Experimental Models to Study Autism Spectrum Disorders: hiPSCs, Rodents and Zebrafish

 ,
,  , ,
, , 
Abstract
1. Introduction
1.1. Definition and Epidemiology of Autism Spectrum Disorders
1.2. Aetiology of Autism Spectrum Disorders
1.3. Diagnostic of Autism Spectrum Disorders
1.4. Treatment of Autism Spectrum Disorders
2. Genome Editing Systems, a Promising Tool for Modeling Human Disorders
Fundamentals of Genomic Editing
3. In Vitro Models of ASD: The Stem Cell Revolution
4. Animal Models in ASD Research
4.1. Rodents and the Modelling of Human Disorders
4.1.1. Mus Musculus in ASD Research
4.1.2. Rattus norvegicus in ASD Research
4.2. Zebrafish and the Modeling of Human Disorders
4.2.1. Zebrafish and Mammals: Conservation throughout Evolution
4.2.2. Gene Targeting in Zebrafish
4.2.3. Characterization of Zebrafish Models
4.2.4. Limitations of Zebrafish to Model Human Disorders
5. Future Challenges
5.1. In Vitro Modelling
5.2. In Vitro Modelling
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| ABA | Applied Behavior Analysis |
| ADNP/Adnp | Activity dependent neuroprotector homeobox |
| AFF2 | AF4/FMR2 family, member 2 |
| ARHGEF9 | Cdc42 guanine nucleotide exchange factor 9 |
| ARID1B/Arid1b/arid1b | AT-rich interaction domain 1B |
| ARRIVE | Animals in Research: Reporting In Vivo Experiments |
| ARX/arxa | Aristaless related homeobox |
| ASD | Autism Spectrum Disorders |
| ASH1L/Ash1l | ASH1 like histone lysine methyltransferase |
| ASTN2 | Astrotactin 2 |
| atoh1 | Atonal bHLH transcription factor 1 |
| ath5 | Atonal bHLH transcription factor 7 |
| ATRX | α thalassemia/mental retardation syndrome X-linked |
| AUTS2/auts2a and auts2b | Autism susceptibility candidate 2 |
| BCKDK/Bckdk | Branched chain ketoacid dehydrogenase kinase |
| brn3c | POU class 4 homeobox 3 |
| CACNA1C/Cacna1c/cacna1c | Calcium channel voltage-dependent, L type, α 1C subunit |
| Cas | CRISPR-associated genes |
| Cas13 | CRISPR-associated endoribonuclease Cas13 |
| Cas9 | CRISPR associated endonuclease Cas9 |
| CDKL5 | Cyclin-dependent kinase-like 5 |
| CEP41/cep41 | Testis specific, 14 |
| CHD2/Chd2/chd2 | Chromodomain helicase DNA binding protein 2 |
| CHD8/Chd8/chd8 | Chromodomain helicase DNA binding protein 8 |
| CIC/Cic | Capicua transcriptional repressor |
| c-Myc | MYC proto-oncogene |
| CNTN5 | Contactin 5 |
| CNTNAP2/Cntnap2/cntnap2a and cntap2b | Contactin associated protein-like 2 |
| CNVs | Copy Number Variations |
| Cre | Cre recombinase |
| crh | Corticotropin releasing hormone |
| CRISPR | Clustered Regularly Interspaced Short Palindromic Repeats |
| CRISPRa | CRISPR activation |
| CRISPRi | CRISPR interference |
| crRNA | CRISPR RNA |
| CTNND2/ctnnd2b | Catenin (cadherin-associated protein), delta 2 |
| CYFIP1/Cyfip1 | Cytoplasmic FMR1 interacting protein 1 |
| D. rerio | Danio rerio |
| dat | Dopamine transporter/Solute carrier family 6 member 3 |
| dCas9 | Catalytically dead Cas9 |
| Disc1 | Disrupted in schizophrenia 1 |
| DNA | Deoxyribonucleic acid |
| dpf | Days post-fertilization |
| DSBs | Double-Strand Breaks |
| DSM-5 | Diagnostic and statistical manual of mental disorders, 5th edition |
| DYRK1A/dyrk1a | Dual-specificity tyrosine-(Y)-phosphorylation regulated kinase 1A |
| EHMT1 | Euchromatic histone-lysine N-methyltransferase 1 |
| elavl3 | ELAV like neuron-specific RNA binding protein 3 |
| emx1 | Empty spiracles homeobox 1 |
| En-1 | Engrailed homeobox 1 |
| ENU | N-ethyl-N-nitrosourea |
| Ext1 | Exostosin glycosyltransferase 1 |
| FMR1/Fmr1/fmr1 | Fragile X mental retardation 1 |
| FokI | Type IIS restriction endonuclease from Flavobacterium okeanokoites |
| GABA | γ-aminobutyric acid |
| GABRB3/Gabrb3 | γ-aminobutyric acid type A receptor, subunit beta3 |
| gad1b | Glutamate decarboxylase 1b |
| GFAP/gfap | Glial fibrillary acidic protein |
| glyt2 | Sodium and chloride dependent glycine transporter 2 |
| gsx1 | GS homeobox 1 |
| GWAS | Genome-Wide Association Studies |
| HDR | Homology-directed repair |
| hiPSCs | Human induced pluripotent stem cells |
| HNH | Endonuclease domain characterized by histidine and asparagine residues |
| hpf | Hours post-fertilization |
| Indel | Insertion and/or deletion |
| isl1 | ISL LIM homeobox 1 |
| KCNJ10/kcnj10 | Potassium voltage-gated channel subfamily J, member 10 |
| KCNQ2 | Potassium voltage-gated channel subfamily Q, member 2 |
| kctd15a | Potassium channel tetramerization domain containing 15a |
| KDM6A/kdm6a | Lysine demethylase 6A |
| KI | Knock-in |
| Klf4 | Kruppel like factor 4 |
| KO | Knockout |
| lncRNA | Long non-coding RNA |
| LOF | Loss of function |
| M. musculus | Mus musculus |
| MAPK | Mitogen-activated protein kinase |
| MECP2/Mecp2/mecp2 | Methyl CpG binding protein 2 |
| MET/met | Met proto-oncogene |
| MGE | Medial ganglionic eminence |
| mnx1 | Motor neuron and pancreas homeobox 1 |
| MOs | Morpholinos |
| mRNA | Messenger RNA |
| mTORC1 | Mammalian target of rapamycin complex 1 |
| MYT1L/mytl1a and mytl1b | Myelin transcription factor 1-like |
| NBEA/nbea | Neurobeachin |
| nCas9 | Cas9 nickase |
| NDDs | Neurodevelopmental disorders |
| neurod | Neurogenic differentiation factor 1 |
| neurog1 | Neurogenin 1 |
| NHEJ | Non-homologous end joining |
| NLGN2/Nlgn2 | Neuroligin 2 |
| NLGN3/Nlgn3 | Neuroligin 3 |
| NMDARs | N-methyl-D-aspartate receptors |
| NR3C2/nr3c2 | Nuclear receptor subfamily 3, group C, member 2 |
| NRXN1/Nrxn1 | Neurexin 1 |
| NS | Non specified |
| Oct3/4 | Octamer binding transcription factor 3/4 |
| olig2 | Oligodendrocyte lineage transcription factor 2 |
| otx2a | Orthodenticle homeobox 2a |
| OXTR/oxtr | Oxytocin receptor |
| p53 | Tumor protein p53 |
| PAM | Protospacer adjacent motif |
| PCNA | Proliferating cell nuclear antigen |
| PDD-NOS | Pervasive developmental disorder not otherwise specified |
| pet1 | FEV transcription factor, |
| PREPARE | Planning Research and Experimental Procedures on Animals: Recommendations for Excellence |
| PRT | Pivotal Response Treatment |
| PTCHD1 | Patched domain containing 1 |
| PTCHD1-AS | PTCHD1 antisense RNA |
| PTEN/Pten | Phosphatase and tensin homolog |
| ptf1a | Pancreas associated transcription factor 1a |
| R. norvegicus | Rattus norvegicus |
| RELN/Reln/reln | Reelin |
| RERE/rerea and rereb | Arginine-glutamic acid dipeptide repeats |
| RNA | Ribonucleic acid |
| RNA-seq | RNA sequencing |
| RORα | Nuclear receptor ROR-α |
| RuvC | Endonuclease domain involved in DNA repair |
| SCN1A/Scn1a | Sodium channel, voltage-gated, type I, α subunit |
| SCN2A/Scn2a | Sodium channel, voltage-gated, type II, α subunit |
| SFARI | Simons Foundation Autism Research Initiative |
| sgRNA | Single guide RNA |
| SHANK2/Shank2 | SH3 and multiple ankyrin repeat domains 2 |
| SHANK3/Shank3/shank3a and shankb | SH3 and multiple ankyrin repeat domains 3 |
| shRNA | Short hairpin RNA |
| Sox2/sox2-sox2 | SRY-box transcription factor 2 |
| sox10 | SRY-box transcription factor 10 |
| SVZ | Subventricular zone |
| SYNGAP1/syngap1a and syngap1b | Synaptic Ras GTPase activating protein 1 |
| TALENs | Transcription Activator–Like Effector Nucleases |
| TALEs | Transcription Activator-Like Effectors |
| TAOK2/Taok2 | TAO kinase 2 |
| TBR1/Tbr1 | T-box brain transcription factor 1 |
| tbx2b | T-box transcription factor 2b |
| TCF4/Tcf4 | Transcription factor 4 |
| th1 | Tyrosine hydroxylase 1 |
| TILLING | Targeting Induced Local Lesions in Genomes |
| tracrRNA | Trans-activating crRNA |
| tRNA | Transfer ribonucleic acid |
| TRPC6 | Transient receptor potential cation channel, subfamily C, member 6 |
| TSC2/Tsc2 | Tuberous sclerosis 2 |
| UBE3A/Ube3a | Ubiquitin protein ligase E3A |
| UPF3B/Upf3b | UPF3B regulator of nonsense mediated mRNA decay |
| USVs | Ultrasonic vocalizations |
| vglut2.2 | Vesicular glutamate transporter 2.2 |
| vglut2a | Vesicular glutamate transporter 2.1 |
| vmat2 | Vesicular monoamine transporter 2 |
| WES | Whole exome sequencing |
| WGS | Whole genome sequencing |
| ZFNs | Zinc Finger Nucleases |
| ZNF804A | Zinc finger protein 804A |
References
- American Psychiatric Association. Diagnostic and Statistical Manual of Mental Disorders (DSM-5), 5th ed.; American Psychiatric Publishing: Washington, DC, USA, 2013. [Google Scholar]
- IHME, Global Burden of Disease. Prevalence of Autistic Spectrum Disorder. 2017. Available online: https://ourworldindata.org/grapher/prevalence-of-autistic-spectrum (accessed on 20 March 2020).
- Lyall, K.; Croen, L.; Daniels, J.; Fallin, M.D.; Ladd-Acosta, C.; Lee, B.K.; Park, B.Y.; Snyder, N.W.; Schendel, D.; Volk, H.; et al. The changing epidemiology of Autism Spectrum Disorders. Annu. Rev. Public Health 2017, 38, 81–102. [Google Scholar] [CrossRef]
- Global Burden of Disease Collaborative Network. Global Burden of Disease Study 2016 (GBD 2016) Results. Available online: http://ghdx.healthdata.org/gbd-results-tool (accessed on 20 March 2020).
