | 0 | 99,900 | 0 | 99,900 | 0 |
| selected | 0 | 100 | 0 | 100 | 0 | 100 | 0 | 100 |
| p | 0.1 | 0.05 | 0.01 | 0.001 | ||||
|---|---|---|---|---|---|---|---|---|
| not selected | 99,900 | 0 | 99,900 | 0 | 99,900 | 0 | 99,900 | 0 |
| selected | 0 | 100 | 0 | 100 | 0 | 100 | 0 | 100 |
| Methods | PCA Based Unsupervised FE | TD Based Unsupervised FE | Categorical Regression | Random Forest | Penalized LDA | MNMF | |
|---|---|---|---|---|---|---|---|
| cpu time [s] | synthetic data | 1.55 | 3.5 | 51.5 | 118.2 | 283 (18) | 290 |
| real data | 17 | 20 | 87 | 321 | — | 223 |
| AR | ATAC | FOXA1 | H3K27AC | H3K27me3 | H3K4me3 | HOXB13 | K4me2 | Class | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| N | T | N | T | N | T | N | T | N | T | N | T | N | T | N | T | Error | |
| AR_N | 5 | 0 | 0 | 0 | 1 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1/6 |
| AR_T | 0 | 0 | 0 | 0 | 1 | 3 | 1 | 0 | 0 | 0 | 0 | 0 | 0 | 1 | 0 | 0 | 1 |
| ATAC_N | 0 | 0 | 0 | 2 | 0 | 0 | 0 | 1 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1 |
| ATAC_T | 0 | 0 | 2 | 1 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 2/3 |
| FOXA1_N | 1 | 0 | 0 | 0 | 9 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 2 | 0 | 0 | 0 | 1/4 |
| FOXA1_T | 0 | 0 | 0 | 0 | 4 | 5 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 3 | 0 | 0 | 7/12 |
| H3K27AC_N | 0 | 0 | 0 | 0 | 0 | 0 | 26 | 4 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 2/15 |
| H3K27AC_T | 0 | 0 | 0 | 0 | 0 | 0 | 6 | 24 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1/5 |
| H3K27me3_N | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1 | 2 | 0 | 0 | 0 | 0 | 0 | 0 | 2/3 |
| H3K27me3_T | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1 | 2 | 0 | 0 | 0 | 0 | 0 | 0 | 1/3 |
| H3K4me3_N | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 3 | 0 | 0 | 0 | 0 | 1 |
| H3K4me3_T | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 3 | 0 | 0 | 0 | 0 | 0 | 1 |
| HOXB13_N | 0 | 0 | 0 | 0 | 2 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 9 | 1 | 0 | 0 | 1/4 |
| HOXB13_T | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1 | 11 | 0 | 0 | 1/12 |
| K4me2_N | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 3 | 1 |
| K4me2_T | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1 | 0 | 0 | 0 | 0 | 0 | 0 | 2 | 0 | 1 |
| AR | ATAC | FOXA1 | H3K27AC | H3K27me3 | H3K4me3 | HOXB13 | K4me2 | Class | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| N | T | N | T | N | T | N | T | N | T | N | T | N | T | N | T | Error | |
| AR_N | 0 | 0 | 0 | 0 | 5 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1 | 0 | 0 | 0 | 1 |
| AR_T | 0 | 0 | 0 | 0 | 0 | 5 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1 | 0 | 0 | 1 |
| ATAC_N | 0 | 0 | 0 | 0 | 0 | 0 | 2 | 0 | 0 | 0 | 1 | 0 | 0 | 0 | 0 | 0 | 1 |
| ATAC_T | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 2 | 0 | 0 | 0 | 1 | 0 | 0 | 0 | 0 | 1 |
| FOXA1_N | 0 | 0 | 0 | 0 | 9 | 0 | 1 | 0 | 0 | 0 | 0 | 0 | 2 | 0 | 0 | 0 | 1/4 |
| FOXA1_T | 0 | 0 | 0 | 0 | 0 | 9 | 0 | 1 | 0 | 0 | 0 | 0 | 0 | 2 | 0 | 0 | 1/4 |
| H3K27AC_N | 0 | 0 | 2 | 0 | 0 | 0 | 25 | 0 | 0 | 0 | 1 | 0 | 2 | 0 | 0 | 0 | 1/6 |
| H3K27AC_T | 0 | 0 | 0 | 2 | 0 | 0 | 0 | 25 | 0 | 0 | 0 | 1 | 0 | 2 | 0 | 0 | 1/6 |
| H3K27me3_N | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 3 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| H3K27me3_T | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 3 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| H3K4me3_N | 0 | 0 | 1 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 2 | 0 | 0 | 0 | 0 | 0 | 1/3 |
| H3K4me3_T | 0 | 0 | 0 | 1 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 2 | 0 | 0 | 0 | 0 | 1/3 |
| HOXB13_N | 0 | 0 | 0 | 0 | 4 | 0 | 2 | 0 | 0 | 0 | 0 | 0 | 6 | 0 | 0 | 0 | 1/2 |
| HOXB13_T | 0 | 0 | 0 | 0 | 0 | 4 | 0 | 2 | 0 | 0 | 0 | 0 | 0 | 6 | 0 | 0 | 1/2 |
| K4me2_N | 0 | 0 | 0 | 0 | 0 | 0 | 3 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1 |
| K4me2_T | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 3 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Taguchi, Y.-h.; Turki, T. Tensor-Decomposition-Based Unsupervised Feature Extraction Applied to Prostate Cancer Multiomics Data. Genes 2020, 11, 1493. https://doi.org/10.3390/genes11121493
Taguchi Y-h, Turki T. Tensor-Decomposition-Based Unsupervised Feature Extraction Applied to Prostate Cancer Multiomics Data. Genes. 2020; 11(12):1493. https://doi.org/10.3390/genes11121493
Chicago/Turabian StyleTaguchi, Y-h., and Turki Turki. 2020. "Tensor-Decomposition-Based Unsupervised Feature Extraction Applied to Prostate Cancer Multiomics Data" Genes 11, no. 12: 1493. https://doi.org/10.3390/genes11121493
APA StyleTaguchi, Y.-h., & Turki, T. (2020). Tensor-Decomposition-Based Unsupervised Feature Extraction Applied to Prostate Cancer Multiomics Data. Genes, 11(12), 1493. https://doi.org/10.3390/genes11121493