- Christensen, D.L.; Baio, J.; Van Naarden Braun, K.; Bilder, D.; Charles, J.; Constantino, J.N.; Daniels, J.; Durkin, M.S.; Fitzgerald, R.T.; Kurzius-Spencer, M.; et al. Prevalence and characteristics of Autism Spectrum Disorder among children aged 8 years—Autism and developmental disabilities monitoring network, 11 sites, United States, 2012. MMWR Surveill. Summ. 2018, 65, 1–23. [Google Scholar] [CrossRef]
- R Core Team. R: A Language and Environment for Statistical Computing; R Foundation for Statistical Computing: Vienna, Austria, 2020. [Google Scholar]
- Mitchell, K.J. (Ed.) The Genetics of Neurodevelopmental Disorders, 1st ed.; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 2015. [Google Scholar]
- Ornoy, A.; Weinstein-Fudim, L.; Ergaz, Z. Prenatal factors associated with Autism Spectrum Disorder (ASD). Reprod. Toxicol. 2015, 56, 155–169. [Google Scholar] [CrossRef]
- Kinney, D.K.; Barch, D.H.; Chayka, B.; Napoleon, S.; Munir, K.M. Environmental risk factors for autism: Do they help cause de novo genetic mutations that contribute to the disorder? Med. Hypotheses 2010, 74, 102–106. [Google Scholar] [CrossRef]
- Modabbernia, A.; Velthorst, E.; Reichenberg, A. Environmental risk factors for autism: An evidence-based review of systematic reviews and meta-analyses. Mol. Autism 2017, 8, 13. [Google Scholar] [CrossRef]
- Folstein, S.E.; Rosen-Sheidley, B. Genetics of austim: Complex aetiology for a heterogeneous disorder. Nat. Rev. Genet. 2001, 2, 943–955. [Google Scholar] [CrossRef] [PubMed]
- Rosenberg, R.E.; Law, J.K.; Yenokyan, G.; McGready, J.; Kaufmann, W.E.; Law, P.A. Characteristics and concordance of Autism Spectrum Disorders among 277 twin pairs. Arch. Pediatr. Adolesc. Med. 2009, 163, 907–914. [Google Scholar] [CrossRef] [PubMed]
- Geschwind, D.H. Genetics of autism spectrum disorders. Trends Cogn. Sci. 2011, 15, 409–416. [Google Scholar] [CrossRef] [PubMed]
- Canitano, R.; Bozzi, Y. Editorial: Autism spectrum disorders: Developmental trajectories, neurobiological basis, treatment update. Front Psychiatry 2017, 8, 125. [Google Scholar] [CrossRef] [PubMed]
- Giovedí, S.; Corradi, A.; Fassio, A.; Benfenati, F. Involvement of synaptic genes in the pathogenesis of Autism Spectrum Disorders: The case of synapsins. Front. Pediatr. 2014, 2, 94. [Google Scholar] [CrossRef]
- Iossifov, I.; O’Roak, B.J.; Sanders, S.J.; Ronemus, M.; Krumm, N.; Levy, D.; Stessman, H.A.; Witherspoon, K.T.; Vives, L.; Patterson, K.E.; et al. The contribution of de novo coding mutations to Autism Spectrum Disorder. Nature 2014, 515, 216–221. [Google Scholar] [CrossRef] [PubMed]
- Velmeshev, D.; Schirmer, L.; Jung, D.; Haeussler, M.; Perez, Y.; Mayer, S.; Bhaduri, A.; Goyal, N.; Rowitch, D.H.; Kriegstein, A.R. Single-cell genomics identifies cell type–specific molecular changes in autism. Science 2019, 364, 685–689. [Google Scholar] [CrossRef] [PubMed]
- Woodbury-Smith, M.; Scherer, S.W. Progress in the genetics of Autism Spectrum Disorder. Dev. Med. Child. Neurol. 2018, 60, 445–451. [Google Scholar] [CrossRef] [PubMed]
- Alonso-Gonzalez, A.; Rodriguez-Fontenla, C.; Carracedo, A. De novo mutations (DNMs) in autism spectrum disorder (ASD): Pathway and network analysis. Front. Genet. 2018, 9, 406. [Google Scholar] [CrossRef] [PubMed]
- Abrahams, B.S.; Arking, D.E.; Campbell, D.B.; Mefford, H.C.; Morrow, E.M.; Weiss, L.A.; Menashe, I.; Wadkins, T.; Banerjee-Basu, S.; Packer, A. SFARI Gene 2.0: A community-driven knowledgebase for the autism spectrum disorders (ASDs). Mol. Autism 2013, 4, 36. [Google Scholar] [CrossRef] [PubMed]
- SFARI Gene. Available online: https://gene.sfari.org/ (accessed on 16 May 2019).
- Loomes, R.; Hull, L.; Mandy, W.P.L. What is the male-to-female ratio in Autism Spectrum Disorder? A systematic review and meta-analysis. J. Am. Acad. Child Adolesc. Psychiatry 2017, 56, 466–474. [Google Scholar] [CrossRef]
- Mullin, A.P.; Gokhale, A.; Moreno-De-Luca, A.; Sanyal, S.; Waddington, J.L.; Faundez, V. Neurodevelopmental disorders: Mechanisms and boundary definitions from genomes, interactomes and proteomes. Transl. Psychiatry 2013, 3, e329. [Google Scholar] [CrossRef]
- DeFilippis, M.; Wagner, K.D. Treatment of Autism Spectrum Disorder in children and adolescents. Psychopharmacol. Bull. 2016, 46, 18–41. [Google Scholar]
- Farmer, C.; Thurm, A.; Grant, P. Pharmacotherapy for the core symptoms in autistic disorder: Current status of the research. Drugs 2013, 73, 303–314. [Google Scholar] [CrossRef]
- Kim, Y.G.; Cha, J.; Chandrasegaran, S. Hybrid restriction enzymes: Zinc finger fusions to Fok I cleavage domain. Proc. Natl. Acad. Sci. USA 1996, 93, 1156–1160. [Google Scholar] [CrossRef]
- Christian, M.; Cermak, T.; Doyle, E.L.; Schmidt, C.; Zhang, F.; Hummel, A.; Bogdanove, A.J.; Voytas, D.F. Targeting DNA double-strand breaks with TAL effector nucleases. Genetics 2010, 186, 757–761. [Google Scholar] [CrossRef] [PubMed]
- Jinek, M.; East, A.; Cheng, A.; Lin, S.; Ma, E.; Doudna, J. RNA-programmed genome editing in human cells. Elife 2013, 2, e00471. [Google Scholar] [CrossRef] [PubMed]
- Doudna, J.A.; Charpentier, E. Genome Editing. The new frontier of genome engineering with CRISPR-Cas9. Science 2014, 346, 1258096. [Google Scholar] [CrossRef]
- Hsu, P.D.; Lander, E.S.; Zhang, F. Development and applications of CRISPR-Cas9 for genome engineering. Cell 2014, 157, 1262–1278. [Google Scholar] [CrossRef]
- Mojica, F.J.M.; Montoliu, L. On the origin of CRISPR-Cas technology: From prokaryotes to mammals. Trends Microbiol. 2016, 24, 811–820. [Google Scholar] [CrossRef] [PubMed]
- Huertas, P. DNA resection in eukaryotes: Deciding how to fix the break. Nat. Struct. Mol. Biol. 2010, 17, 11–16. [Google Scholar] [CrossRef] [PubMed]
- Carroll, D. Genome engineering with zinc-finger nucleases. Genetics 2011, 188, 773–782. [Google Scholar] [CrossRef]
- Carbery, I.D.; Ji, D.; Harrington, A.; Brown, V.; Weinstein, E.J.; Liaw, L.; Cui, X. Targeted genome modification in mice using zinc-finger nucleases. Genetics 2010, 186, 451–459. [Google Scholar] [CrossRef]
- Hockemeyer, D.; Soldner, F.; Beard, C.; Gao, Q.; Mitalipova, M.; DeKelver, R.C.; Katibah, G.E.; Amora, R.; Boydston, E.A.; Zeitler, B.; et al. Efficient targeting of expressed and silent genes in human ESCs and iPSCs using zinc-finger nucleases. Nat. Biotechnol. 2009, 27, 851–857. [Google Scholar] [CrossRef]
- Hoffman, E.J.; Turner, K.J.; Fernandez, J.M.; Cifuentes, D.; Ghosh, M.; Ijaz, S.; Jain, R.A.; Kubo, F.; Bill, B.R.; Baier, H.; et al. Estrogens suppress a behavioral phenotype in zebrafish mutants of the autism risk gene, CNTNAP2. Neuron 2016, 89, 725–733. [Google Scholar] [CrossRef]
- Amacher, S.L. Emerging gene knockout technology in zebrafish: Zinc-finger nucleases. Br. Funct Genom. Proteom. 2008, 7, 460–464. [Google Scholar] [CrossRef]
- Joung, J.K.; Sander, J.D. TALENs: A widely applicable technology for targeted genome editing. Nat. Rev. Mol. Cell Biol. 2013, 14, 49–55. [Google Scholar] [CrossRef] [PubMed]
- Wright, A.V.; Nuñez, J.K.; Doudna, J.A. Biology and applications of CRISPR systems: Harnessing nature’s toolbox for genome engineering. Cell 2016, 164, 29–44. [Google Scholar] [CrossRef] [PubMed]
- Abudayyeh, O.O.; Gootenberg, J.S.; Essletzbichler, P.; Han, S.; Joung, J.; Belanto, J.J.; Verdine, V.; Cox, D.B.T.; Kellner, M.J.; Regev, A.; et al. RNA targeting with CRISPR–Cas13. Nature 2017, 550, 280–284. [Google Scholar] [CrossRef]
- Konermann, S.; Lotfy, P.; Brideau, N.J.; Oki, J.; Shokhirev, M.N.; Hsu, P.D. Transcriptome engineering with RNA-Targeting Type VI-D CRISPR effectors. Cell 2018, 173, 665–676.e14. [Google Scholar] [CrossRef] [PubMed]
- Hu, J.H.; Miller, S.M.; Geurts, M.H.; Tang, W.; Chen, L.; Sun, N.; Zeina, C.M.; Gao, X.; Rees, H.A.; Lin, Z.; et al. Evolved Cas9 variants with broad PAM compatibility and high DNA specificity. Nature 2018, 556, 57–63. [Google Scholar] [CrossRef]
- Slaymaker, I.M.; Gao, L.; Zetsche, B.; Scott, D.A.; Yan, W.X.; Zhang, F. Rationally engineered Cas9 nucleases with improved specificity. Science 2016, 351, 84–88. [Google Scholar] [CrossRef]
- Qi, L.S.; Larson, M.H.; Gilbert, L.A.; Doudna, J.A.; Weissman, J.S.; Arkin, A.P.; Lim, W.A. Repurposing CRISPR as an RNA-guided platform for sequence-specific control of gene expression. Cell 2013, 152, 1173–1183. [Google Scholar] [CrossRef]
- Perez-Pinera, P.; Kocak, D.D.; Vockley, C.M.; Adler, A.F.; Kabadi, A.M.; Polstein, L.R.; Thakore, P.I.; Glass, K.A.; Ousterout, D.G.; Leong, K.W.; et al. RNA-guided gene activation by CRISPR-Cas9-based transcription factors. Nat. Methods 2013, 10, 973–976. [Google Scholar] [CrossRef]
- Gaudelli, N.M.; Komor, A.C.; Rees, H.A.; Packer, M.S.; Badran, A.H.; Bryson, D.I.; Liu, D.R. Programmable base editing of A•T to G•C in genomic DNA without DNA cleavage. Nature 2017, 551, 464–471. [Google Scholar] [CrossRef]
- Anzalone, A.V.; Randolph, P.B.; Davis, J.R.; Sousa, A.A.; Koblan, L.W.; Levy, J.M.; Chen, P.J.; Wilson, C.; Newby, G.A.; Raguram, A.; et al. Search-and-replace genome editing without double-strand breaks or donor DNA. Nature 2019, 576, 149–157. [Google Scholar] [CrossRef] [PubMed]
- Smith, C.; Gore, A.; Yan, W.; Abalde-Atristain, L.; Li, Z.; He, C.; Wang, Y.; Brodsky, R.A.; Zhang, K.; Cheng, L.; et al. Whole-Genome Sequencing analysis reveals high specificity of CRISPR/Cas9 and TALEN-based genome editing in human iPSCs. Cell Stem Cell 2014, 15, 12–13. [Google Scholar] [CrossRef] [PubMed]
- Cho, S.W.; Kim, S.; Kim, Y.; Kweon, J.; Kim, H.S.; Bae, S.; Kim, J.-S. Analysis of off-target effects of CRISPR/Cas-derived RNA-guided endonucleases and nickases. Genome Res. 2014, 24, 132–141. [Google Scholar] [CrossRef] [PubMed]
- Friedland, A.E.; Tzur, Y.B.; Esvelt, K.M.; Colaiácovo, M.P.; Church, G.M.; Calarco, J.A. Heritable genome editing in C. elegans via a CRISPR-Cas9 system. Nat. Methods 2013, 10, 741–743. [Google Scholar] [CrossRef] [PubMed]
- Bassett, A.R.; Tibbit, C.; Ponting, C.P.; Liu, J.-L. Highly efficient targeted mutagenesis of Drosophila with the CRISPR/Cas9 System. Cell Rep. 2013, 4, 220–228. [Google Scholar] [CrossRef] [PubMed]
- Hwang, W.Y.; Fu, Y.; Reyon, D.; Maeder, M.L.; Tsai, S.Q.; Sander, J.D.; Peterson, R.T.; Yeh, J.-R.J.; Joung, J.K. Efficient genome editing in zebrafish using a CRISPR-Cas system. Nat. Biotechnol. 2013, 31, 227–229. [Google Scholar] [CrossRef] [PubMed]
- Li, D.; Qiu, Z.; Shao, Y.; Chen, Y.; Guan, Y.; Liu, M.; Li, Y.; Gao, N.; Wang, L.; Lu, X.; et al. Heritable gene targeting in the mouse and rat using a CRISPR-Cas system. Nat. Biotechnol. 2013, 31, 681–683. [Google Scholar] [CrossRef]
- Zhou, Y.; Sharma, J.; Ke, Q.; Landman, R.; Yuan, J.; Chen, H.; Hayden, D.S.; Fisher, J.W.; Jiang, M.; Menegas, W.; et al. Atypical behaviour and connectivity in SHANK3-mutant macaques. Nature 2019, 570, 326–331. [Google Scholar] [CrossRef]
- Baltimore, D.; Berg, P.; Botchan, M.; Carroll, D.; Charo, R.A.; Church, G.; Corn, J.E.; Daley, G.Q.; Doudna, J.A.; Fenner, M.; et al. Biotechnology. A prudent path forward for genomic engineering and germline gene modification. Science 2015, 348, 36–38. [Google Scholar] [CrossRef]
- Boissart, C.; Poulet, A.; Georges, P.; Darville, H.; Julita, E.; Delorme, R.; Bourgeron, T.; Peschanski, M.; Benchoua, A. Differentiation from human pluripotent stem cells of cortical neurons of the superficial layers amenable to psychiatric disease modeling and high-throughput drug screening. Transl. Psychiatry 2013, 3, e294. [Google Scholar] [CrossRef]
- Shi, Y.; Inoue, H.; Wu, J.C.; Yamanaka, S. Induced pluripotent stem cell technology: A decade of progress. Nat. Rev. Drug Discov. 2017, 16, 115–130. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, K.; Yamanaka, S. Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. Cell 2006, 126, 663–676. [Google Scholar] [CrossRef] [PubMed]
- Juopperi, T.A.; Song, H.; Ming, G.-L. Modeling neurological diseases using patient-derived induced pluripotent stem cells. Future Neurol. 2011, 6, 363–373. [Google Scholar] [CrossRef] [PubMed]
- Brennand, K.J.; Simone, A.; Tran, N.; Gage, F.H. Modeling psychiatric disorders at the cellular and network levels. Mol. Psychiatry 2012, 17, 1239–1253. [Google Scholar] [CrossRef]
- Ross, P.J.; Zhang, W.B.; Mok, R.S.F.; Zaslavsky, K.; Deneault, E.; D’Abate, L.; Rodrigues, D.C.; Yuen, R.K.C.; Faheem, M.; Mufteev, M.; et al. Synaptic dysfunction in human neurons with autism-associated deletions in PTCHD1-AS. Biol. Psychiatry 2020, 87, 139–149. [Google Scholar] [CrossRef]
- Rontani, P.; Perche, O.; Greetham, L.; Jullien, N.; Gepner, B.; Féron, F.; Nivet, E.; Erard-Garcia, M. Impaired expression of the COSMOC/MOCOS gene unit in ASD patient stem cells. Mol. Psychiatry 2020. [Google Scholar] [CrossRef]
- Ross, P.J.; Mok, R.S.F.; Smith, B.S.; Rodrigues, D.C.; Mufteev, M.; Scherer, S.W.; Ellis, J. Modeling neuronal consequences of autism-associated gene regulatory variants with human induced pluripotent stem cells. Mol. Autism 2020, 11, 33. [Google Scholar] [CrossRef]
- Nagy, J.; Kobolák, J.; Berzsenyi, S.; Ábrahám, Z.; Avci, H.X.; Bock, I.; Bekes, Z.; Hodoscsek, B.; Chandrasekaran, A.; Téglási, A.; et al. Altered neurite morphology and cholinergic function of induced pluripotent stem cell-derived neurons from a patient with Kleefstra Syndrome and autism. Transl. Psychiatry 2017, 7, e1179. [Google Scholar] [CrossRef]
- Nageshappa, S.; Carromeu, C.; Trujillo, C.A.; Mesci, P.; Espuny-Camacho, I.; Pasciuto, E.; Vanderhaeghen, P.; Verfaillie, C.M.; Raitano, S.; Kumar, A.; et al. Altered neuronal network and rescue in a human MECP2 duplication model. Mol. Psychiatry 2016, 21, 178–188. [Google Scholar] [CrossRef]
- Avazzadeh, S.; McDonagh, K.; Reilly, J.; Wang, Y.; Boomkamp, S.D.; McInerney, V.; Krawczyk, J.; Fitzgerald, J.; Feerick, N.; O’Sullivan, M.; et al. Increased Ca2+ signaling in NRXN1α +/- neurons derived from ASD induced pluripotent stem cells. Mol. Autism 2019, 10, 52. [Google Scholar] [CrossRef]
- Zaslavsky, K.; Zhang, W.-B.; McCready, F.P.; Rodrigues, D.C.; Deneault, E.; Loo, C.; Zhao, M.; Ross, P.J.; El Hajjar, J.; Romm, A.; et al. SHANK2 mutations associated with autism spectrum disorder cause hyperconnectivity of human neurons. Nat. Neurosci. 2019, 22, 556–564. [Google Scholar] [CrossRef]
- Gouder, L.; Vitrac, A.; Goubran-Botros, H.; Danckaert, A.; Tinevez, J.Y.; André-Leroux, G.; Atanasova, E.; Lemière, N.; Biton, A.; Leblond, C.S.; et al. Altered spinogenesis in iPSC-derived cortical neurons from patients with autism carrying de novo SHANK3 mutations. Sci. Rep. 2019, 9, 94. [Google Scholar] [CrossRef]
- Darville, H.; Poulet, A.; Rodet-Amsellem, F.; Chatrousse, L.; Pernelle, J.; Boissart, C.; Héron, D.; Nava, C.; Perrier, A.; Jarrige, M.; et al. Human pluripotent stem cell-derived cortical neurons for high throughput medication screening in autism: A proof of concept study in SHANK3 haploinsufficiency syndrome. EBioMedicine 2016, 9, 293–305. [Google Scholar] [CrossRef] [PubMed]
- Ebrahimi-Fakhari, D.; Saffari, A.; Wahlster, L.; Di Nardo, A.; Turner, D.; Lewis, T.L.; Conrad, C.; Rothberg, J.M.; Lipton, J.O.; Kölker, S.; et al. Impaired mitochondrial dynamics and mitophagy in neuronal models of tuberous sclerosis complex. Cell Rep. 2016, 17, 1053–1070. [Google Scholar] [CrossRef] [PubMed]
- Winden, K.D.; Sundberg, M.; Yang, C.; Wafa, S.M.A.; Dwyer, S.; Chen, P.F.; Buttermore, E.D.; Sahin, M. Biallelic mutations in TSC2 lead to abnormalities associated with cortical tubers in human iPSC-derived neurons. J. Neurosci. 2019, 39, 9294–9305. [Google Scholar] [CrossRef] [PubMed]
- Arioka, Y.; Shishido, E.; Kubo, H.; Kushima, I.; Yoshimi, A.; Kimura, H.; Ishizuka, K.; Aleksic, B.; Maeda, T.; Ishikawa, M.; et al. Single-cell trajectory analysis of human homogenous neurons carrying a rare RELN variant. Transl. Psychiatry 2018, 8, 129. [Google Scholar] [CrossRef]
- Deneault, E.; White, S.H.; Rodrigues, D.C.; Ross, P.J.; Faheem, M.; Zaslavsky, K.; Wang, Z.; Alexandrova, R.; Pellecchia, G.; Wei, W.; et al. Complete disruption of autism-susceptibility genes by gene editing predominantly reduces functional connectivity of isogenic human neurons. Stem Cell Rep. 2018, 11, 1211–1225. [Google Scholar] [CrossRef]
- Deneault, E.; Faheem, M.; White, S.H.; Rodrigues, D.C.; Sun, S.; Wei, W.; Piekna, A.; Thompson, T.; Howe, J.L.; Chalil, L.; et al. CNTN5−/+ or EHMT2−/+ human iPSC-derived neurons from individuals with autism develop hyperactive neuronal networks. Elife 2019, 8, e40092. [Google Scholar] [CrossRef]
- Machado, C.O.F.; Griesi-Oliveira, K.; Rosenberg, C.; Kok, F.; Martins, S.; Rita Passos-Bueno, M.; Sertie, A.L. Collybistin binds and inhibits mTORC1 signaling: A potential novel mechanism contributing to intellectual disability and autism. Eur. J. Hum. Genet. 2016, 24, 59–65. [Google Scholar] [CrossRef]
- Paşca, S.P.; Portmann, T.; Voineagu, I.; Yazawa, M.; Shcheglovitov, A.; Paşca, A.M.; Cord, B.; Palmer, T.D.; Chikahisa, S.; Nishino, S.; et al. Using iPSC-derived neurons to uncover cellular phenotypes associated with Timothy Syndrome. Nat. Med. 2011, 17, 1657–1662. [Google Scholar] [CrossRef]
- Tian, Y.; Voineagu, I.; Paşca, S.P.; Won, H.; Chandran, V.; Horvath, S.; Dolmetsch, R.E.; Geschwind, D.H. Alteration in basal and depolarization induced transcriptional network in iPSC derived neurons from Timothy Syndrome. Genome Med. 2014, 6, 75. [Google Scholar] [CrossRef] [PubMed]
- Gao, Y.; Irvine, E.E.; Eleftheriadou, I.; Naranjo, C.J.; Hearn-Yeates, F.; Bosch, L.; Glegola, J.A.; Murdoch, L.; Czerniak, A.; Meloni, I.; et al. Gene replacement ameliorates deficits in mouse and human models of cyclin-dependent kinase-like 5 disorder. Brain 2020, 143, 811–832. [Google Scholar] [CrossRef] [PubMed]
- Wang, P.; Lin, M.; Pedrosa, E.; Hrabovsky, A.; Zhang, Z.; Guo, W.; Lachman, H.M.; Zheng, D. CRISPR/Cas9-mediated heterozygous knockout of the autism gene CHD8 and characterization of its transcriptional networks in neurodevelopment. Mol. Autism 2015, 6, 55. [Google Scholar] [CrossRef] [PubMed]
- Boland, M.J.; Nazor, K.L.; Tran, H.T.; Szücs, A.; Lynch, C.L.; Paredes, R.; Tassone, F.; Sanna, P.P.; Hagerman, R.J.; Loring, J.F. Molecular analyses of neurogenic defects in a human pluripotent stem cell model of Fragile X Syndrome. Brain 2017, 140, 582–598. [Google Scholar] [CrossRef] [PubMed]
- Doers, M.E.; Musser, M.T.; Nichol, R.; Berndt, E.R.; Baker, M.; Gomez, T.M.; Zhang, S.C.; Abbeduto, L.; Bhattacharyya, A. iPSC-derived forebrain neurons from FXS individuals show defects in initial neurite outgrowth. Stem Cells Dev. 2014, 23, 1777–1787. [Google Scholar] [CrossRef] [PubMed]
- Sheridan, S.D.; Theriault, K.M.; Reis, S.A.; Zhou, F.; Madison, J.M.; Daheron, L.; Loring, J.F.; Haggarty, S.J. Epigenetic characterization of the FMR1 gene and aberrant neurodevelopment in human induced pluripotent stem cell models of Fragile X Syndrome. PLoS ONE 2011, 6, e26203. [Google Scholar] [CrossRef] [PubMed]
- Huang, G.; Chen, S.; Chen, X.; Zheng, J.; Xu, Z.; Torshizi, A.D.; Gong, S.; Chen, Q.; Ma, X.; Yu, J.; et al. Uncovering the functional link between SHANK3 deletions and deficiency in neurodevelopment using iPSC-derived human neurons. Front. Neuroanat. 2019, 13, 23. [Google Scholar] [CrossRef] [PubMed]
- Shcheglovitov, A.; Shcheglovitova, O.; Yazawa, M.; Portmann, T.; Shu, R.; Sebastiano, V.; Krawisz, A.; Froehlich, W.; Bernstein, J.A.; Hallmayer, J.F.; et al. SHANK3 and IGF1 restore synaptic deficits in neurons from 22q13 deletion syndrome patients. Nature 2013, 503, 267–271. [Google Scholar] [CrossRef]
- Griesi-Oliveira, K.; Acab, A.; Gupta, A.R.; Sunaga, D.Y.; Chailangkarn, T.; Nicol, X.; Nunez, Y.; Walker, M.F.; Murdoch, J.D.; Sanders, S.J.; et al. Modeling non-syndromic autism and the impact of TRPC6 disruption in human neurons. Mol. Psychiatry 2015, 20, 1350–1365. [Google Scholar] [CrossRef]
- Wang, P.; Mokhtari, R.; Pedrosa, E.; Kirschenbaum, M.; Bayrak, C.; Zheng, D.; Lachman, H.M. CRISPR/Cas9-mediated heterozygous knockout of the autism gene CHD8 and characterization of its transcriptional networks in cerebral organoids derived from iPS cells. Mol. Autism 2017, 8, 11. [Google Scholar] [CrossRef]
- Lam, M.; Moslem, M.; Bryois, J.; Pronk, R.J.; Uhlin, E.; Ellström, I.D.; Laan, L.; Olive, J.; Morse, R.; Rönnholm, H.; et al. Single cell analysis of autism patient with bi-allelic NRXN1-alpha deletion reveals skewed fate choice in neural progenitors and impaired neuronal functionality. Exp. Cell Res. 2019, 383, 111469. [Google Scholar] [CrossRef] [PubMed]
- Zeng, L.; Zhang, P.; Shi, L.; Yamamoto, V.; Lu, W.; Wang, K. Functional impacts of NRXN1 knockdown on neurodevelopment in stem cell models. PLoS ONE 2013, 8, e59685. [Google Scholar] [CrossRef] [PubMed]
- Sánchez-Sánchez, S.M.; Magdalon, J.; Griesi-Oliveira, K.; Yamamoto, G.L.; Santacruz-Perez, C.; Fogo, M.; Passos-Bueno, M.R.; Sertié, A.L. Rare RELN variants affect Reelin-DAB1 signal transduction in Autism Spectrum Disorder. Hum. Mutat. 2018, 39, 1372–1383. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Lin, M.; Hrabovsky, A.; Pedrosa, E.; Dean, J.; Jain, S.; Zheng, D.; Lachman, H.M. ZNF804A transcriptional networks in differentiating neurons derived from induced pluripotent stem cells of human origin. PLoS ONE 2015, 10, e0124597. [Google Scholar] [CrossRef] [PubMed]
- Kathuria, A.; Nowosiad, P.; Jagasia, R.; Aigner, S.; Taylor, R.D.; Andreae, L.C.; Gatford, N.J.F.; Lucchesi, W.; Srivastava, D.P.; Price, J. Stem cell-derived neurons from autistic individuals with SHANK3 mutation show morphogenetic abnormalities during early development. Mol. Psychiatry 2018, 23, 735–746. [Google Scholar] [CrossRef]
- Sundberg, M.; Tochitsky, I.; Buchholz, D.E.; Winden, K.; Kujala, V.; Kapur, K.; Cataltepe, D.; Turner, D.; Han, M.J.; Woolf, C.J.; et al. Purkinje cells derived from TSC patients display hypoexcitability and synaptic deficits associated with reduced FMRP levels and reversed by rapamycin. Mol. Psychiatry 2018, 23, 2167–2183. [Google Scholar] [CrossRef]
- Silverman, J.L.; Yang, M.; Lord, C.; Crawley, J.N. Behavioural phenotyping assays for mouse models of autism. Nat. Rev. Neurosci. 2010, 11, 490–502. [Google Scholar] [CrossRef] [PubMed]
- Patterson, P.H. Modeling autistic features in animals. Pediatr. Res. 2011, 69, 34R–40R. [Google Scholar] [CrossRef]
- Norton, W.H.J. Toward developmental models of psychiatric disorders in zebrafish. Front. Neural Circuits 2013, 7, 79. [Google Scholar] [CrossRef]
- Sakai, C.; Ijaz, S.; Hoffman, E.J. Zebrafish models of neurodevelopmental disorders: Past, present, and future. Front. Mol. Neurosci. 2018, 11, 294. [Google Scholar] [CrossRef]
- Ellenbroek, B.; Youn, J. Rodent models in neuroscience research: Is it a rat race? Dis. Model. Mech. 2016, 9, 1079–1087. [Google Scholar] [CrossRef] [PubMed]
- Wöhr, M.; Scattoni, M.L. Behavioural methods used in rodent models of Autism Spectrum Disorders: Current standards and new developments. Behav. Brain Res. 2013, 251, 5–17. [Google Scholar] [CrossRef] [PubMed]
- Meshalkina, D.A.; Kizlyk, M.N.; Kysil, E.V.; Collier, A.D.; Echevarria, D.J.; Abreu, M.S.; Barcellos, L.J.G.; Song, C.; Warnick, J.E.; Kyzar, E.J.; et al. Zebrafish models of Autism Spectrum Disorder. Exp. Neurol. 2018, 299, 207–216. [Google Scholar] [CrossRef] [PubMed]
- Meyza, K.Z.; Defensor, E.B.; Jensen, A.L.; Corley, M.J.; Pearson, B.L.; Pobbe, R.L.H.; Bolivar, V.J.; Blanchard, D.C.; Blanchard, R.J. The BTBR T+tf/J mouse model for Autism Spectrum Disorders–in search of biomarkers. Behav. Brain Res. 2013, 251, 25–34. [Google Scholar] [CrossRef]
- Amram, N.; Hacohen-Kleiman, G.; Sragovich, S.; Malishkevich, A.; Katz, J.; Touloumi, O.; Lagoudaki, R.; Grigoriadis, N.C.; Giladi, E.; Yeheskel, A.; et al. Sexual divergence in microtubule function: The novel intranasal microtubule targeting SKIP normalizes axonal transport and enhances memory. Mol. Psychiatry 2016, 21, 1467–1476. [Google Scholar] [CrossRef]
- Sragovich, S.; Malishkevich, A.; Piontkewitz, Y.; Giladi, E.; Touloumi, O.; Lagoudaki, R.; Grigoriadis, N.; Gozes, I. The autism/neuroprotection-linked ADNP/NAP regulate the excitatory glutamatergic synapse. Transl. Psychiatry 2019, 9, 2. [Google Scholar] [CrossRef]
- Vulih-Shultzman, I.; Pinhasov, A.; Mandel, S.; Grigoriadis, N.; Touloumi, O.; Pittel, Z.; Gozes, I. Activity-dependent neuroprotective protein snippet NAP reduces tau hyperphosphorylation and enhances learning in a novel transgenic mouse model. J. Pharmacol. Exp. Ther. 2007, 323, 438–449. [Google Scholar] [CrossRef]
- Celen, C.; Chuang, J.-C.; Luo, X.; Nijem, N.; Walker, A.K.; Chen, F.; Zhang, S.; Chung, A.S.; Nguyen, L.H.; Nassour, I.; et al. Arid1b haploinsufficient mice reveal neuropsychiatric phenotypes and reversible causes of growth impairment. Elife 2017, 6, e25730. [Google Scholar] [CrossRef]
- Shibutani, M.; Horii, T.; Shoji, H.; Morita, S.; Kimura, M.; Terawaki, N.; Miyakawa, T.; Hatada, I.; Shibutani, M.; Horii, T.; et al. Arid1b Haploinsufficiency Causes Abnormal Brain Gene Expression and Autism-Related Behaviors in Mice. Int. J. Mol. Sci. 2017, 18, 1872. [Google Scholar] [CrossRef]
- Brinkmeier, M.L.; Geister, K.A.; Jones, M.; Waqas, M.; Maillard, I.; Camper, S.A. The histone methyltransferase gene Absent, Small, or Homeotic Discs-1 Like is required for normal hox gene expression and fertility in mice. Biol. Reprod. 2015, 93, 121. [Google Scholar] [CrossRef]
- Xia, M.; Liu, J.; Wu, X.; Liu, S.; Li, G.; Han, C.; Song, L.; Li, Z.; Wang, Q.; Wang, J.; et al. Histone Methyltransferase Ash1l Suppresses Interleukin-6 Production and Inflammatory Autoimmune Diseases by Inducing the Ubiquitin-Editing Enzyme A20. Immunity 2013, 39, 470–481. [Google Scholar] [CrossRef] [PubMed]
- Zhu, T.; Liang, C.; Li, D.; Tian, M.; Liu, S.; Gao, G.; Guan, J.S. Histone methyltransferase Ash1L mediates activity-dependent repression of neurexin-1α. Sci. Rep. 2016, 6, 26597. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.J.; Khoshkhoo, S.; Frankowski, J.C.; Zhu, B.; Abbasi, S.; Lee, S.; Wu, Y.E.; Hunt, R.F. Chd2 Is Necessary for Neural Circuit Development and Long-Term Memory. Neuron 2018, 100, 1180–1193.e6. [Google Scholar] [CrossRef] [PubMed]
- Nagarajan, P.; Onami, T.M.; Rajagopalan, S.; Kania, S.; Donnell, R.; Venkatachalam, S. Role of chromodomain helicase DNA-binding protein 2 in DNA damage response signaling and tumorigenesis. Oncogene 2009, 28, 1053–1062. [Google Scholar] [CrossRef][Green Version]
- Durak, O.; Gao, F.; Kaeser-Woo, Y.J.; Rueda, R.; Martorell, A.J.; Nott, A.; Liu, C.Y.; Watson, L.A.; Tsai, L.-H. Chd8 mediates cortical neurogenesis via transcriptional regulation of cell cycle and Wnt signaling. Nat. Neurosci. 2016, 19, 1477–1488. [Google Scholar] [CrossRef] [PubMed]
- Gompers, A.L.; Su-Feher, L.; Ellegood, J.; Copping, N.A.; Riyadh, M.A.; Stradleigh, T.W.; Pride, M.C.; Schaffler, M.D.; Wade, A.A.; Catta-Preta, R.; et al. Germline Chd8 haploinsufficiency alters brain development in mouse. Nat. Neurosci. 2017, 20, 1062–1073. [Google Scholar] [CrossRef] [PubMed]
- Jung, H.; Park, H.; Choi, Y.; Kang, H.; Lee, E.; Kweon, H.; Roh, J.D.; Ellegood, J.; Choi, W.; Kang, J.; et al. Sexually dimorphic behavior, neuronal activity, and gene expression in Chd8-mutant mice. Nat. Neurosci. 2018, 21, 1218–1228. [Google Scholar] [CrossRef]
- Katayama, Y.; Nishiyama, M.; Shoji, H.; Ohkawa, Y.; Kawamura, A.; Sato, T.; Suyama, M.; Takumi, T.; Miyakawa, T.; Nakayama, K.I. CHD8 haploinsufficiency results in autistic-like phenotypes in mice. Nature 2016, 537, 675–679. [Google Scholar] [CrossRef]
- Nishiyama, M.; Oshikawa, K.; Tsukada, Y.; Nakagawa, T.; Iemura, S.; Natsume, T.; Fan, Y.; Kikuchi, A.; Skoultchi, A.I.; Nakayama, K.I. CHD8 suppresses p53-mediated apoptosis through histone H1 recruitment during early embryogenesis. Nat. Cell Biol. 2009, 11, 172–182. [Google Scholar] [CrossRef]
- Platt, R.J.; Zhou, Y.; Slaymaker, I.M.; Shetty, A.S.; Weisbach, N.R.; Kim, J.-A.; Sharma, J.; Desai, M.; Sood, S.; Kempton, H.R.; et al. Chd8 Mutation Leads to Autistic-like Behaviors and Impaired Striatal Circuits. Cell Rep. 2017, 19, 335–350. [Google Scholar] [CrossRef]
- Suetterlin, P.; Hurley, S.; Mohan, C.; Riegman, K.L.H.; Pagani, M.; Caruso, A.; Ellegood, J.; Galbusera, A.; Crespo-Enriquez, I.; Michetti, C.; et al. Altered neocortical gene expression, brain overgrowth and functional over-connectivity in chd8 haploinsufficient mice. Cereb. Cortex 2018, 28, 2192–2206. [Google Scholar] [CrossRef] [PubMed]
- Zhao, C.; Dong, C.; Frah, M.; Deng, Y.; Marie, C.; Zhang, F.; Xu, L.; Ma, Z.; Dong, X.; Lin, Y.; et al. Dual Requirement of CHD8 for Chromatin Landscape Establishment and Histone Methyltransferase Recruitment to Promote CNS Myelination and Repair. Dev. Cell 2018, 45, 753–768.e8. [Google Scholar] [CrossRef] [PubMed]
- Lu, H.-C.; Tan, Q.; Rousseaux, M.W.C.; Wang, W.; Kim, J.-Y.; Richman, R.; Wan, Y.-W.; Yeh, S.-Y.; Patel, J.M.; Liu, X.; et al. Disruption of the ATXN1–CIC complex causes a spectrum of neurobehavioral phenotypes in mice and humans. Nat. Genet. 2017, 49, 527–536. [Google Scholar] [CrossRef] [PubMed]
- Horresh, I.; Bar, V.; Kissil, J.L.; Peles, E. Organization of myelinated axons by Caspr and Caspr2 requires the cytoskeletal adapter protein 4.1B. J. Neurosci. 2010, 30, 2480–2489. [Google Scholar] [CrossRef] [PubMed]
- Peñagarikano, O.; Abrahams, B.S.; Herman, E.I.; Winden, K.D.; Gdalyahu, A.; Dong, H.; Sonnenblick, L.I.; Gruver, R.; Almajano, J.; Bragin, A.; et al. Absence of CNTNAP2 leads to epilepsy, neuronal migration abnormalities, and core autism-related deficits. Cell 2011, 147, 235–246. [Google Scholar] [CrossRef]
- Peñagarikano, O.; Lázaro, M.T.; Lu, X.-H.; Gordon, A.; Dong, H.; Lam, H.A.; Peles, E.; Maidment, N.T.; Murphy, N.P.; Yang, X.W.; et al. Exogenous and evoked oxytocin restores social behavior in the Cntnap2 mouse model of autism. Sci. Transl. Med. 2015, 7, ra8–ra271. [Google Scholar] [CrossRef]
- Poliak, S.; Salomon, D.; Elhanany, H.; Sabanay, H.; Kiernan, B.; Pevny, L.; Stewart, C.L.; Xu, X.; Chiu, S.-Y.; Shrager, P.; et al. Juxtaparanodal clustering of Shaker-like K+ channels in myelinated axons depends on Caspr2 and TAG-1. J. Cell Biol. 2003, 162, 1149–1160. [Google Scholar] [CrossRef]
- Schaafsma, S.M.; Gagnidze, K.; Reyes, A.; Norstedt, N.; Månsson, K.; Francis, K.; Pfaff, D.W. Sex-specific gene–environment interactions underlying ASD-like behaviors. Proc. Natl. Acad. Sci. USA 2017, 114, 1383–1388. [Google Scholar] [CrossRef]
- Selimbeyoglu, A.; Kim, C.K.; Inoue, M.; Lee, S.Y.; Hong, A.S.O.; Kauvar, I.; Ramakrishnan, C.; Fenno, L.E.; Davidson, T.J.; Wright, M.; et al. Modulation of prefrontal cortex excitation/inhibition balance rescues social behavior in CNTNAP2 -deficient mice. Sci. Transl. Med. 2017, 9, eaah6733. [Google Scholar] [CrossRef]
- DeLorey, T.M.; Handforth, A.; Anagnostaras, S.G.; Homanics, G.E.; Minassian, B.A.; Asatourian, A.; Fanselow, M.S.; Delgado-Escueta, A.; Ellison, G.D.; Olsen, R.W. Mice lacking the β3 subunit of the GABA(A) receptor have the epilepsy phenotype and many of the behavioral characteristics of Angelman syndrome. J. Neurosci. 1998, 18, 8505–8514. [Google Scholar] [CrossRef]
- DeLorey, T.M.; Sahbaie, P.; Hashemi, E.; Li, W.-W.; Salehi, A.; Clark, D.J. Somatosensory and sensorimotor consequences associated with the heterozygous disruption of the autism candidate gene, Gabrb3. Behav. Brain Res. 2011, 216, 36–45. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Kumar, T.P.; Joshee, S.; Kirschstein, T.; Subburaju, S.; Khalili, J.S.; Kloepper, J.; Du, C.; Elkhal, A.; Szabó, G.; et al. Endothelial cell-derived GABA signaling modulates neuronal migration and postnatal behavior. Cell Res. 2018, 28, 221–248. [Google Scholar] [CrossRef] [PubMed]
- Liljelund, P.; Handforth, A.; Homanics, G.E.; Olsen, R.W. GABAA receptor β3 subunit gene-deficient heterozygous mice show parent-of-origin and gender-related differences in β3 subunit levels, EEG, and behavior. Dev. Brain Res. 2005, 157, 150–161. [Google Scholar] [CrossRef] [PubMed]
- Orefice, L.L.; Zimmerman, A.L.; Chirila, A.M.; Sleboda, S.J.; Head, J.P.; Ginty, D.D. Peripheral Mechanosensory Neuron Dysfunction Underlies Tactile and Behavioral Deficits in Mouse Models of ASDs. Cell 2016, 166, 299–313. [Google Scholar] [CrossRef]
- Cabral-Costa, J.V.; Andreotti, D.Z.; Mello, N.P.; Scavone, C.; Camandola, S.; Kawamoto, E.M. Intermittent fasting uncovers and rescues cognitive phenotypes in PTEN neuronal haploinsufficient mice. Sci. Rep. 2018, 8, 8595. [Google Scholar] [CrossRef]
- Clipperton-Allen, A.E.; Page, D.T. Decreased aggression and increased repetitive behavior in Pten haploinsufficient mice. Genes Brain Behav. 2015, 14, 145–157. [Google Scholar] [CrossRef]
- Cupolillo, D.; Hoxha, E.; Faralli, A.; De Luca, A.; Rossi, F.; Tempia, F.; Carulli, D. Autistic-like traits and cerebellar dysfunction in purkinje cell PTEN knock-out mice. Neuropsychopharmacology 2016, 41, 1457–1466. [Google Scholar] [CrossRef]
- Kwon, C.-H.; Luikart, B.W.; Powell, C.M.; Zhou, J.; Matheny, S.A.; Zhang, W.; Li, Y.; Baker, S.J.; Parada, L.F. Pten Regulates Neuronal Arborization and Social Interaction in Mice. Neuron 2006, 50, 377–388. [Google Scholar] [CrossRef]
- Vogt, D.; Cho, K.K.A.; Lee, A.T.; Sohal, V.S.; Rubenstein, J.L.R. The Parvalbumin/Somatostatin Ratio Is Increased in Pten Mutant Mice and by Human PTEN ASD Alleles. Cell Rep. 2015, 11, 944–956. [Google Scholar] [CrossRef]
- Williams, M.R.; DeSpenza, T.; Li, M.; Gulledge, A.T.; Luikart, B.W. Hyperactivity of Newborn Pten Knock-out Neurons Results from Increased Excitatory Synaptic Drive. J. Neurosci. 2015, 35, 943–959. [Google Scholar] [CrossRef]
- Zhou, J.; Blundell, J.; Ogawa, S.; Kwon, C.H.; Zhang, W.; Sinton, C.; Powell, C.M.; Parada, L.F. Pharmacological inhibition of mTORCl suppresses anatomical, cellular, and behavioral abnormalities in neural-specific PTEN knock-out mice. J. Neurosci. 2009, 29, 1773–1783. [Google Scholar] [CrossRef] [PubMed]
- Mullen, B.R.; Khialeeva, E.; Hoffman, D.B.; Ghiani, C.A.; Carpenter, E.M. Decreased reelin expression and organophosphate pesticide exposure alters mouse behaviour and brain morphology. ASN Neuro 2013, 5, 27–42. [Google Scholar] [CrossRef] [PubMed]
- Nyarenchi, O.M.; Scherer, A.; Wilson, S.; Fulkerson, D.H. Cloacal exstrophy with extensive Chiari II malformation: Case report and review of the literature. Child’s Nerv. Syst. 2014, 30, 337–343. [Google Scholar] [CrossRef] [PubMed]
- Rice, D.S.; Nusinowitz, S.; Azimi, A.M.; Martínez, A.; Soriano, E.; Curran, T. The Reelin Pathway Modulates the Structure and Function of Retinal Synaptic Circuitry. Neuron 2001, 31, 929–941. [Google Scholar] [CrossRef]
- Hawkins, N.A.; Martin, M.S.; Frankel, W.N.; Kearney, J.A.; Escayg, A. Neuronal voltage-gated ion channels are genetic modifiers of generalized epilepsy with febrile seizures plus. Neurobiol. Dis. 2011, 41, 655–660. [Google Scholar] [CrossRef] [PubMed]
- Planells-Cases, R.; Caprini, M.; Zhang, J.; Rockenstein, E.M.; Rivera, R.R.; Murre, C.; Masliah, E.; Montal, M. Neuronal death and perinatal lethality in voltage-gated sodium channel α(II)-deficient mice. Biophys. J. 2000, 78, 2878–2891. [Google Scholar] [CrossRef]
- Tatsukawa, T.; Raveau, M.; Ogiwara, I.; Hattori, S.; Miyamoto, H.; Mazaki, E.; Itohara, S.; Miyakawa, T.; Montal, M.; Yamakawa, K. Scn2a haploinsufficient mice display a spectrum of phenotypes affecting anxiety, sociability, memory flexibility and ampakine CX516 rescues their hyperactivity. Mol. Autism 2019, 10, 15. [Google Scholar] [CrossRef] [PubMed]
- Chung, C.; Ha, S.; Kang, H.; Lee, J.; Um, S.M.; Yan, H.; Yoo, Y.-E.; Yoo, T.; Jung, H.; Lee, D.; et al. Early Correction of N-Methyl-D-Aspartate Receptor Function Improves Autistic-like Social Behaviors in Adult Shank2−/− Mice. Biol. Psychiatry 2019, 85, 534–543. [Google Scholar] [CrossRef]
- Ha, S.; Lee, D.; Cho, Y.S.; Chung, C.; Yoo, Y.-E.; Kim, J.; Lee, J.; Kim, W.; Kim, H.; Bae, Y.C.; et al. Cerebellar Shank2 Regulates Excitatory Synapse Density, Motor Coordination, and Specific Repetitive and Anxiety-Like Behaviors. J. Neurosci. 2016, 36, 12129–12143. [Google Scholar] [CrossRef]
- Lee, E.-J.; Lee, H.; Huang, T.-N.; Chung, C.; Shin, W.; Kim, K.; Koh, J.-Y.; Hsueh, Y.-P.; Kim, E. Trans-synaptic zinc mobilization improves social interaction in two mouse models of autism through NMDAR activation. Nat. Commun. 2015, 6, 7168. [Google Scholar] [CrossRef]
- Lim, C.-S.; Kim, H.; Yu, N.-K.; Kang, S.J.; Kim, T.; Ko, H.-G.; Lee, J.; Yang, J.; Ryu, H.-H.; Park, T.; et al. Enhancing inhibitory synaptic function reverses spatial memory deficits in Shank2 mutant mice. Neuropharmacology 2017, 112, 104–112. [Google Scholar] [CrossRef] [PubMed]
- Won, H.; Lee, H.-R.; Gee, H.Y.; Mah, W.; Kim, J.-I.; Lee, J.; Ha, S.; Chung, C.; Jung, E.S.; Cho, Y.S.; et al. Autistic-like social behaviour in Shank2-mutant mice improved by restoring NMDA receptor function. Nature 2012, 486, 261–265. [Google Scholar] [CrossRef] [PubMed]
- Richter, M.; Murtaza, N.; Scharrenberg, R.; White, S.H.; Johanns, O.; Walker, S.; Yuen, R.K.C.; Schwanke, B.; Bedürftig, B.; Henis, M.; et al. Altered TAOK2 activity causes autism-related neurodevelopmental and cognitive abnormalities through RhoA signaling. Mol. Psychiatry 2019, 24, 1329–1350. [Google Scholar] [CrossRef] [PubMed]
- Fazel Darbandi, S.; Robinson Schwartz, S.E.; Qi, Q.; Catta-Preta, R.; Pai, E.L.-L.; Mandell, J.D.; Everitt, A.; Rubin, A.; Krasnoff, R.A.; Katzman, S.; et al. Neonatal Tbr1 Dosage Controls Cortical Layer 6 Connectivity. Neuron 2018, 100, 831–845.e7. [Google Scholar] [CrossRef] [PubMed]
- Hevner, R.F.; Shi, L.; Justice, N.; Hsueh, Y.; Sheng, M.; Smiga, S.; Bulfone, A.; Goffinet, A.M.; Campagnoni, A.T.; Rubenstein, J.L. Tbr1 regulates differentiation of the preplate and layer 6. Neuron 2001, 29, 353–366. [Google Scholar] [CrossRef]
- Huang, T.-N.; Yen, T.-L.; Qiu, L.R.; Chuang, H.-C.; Lerch, J.P.; Hsueh, Y.-P. Haploinsufficiency of autism causative gene Tbr1 impairs olfactory discrimination and neuronal activation of the olfactory system in mice. Mol. Autism 2019, 10, 5. [Google Scholar] [CrossRef]
- Huang, T.-N.; Chuang, H.-C.; Chou, W.-H.; Chen, C.-Y.; Wang, H.-F.; Chou, S.-J.; Hsueh, Y.-P. Tbr1 haploinsufficiency impairs amygdalar axonal projections and results in cognitive abnormality. Nat. Neurosci. 2014, 17, 240–247. [Google Scholar] [CrossRef]
- Huang, L.; Shum, E.Y.; Jones, S.H.; Lou, C.-H.; Dumdie, J.; Kim, H.; Roberts, A.J.; Jolly, L.A.; Espinoza, J.L.; Skarbrevik, D.M.; et al. A Upf3b-mutant mouse model with behavioral and neurogenesis defects. Mol. Psychiatry 2018, 23, 1773–1786. [Google Scholar] [CrossRef]
- Zigler, J.S.; Hodgkinson, C.A.; Wright, M.; Klise, A.; Sundin, O.; Broman, K.W.; Hejtmancik, F.; Huang, H.; Patek, B.; Sergeev, Y.; et al. A Spontaneous missense mutation in branched chain keto acid dehydrogenase kinase in the rat affects both the central and peripheral nervous systems. PLoS ONE 2016, 11, e0160447. [Google Scholar] [CrossRef]
- Kisko, T.M.; Braun, M.D.; Michels, S.; Witt, S.H.; Rietschel, M.; Culmsee, C.; Schwarting, R.K.W.; Wöhr, M. Cacna1c haploinsufficiency leads to pro-social 50-kHz ultrasonic communication deficits in rats. Dis. Model. Mech. 2018, 11, dmm034116. [Google Scholar] [CrossRef]
- Wöhr, M.; Willadsen, M.; Kisko, T.M.; Schwarting, R.K.W.; Fendt, M. Sex-dependent effects of Cacna1c haploinsufficiency on behavioral inhibition evoked by conspecific alarm signals in rats. Prog. Neuro-Psychopharmacol. Biol. Psychiatry 2020, 99, 109849. [Google Scholar] [CrossRef] [PubMed]
- Scott, K.E.; Schormans, A.L.; Pacoli, K.Y.; De Oliveira, C.; Allman, B.L.; Schmid, S. Altered auditory processing, filtering, and reactivity in the Cntnap2 knock-out rat model for neurodevelopmental disorders. J. Neurosci. 2018, 38, 8588–8604. [Google Scholar] [CrossRef] [PubMed]
- Thomas, A.M.; Schwartz, M.D.; Saxe, M.D.; Kilduff, T.S. Cntnap2 knockout rats and mice exhibit epileptiform activity and abnormal sleep–wake physiology. Sleep 2017, 40. [Google Scholar] [CrossRef] [PubMed]
- Silva, A.I.; Haddon, J.E.; Ahmed Syed, Y.; Trent, S.; Lin, T.C.E.; Patel, Y.; Carter, J.; Haan, N.; Honey, R.C.; Humby, T.; et al. Cyfip1 haploinsufficient rats show white matter changes, myelin thinning, abnormal oligodendrocytes and behavioural inflexibility. Nat. Commun. 2019, 10, 3455. [Google Scholar] [CrossRef] [PubMed]
- Asiminas, A.; Jackson, A.D.; Louros, S.R.; Till, S.M.; Spano, T.; Dando, O.; Bear, M.F.; Chattarji, S.; Hardingham, G.E.; Osterweil, E.K.; et al. Sustained correction of associative learning deficits after brief, early treatment in a rat model of Fragile X Syndrome. Sci. Transl. Med. 2019, 11. [Google Scholar] [CrossRef] [PubMed]
- Hamilton, S.M.; Green, J.R.; Veeraragavan, S.; Yuva, L.; McCoy, A.; Wu, Y.; Warren, J.; Little, L.; Ji, D.; Cui, X.; et al. Fmr1 and Nlgn3 knockout rats: Novel tools for investigating autism spectrum disorders. Behav. Neurosci. 2014, 128, 103–109. [Google Scholar] [CrossRef] [PubMed]
- Ruby, K.; Falvey, K.; Kulesza, R.J. Abnormal neuronal morphology and neurochemistry in the auditory brainstem of Fmr1 knockout rats. Neuroscience 2015, 303, 285–298. [Google Scholar] [CrossRef]
- Engineer, C.T.; Rahebi, K.C.; Borland, M.S.; Buell, E.P.; Centanni, T.M.; Fink, M.K.; Im, K.W.; Wilson, L.G.; Kilgard, M.P. Degraded neural and behavioral processing of speech sounds in a rat model of Rett syndrome. Neurobiol. Dis. 2015, 83, 26–34. [Google Scholar] [CrossRef]
- Wu, Y.; Zhong, W.; Cui, N.; Johnson, C.M.; Xing, H.; Zhang, S.; Jiang, C. Characterization of Rett Syndrome-like phenotypes in Mecp2-knockout rats. J. Neurodev. Disord. 2016, 8, 23. [Google Scholar] [CrossRef]
- Kohl, C.; Riccio, O.; Grosse, J.; Zanoletti, O.; Fournier, C.; Schmidt, M.V.; Sandi, C. Hippocampal neuroligin-2 overexpression leads to reduced aggression and inhibited novelty reactivity in rats. PLoS ONE 2013, 8, e56871. [Google Scholar] [CrossRef]
- Thomas, A.M.; Schwartz, M.D.; Saxe, M.D.; Kilduff, T.S. Sleep/wake physiology and quantitative electroencephalogram analysis of the Neuroligin-3 knockout rat model of Autism Spectrum Disorder. Sleep 2017, 40. [Google Scholar] [CrossRef] [PubMed]
- Esclassan, F.; Francois, J.; Phillips, K.G.; Loomis, S.; Gilmour, G. Phenotypic characterization of nonsocial behavioral impairment in neurexin 1α knockout rats. Behav. Neurosci. 2015, 129, 74–85. [Google Scholar] [CrossRef] [PubMed]
- Rowley, P.A.; Guerrero-Gonzalez, J.; Alexander, A.L.; Yu, J.-P.J. Convergent microstructural brain changes across genetic models of Autism Spectrum Disorder—A pilot study. Psychiatry Res. Neuroimaging 2019, 283, 83–91. [Google Scholar] [CrossRef]
- Ohmori, I.; Kawakami, N.; Liu, S.; Wang, H.; Miyazaki, I.; Asanuma, M.; Michiue, H.; Matsui, H.; Mashimo, T.; Ouchida, M. Methylphenidate improves learning impairments and hyperthermia-induced seizures caused by an Scn1a mutation. Epilepsia 2014, 55, 1558–1567. [Google Scholar] [CrossRef] [PubMed]
- Modi, M.E.; Brooks, J.M.; Guilmette, E.R.; Beyna, M.; Graf, R.; Reim, D.; Schmeisser, M.J.; Boeckers, T.M.; O’Donnell, P.; Buhl, D.L. Hyperactivity and hypermotivation associated with increased striatal mGluR1 signaling in a Shank2 rat model of autism. Front. Mol. Neurosci. 2018, 11, 107. [Google Scholar] [CrossRef]
- Harony-Nicolas, H.; Kay, M.; du Hoffmann, J.; Klein, M.E.; Bozdagi-Gunal, O.; Riad, M.; Daskalakis, N.P.; Sonar, S.; Castillo, P.E.; Hof, P.R.; et al. Oxytocin improves behavioral and electrophysiological deficits in a novel Shank3-deficient rat. Elife 2017, 6, e18904. [Google Scholar] [CrossRef]
- Rannals, M.D.; Page, S.C.; Campbell, M.N.; Gallo, R.A.; Mayfield, B.; Maher, B.J. Neurodevelopmental models of transcription factor 4 deficiency converge on a common ion channel as a potential therapeutic target for Pitt Hopkins Syndrome. Rare Dis. 2016, 4, e1220468. [Google Scholar] [CrossRef]
- Chi, O.Z.; Wu, C.C.; Liu, X.; Rah, K.H.; Jacinto, E.; Weiss, H.R. Restoration of normal cerebral oxygen consumption with rapamycin treatment in a rat model of autism–tuberous sclerosis. NeuroMol. Med. 2015, 17, 305–313. [Google Scholar] [CrossRef]
- Waltereit, R.; Welzl, H.; Dichgans, J.; Lipp, H.P.; Schmidt, W.J.; Weller, M. Enhanced episodic-like memory and kindling epilepsy in a rat model of tuberous sclerosis. J. Neurochem. 2006, 96, 407–413. [Google Scholar] [CrossRef]
- Dodge, A.; Peters, M.M.; Greene, H.E.; Dietrick, C.; Botelho, R.; Chung, D.; Willman, J.; Nenninger, A.W.; Ciarlone, S.; Kamath, S.G.; et al. Generation of a novel rat model of Angelman Syndrome with a complete Ube3a gene deletion. Autism Res. 2020, 13, 397–409. [Google Scholar] [CrossRef]
- Li, J.; Ge, W. Zebrafish as a model for studying ovarian development: Recent advances from targeted gene knockout studies. Mol. Cell. Endocrinol. 2020, 507, 110778. [Google Scholar] [CrossRef] [PubMed]
- Zon, L.I. Zebrafish: A new model for human disease. Genome Res. 1999, 9, 99–100. [Google Scholar] [PubMed]
- Ahrens, M.B.; Orger, M.B.; Robson, D.N.; Li, J.M.; Keller, P.J. Whole-brain functional imaging at cellular resolution using light-sheet microscopy. Nat. Methods 2013, 10, 413–420. [Google Scholar] [CrossRef] [PubMed]
- Rainboth, W. Inland fishes of India and adjacent countries. Rev. Fish Biol. Fish. 1994, 4, 135–136. [Google Scholar] [CrossRef]
- Parichy, D.M. The natural history of model organisms: Advancing biology through a deeper understanding of zebrafish ecology and evolution. Elife 2015, 2015, e05635. [Google Scholar] [CrossRef] [PubMed]
- Westerfield, M. The Zebrafish Book: A Guide for the Laboratory Use of Zebrafish. 2000. Available online: https://zfin.org/zf_info/zfbook/zfbk.html (accessed on 19 September 2020).
- Meshalkina, D.; Kysil, E.; Warnick, J.E.; Demin, K. Adult zebrafish in CNS disease modeling: A tank that’s half-full, not half-empty, and still filling. Lab Anim 2017, 46, 378–387. [Google Scholar] [CrossRef]
- Lam, S.H.; Chua, H.L.; Gong, Z.; Lam, T.J.; Sin, Y.M. Development and maturation of the immune system in zebrafish, Danio rerio: A gene expression profiling, in situ hybridization and immunological study. Dev. Comp. Immunol. 2004, 28, 9–28. [Google Scholar] [CrossRef]
- Patton, E.E.; Zon, L.I. The art and design of genetic screens: Zebrafish. Nat. Rev. Genet. 2001, 2, 956–966. [Google Scholar] [CrossRef]
- Dooley, K.; Zon, L.I. Zebrafish: A model system for the study of human disease. Curr. Opin. Genet. Dev. 2000, 10, 252–256. [Google Scholar] [CrossRef]
- Workman, A.D.; Charvet, C.J.; Clancy, B.; Darlington, R.B.; Finlay, B.L. Modeling transformations of neurodevelopmental sequences across mammalian species. J. Neurosci. 2013, 33, 7368–7383. [Google Scholar] [CrossRef]
- Lumsden, A.; Krumlauf, R. Patterning the vertebrate neuraxis. Science 1996, 274, 1109–1115. [Google Scholar] [CrossRef] [PubMed]
- Kimmel, C.B.; Ballard, W.W.; Kimmel, S.R.; Ullmann, B.; Schilling, T.F. Stages of embryonic development of the zebrafish. Dev. Dyn. 1995, 203, 253–310. [Google Scholar] [CrossRef] [PubMed]
- Guo, S. Using zebrafish to assess the impact of drugs on neural development and function. Expert Opin. Drug Discov. 2009, 4, 715–726. [Google Scholar] [CrossRef]
- Mueller, T.; Vernier, P.; Wullimann, M.F. The adult central nervous cholinergic system of a neurogenetic model animal, the zebrafish Danio rerio. Brain Res. 2004, 1011, 156–169. [Google Scholar] [CrossRef] [PubMed]
- Mueller, T.; Wullimann, M.F. An evolutionary interpretation of teleostean forebrain anatomy. Brain Behav. Evol. 2009, 74, 30–42. [Google Scholar] [CrossRef]
- Mueller, T.; Dong, Z.; Berberoglu, M.A.; Guo, S. The dorsal pallium in zebrafish, Danio rerio (Cyprinidae, Teleostei). Brain Res. 2011, 1381, 95–105. [Google Scholar] [CrossRef] [PubMed]
- Kozol, R.A. Prenatal neuropathologies in autism spectrum disorder and intellectual disability: The gestation of a comprehensive Zebrafish model. J. Dev. Biol. 2018, 6, 29. [Google Scholar] [CrossRef]
- Bae, Y.K.; Kani, S.; Shimizu, T.; Tanabe, K.; Nojima, H.; Kimura, Y.; Shin-ichi, H.; Hibi, M. Anatomy of zebrafish cerebellum and screen for mutations affecting its development. Dev. Biol. 2009, 330, 406–426. [Google Scholar] [CrossRef]
- Kani, S.; Bae, Y.K.; Shimizu, T.; Tanabe, K.; Satou, C.; Parsons, M.J.; Scott, E.; Higashijima, S.I.; Hibi, M. Proneural gene-linked neurogenesis in zebrafish cerebellum. Dev. Biol. 2010, 343, 1–17. [Google Scholar] [CrossRef]
- Sudarov, A. Defining the role of cerebellar purkinje cells in autism spectrum disorders. Cerebellum 2013, 12, 950–955. [Google Scholar] [CrossRef]
- Bauman, M.L.; Kemper, T.L. Neuroanatomic observations of the brain in autism: A review and future directions. Int. J. Dev. Neurosci. 2005, 23, 183–187. [Google Scholar] [CrossRef] [PubMed]
- Jeong, J.W.; Tiwari, V.N.; Behen, M.E.; Chugani, H.T.; Chugani, D.C. In vivo detection of reduced purkinje cell fibers with diffusion MRI tractography in children with autistic spectrum disorders. Front. Hum. Neurosci. 2014, 8, 110. [Google Scholar] [CrossRef] [PubMed]
- Rico, E.P.; Rosemberg, D.B.; Seibt, K.J.; Capiotti, K.M.; Da Silva, R.S.; Bonan, C.D. Zebrafish neurotransmitter systems as potential pharmacological and toxicological targets. Neurotoxicol. Teratol. 2011, 33, 608–617. [Google Scholar] [CrossRef] [PubMed]
- Guissart, C.; Latypova, X.; Rollier, P.; Khan, T.N.; Stamberger, H.; McWalter, K.; Cho, M.T.; Kjaergaard, S.; Weckhuysen, S.; Lesca, G.; et al. Dual Molecular Effects of Dominant RORA Mutations Cause Two Variants of Syndromic Intellectual Disability with Either Autism or Cerebellar Ataxia. Am. J. Hum. Genet. 2018, 102, 744–759. [Google Scholar] [CrossRef] [PubMed]
- Howe, K.; Clark, M.D.; Torroja, C.F.; Torrance, J.; Berthelot, C.; Muffato, M.; Collins, J.E.; Humphray, S.; McLaren, K.; Matthews, L.; et al. The zebrafish reference genome sequence and its relationship to the human genome. Nature 2013, 496, 498–503. [Google Scholar] [CrossRef]
- Souza, B.R.; Tropepe, V. The role of dopaminergic signalling during larval zebrafish brain development: A tool for investigating the developmental basis of neuropsychiatric disorders. Rev. Neurosci. 2011, 22, 107–119. [Google Scholar] [CrossRef]
- Yoshida, M.; Macklin, W.B. Oligodendrocyte development and myelination in GFP-transgenic zebrafish. J. Neurosci. Res. 2005, 81, 1–8. [Google Scholar] [CrossRef]
- Lovett-Barron, M.; Andalman, A.S.; Allen, W.E.; Vesuna, S.; Kauvar, I.; Burns, V.M.; Deisseroth, K. Ancestral Circuits for the Coordinated Modulation of Brain State. Cell 2017, 171, 1411–1423.e17. [Google Scholar] [CrossRef]
- Hisano, Y.; Ota, S.; Kawahara, A. Genome editing using artificial site-specific nucleases in zebrafish. Dev. Growth Differ. 2014, 56, 26–33. [Google Scholar] [CrossRef]
- Nasevicius, A.; Ekker, S.C. Effective targeted gene “knockdown” in zebrafish. Nat. Genet. 2000, 26, 216–220. [Google Scholar] [CrossRef]
- Summerton, J.E. Invention and early history of morpholinos: From pipe dream to practical products. In Methods in Molecular Biology; Humana Press Inc.: Totowa, NJ, USA, 2017; Volume 1565, pp. 1–15. [Google Scholar]
- Bill, B.R.; Petzold, A.M.; Clark, K.J.; Schimmenti, L.A.; Ekker, S.C. A primer for morpholino use in zebrafish. Zebrafish 2009, 6, 69–77. [Google Scholar] [CrossRef] [PubMed]
- Eisen, J.S.; Smith, J.C. Controlling morpholino experiments: Don’t stop making antisense. Development 2008, 135, 1735–1743. [Google Scholar] [CrossRef] [PubMed]
- Stainier, D.Y.R.; Raz, E.; Lawson, N.D.; Ekker, S.C.; Burdine, R.D.; Eisen, J.S.; Ingham, P.W.; Schulte-Merker, S.; Yelon, D.; Weinstein, B.M.; et al. Guidelines for morpholino use in zebrafish. PLoS Genet. 2017, 13. [Google Scholar] [CrossRef] [PubMed]
- Wienholds, E.; van Eeden, F.; Kosters, M.; Mudde, J.; Plasterk, R.H.A.; Cuppen, E. Efficient target-selected mutagenesis in zebrafish. Genome Res. 2003, 13, 2700–2707. [Google Scholar] [CrossRef]
- Kuroyanagi, M.; Katayama, T.; Imai, T.; Yamamoto, Y.; Shin-ichi, C.; Yoshiura, Y.; Ushijima, T.; Matsushita, T.; Fujita, M.; Nozawa, A.; et al. New approach for fish breeding by chemical mutagenesis: Establishment of TILLING method in fugu (Takifugu rubripes) with ENU mutagenesis. BMC Genomics 2013, 14. [Google Scholar] [CrossRef] [PubMed]
- Doyon, Y.; McCammon, J.M.; Miller, J.C.; Faraji, F.; Ngo, C.; Katibah, G.E.; Amora, R.; Hocking, T.D.; Zhang, L.; Rebar, E.J.; et al. Heritable targeted gene disruption in zebrafish using designed zinc-finger nucleases. Nat. Biotechnol. 2008, 26, 702–708. [Google Scholar] [CrossRef] [PubMed]
- Huang, P.; Xiao, A.; Zhou, M.; Zhu, Z.; Lin, S.; Zhang, B. Heritable gene targeting in zebrafish using customized TALENs. Nat. Biotechnol. 2011, 29, 699–700. [Google Scholar] [CrossRef]
- Moreno-Mateos, M.A.; Vejnar, C.E.; Beaudoin, J.D.; Fernandez, J.P.; Mis, E.K.; Khokha, M.K.; Giraldez, A.J. CRISPRscan: Designing highly efficient sgRNAs for CRISPR-Cas9 targeting in vivo. Nat. Methods 2015, 12, 982–988. [Google Scholar] [CrossRef]
- Vejnar, C.E.; Moreno-Mateos, M.A.; Cifuentes, D.; Bazzini, A.A.; Giraldez, A.J. Optimized CRISPR-Cas9 system for genome editing in zebrafish. Cold Spring Harb. Protoc. 2016, 2016, 856–870. [Google Scholar] [CrossRef]
- Thyme, S.B.; Pieper, L.M.; Li, E.H.; Pandey, S.; Wang, Y.; Morris, N.S.; Sha, C.; Choi, J.W.; Herrera, K.J.; Soucy, E.R.; et al. Phenotypic Landscape of Schizophrenia-Associated Genes Defines Candidates and Their Shared Functions. Cell 2019, 177, 478–491.e20. [Google Scholar] [CrossRef]
- Liu, X.; Hu, G.; Ye, J.; Ye, B.; Shen, N.; Tao, Y.; Zhang, X.; Fan, Y.; Liu, H.; Zhang, Z.; et al. De Novo ARID1B mutations cause growth delay associated with aberrant Wnt/β–catenin signaling. Hum. Mutat. 2020, 41, 1012–1024. [Google Scholar] [CrossRef] [PubMed]
- Ishibashi, M.; Manning, E.; Shoubridge, C.; Krecsmarik, M.; Hawkins, T.A.; Giacomotto, J.; Zhao, T.; Mueller, T.; Bader, P.I.; Cheung, S.W.; et al. Copy number variants in patients with intellectual disability affect the regulation of ARX transcription factor gene. Hum. Genet. 2015, 134, 1163–1182. [Google Scholar] [CrossRef] [PubMed]
- Oksenberg, N.; Stevison, L.; Wall, J.D.; Ahituv, N. Function and regulation of AUTS2, a gene implicated in autism and human evolution. PLoS Genet. 2013, 9, e1003221. [Google Scholar] [CrossRef] [PubMed]
- Ramachandran, K.V.; Hennessey, J.A.; Barnett, A.S.; Yin, X.; Stadt, H.A.; Foster, E.; Shah, R.A.; Yazawa, M.; Dolmetsch, R.E.; Kirby, M.L.; et al. Calcium influx through L-type CaV1.2 Ca2+ channels regulates mandibular development. J. Clin. Investig. 2013, 123, 1638–1646. [Google Scholar] [CrossRef] [PubMed]
- Patowary, A.; Won, S.Y.; Oh, S.J.; Nesbitt, R.R.; Archer, M.; Nickerson, D.; Raskind, W.H.; Bernier, R.; Lee, J.E.; Brkanac, Z. Family-based exome sequencing and case-control analysis implicate CEP41 as an ASD gene. Transl. Psychiatry 2019, 9. [Google Scholar] [CrossRef] [PubMed]
- Suls, A.; Jaehn, J.A.; Kecskés, A.; Weber, Y.; Weckhuysen, S.; Craiu, D.C.; Siekierska, A.; Djémie, T.; Afrikanova, T.; Gormley, P.; et al. De novo loss-of-function mutations in CHD2 cause a fever-sensitive myoclonic epileptic encephalopathy sharing features with dravet syndrome. Am. J. Hum. Genet. 2013, 93, 967–975. [Google Scholar] [CrossRef]
- Bernier, R.; Golzio, C.; Xiong, B.; Stessman, H.A.; Coe, B.P.; Penn, O.; Witherspoon, K.; Gerdts, J.; Baker, C.; Vulto-van Silfhout, A.T.; et al. Disruptive CHD8 mutations define a subtype of autism early in development. Cell 2014, 158, 263–276. [Google Scholar] [CrossRef]
- Sugathan, A.; Biagioli, M.; Golzio, C.; Erdin, S.; Blumenthal, I.; Manavalan, P.; Ragavendran, A.; Brand, H.; Lucente, D.; Miles, J.; et al. CHD8 regulates neurodevelopmental pathways associated with autism spectrum disorder in neural progenitors. Proc. Natl. Acad. Sci. USA 2014, 111, E4468–E4477. [Google Scholar] [CrossRef]
- Turner, T.N.; Sharma, K.; Oh, E.C.; Liu, Y.P.; Collins, R.L.; Sosa, M.X.; Auer, D.R.; Brand, H.; Sanders, S.J.; Moreno-De-Luca, D.; et al. Loss of δ-catenin function in severe autism. Nature 2015, 520, 51–56. [Google Scholar] [CrossRef]
- Kim, O.H.; Cho, H.J.; Han, E.; Hong, T.I.; Ariyasiri, K.; Choi, J.H.; Hwang, K.S.; Jeong, Y.M.; Yang, S.Y.; Yu, K.; et al. Zebrafish knockout of Down syndrome gene, DYRK1A, shows social impairments relevant to autism. Mol. Autism 2017, 8. [Google Scholar] [CrossRef]
- Hu, J.; Chen, L.; Yin, J.; Yin, H.; Huang, Y.; Tian, J. Hyperactivity, memory defects, and craniofacial abnormalities in zebrafish fmr1 mutant larvae. Behav. Genet. 2020, 50, 152–160. [Google Scholar] [CrossRef] [PubMed]
- Kim, L.; He, L.; Maaswinkel, H.; Zhu, L.; Sirotkin, H.; Weng, W. Anxiety, hyperactivity and stereotypy in a zebrafish model of Fragile X Syndrome and Autism Spectrum Disorder. Prog. Neuro-Psychopharmacol. Biol. Psychiatry 2014, 55, 40–49. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.J.; Hsu, M.T.; Ng, M.C.; Amstislavskaya, T.G.; Tikhonova, M.A.; Yang, Y.L.; Lu, K.T. Fragile X mental retardation-1 knockout zebrafish shows precocious development in social behavior. Zebrafish 2017, 14, 438–443. [Google Scholar] [CrossRef] [PubMed]
- Sicca, F.; Ambrosini, E.; Marchese, M.; Sforna, L.; Servettini, I.; Valvo, G.; Brignone, M.S.; Lanciotti, A.; Moro, F.; Grottesi, A.; et al. Gain-of-function defects of astrocytic Kir4.1 channels in children with Autism Spectrum Disorders and epilepsy. Sci. Rep. 2016, 6, 34325. [Google Scholar] [CrossRef]
- Bögershausen, N.; Tsai, I.C.; Pohl, E.; Kiper, P.O.S.; Beleggia, F.; Ferda Percin, E.; Keupp, K.; Matchan, A.; Milz, E.; Alanay, Y.; et al. RAP1-mediated MEK/ERK pathway defects in Kabuki syndrome. J. Clin. Investig. 2015, 125, 3585–3599. [Google Scholar] [CrossRef]
- Van Laarhoven, P.M.; Neitzel, L.R.; Quintana, A.M.; Geiger, E.A.; Zackai, E.H.; Clouthier, D.E.; Artinger, K.B.; Ming, J.E.; Shaikh, T.H. Kabuki syndrome genes KMT2D and KDM6A: Functional analyses demonstrate critical roles in craniofacial, heart and brain development. Hum. Mol. Genet. 2015, 24, 4443–4453. [Google Scholar] [CrossRef]
- Leong, W.Y.; Lim, Z.H.; Korzh, V.; Pietri, T.; Goh, E.L.K. Methyl-CpG binding protein 2 (Mecp2) regulates sensory function through Sema5b and Robo2. Front. Cell. Neurosci. 2015, 9. [Google Scholar] [CrossRef]
- Pietri, T.; Roman, A.-C.; Guyon, N.; Romano, S.A.; Washbourne, P.; Moens, C.B.; de Polavieja, G.G.; Sumbre, G. The first mecp2-null zebrafish model shows altered motor behaviors. Front. Neural Circuits 2013, 7, 118. [Google Scholar] [CrossRef]
- Van Der Vaart, M.; Svoboda, O.; Weijts, B.G.; Espín-Palazón, R.; Sapp, V.; Pietri, T.; Bagnat, M.; Muotri, A.R.; Traver, D. Mecp2 regulates tnfa during zebrafish embryonic development and acute inflammation. DMM Dis. Model. Mech. 2017, 10, 1439–1451. [Google Scholar] [CrossRef]
- Elsen, G.E.; Choi, L.Y.; Prince, V.E.; Ho, R.K. The autism susceptibility gene met regulates zebrafish cerebellar development and facial motor neuron migration. Dev. Biol. 2009, 335, 78–92. [Google Scholar] [CrossRef]
- Blanchet, P.; Bebin, M.; Bruet, S.; Cooper, G.M.; Thompson, M.L.; Duban-Bedu, B.; Gerard, B.; Piton, A.; Suckno, S.; Deshpande, C.; et al. MYT1L mutations cause intellectual disability and variable obesity by dysregulating gene expression and development of the neuroendocrine hypothalamus. PLoS Genet. 2017, 13. [Google Scholar] [CrossRef] [PubMed]
- Miller, A.C.; Voelker, L.H.; Shah, A.N.; Moens, C.B. Neurobeachin is required postsynaptically for electrical and chemical synapse formation. Curr. Biol. 2015, 25, 16–28. [Google Scholar] [CrossRef] [PubMed]
- Ruzzo, E.K.; Pérez-Cano, L.; Jung, J.Y.; Wang, L.K.; Kashef-Haghighi, D.; Hartl, C.; Singh, C.; Xu, J.; Hoekstra, J.N.; Leventhal, O.; et al. Inherited and de novo genetic risk for autism impacts shared networks. Cell 2019, 178, 850–866.e26. [Google Scholar] [CrossRef] [PubMed]
- Ribeiro, D.; Nunes, A.R.; Gliksberg, M.; Anbalagan, S.; Levkowitz, G.; Oliveira, R.F. Oxytocin receptor signalling modulates novelty recognition but not social preference in zebrafish. J. Neuroendocrinol. 2020, 32, e12834. [Google Scholar] [CrossRef]
- Vecchia, E.D.; Di Donato, V.; Young, A.M.J.; Del Bene, F.; Norton, W.H.J. Reelin signaling controls the preference for social novelty in zebrafish. Front. Behav. Neurosci. 2019, 13, 214. [Google Scholar] [CrossRef]
- Plaster, N.; Sonntag, C.; Schilling, T.F.; Hammerschmidt, M. REREa/Atrophin-2 interacts with histone deacetylase and Fgf8 signaling to regulate multiple processes of zebrafish development. Dev. Dyn. 2007, 236, 1891–1904. [Google Scholar] [CrossRef]
- Kozol, R.A.; Cukier, H.N.; Zou, B.; Mayo, V.; De Rubeis, S.; Cai, G.; Griswold, A.J.; Whitehead, P.L.; Haines, J.L.; Gilbert, J.R.; et al. Two knockdown models of the autism genes SYNGAP1 and SHANK3 in zebrafish produce similar behavioral phenotypes associated with embryonic disruptions of brain morphogenesis. Hum. Mol. Genet. 2015, 24, 4006–4023. [Google Scholar] [CrossRef]
- Liu, C.X.; Li, C.Y.; Hu, C.C.; Wang, Y.; Lin, J.; Jiang, Y.H.; Li, Q.; Xu, X. CRISPR/Cas9-induced shank3b mutant zebrafish display autism-like behaviors. Mol. Autism 2018, 9. [Google Scholar] [CrossRef]
- Masai, I.; Lele, Z.; Yamaguchi, M.; Komori, A.; Nakata, A.; Nishiwaki, Y.; Wada, H.; Tanaka, H.; Nojima, Y.; Hammerschmidt, M.; et al. N-cadherin mediates retinal lamination, maintenance of forebrain compartments and patterning of retinal neurites. Development 2003, 130, 2479–2494. [Google Scholar] [CrossRef]
- Xiao, T.; Roeser, T.; Staub, W.; Baier, H. A GFP-based genetic screen reveals mutations that disrupt the architecture of the zebrafish retinotectal projection. Development 2005, 132, 2955–2967. [Google Scholar] [CrossRef]
- Xi, Y.; Yu, M.; Godoy, R.; Hatch, G.; Poitras, L.; Ekker, M. Transgenic zebrafish expressing green fluorescent protein in dopaminergic neurons of the ventral diencephalon. Dev. Dyn. 2011, 240, 2539–2547. [Google Scholar] [CrossRef] [PubMed]
- dal Maschio, M.; Donovan, J.C.; Helmbrecht, T.O.; Baier, H. Linking Neurons to Network Function and Behavior by Two-Photon Holographic Optogenetics and Volumetric Imaging. Neuron 2017, 94, 774–789.e5. [Google Scholar] [CrossRef] [PubMed]
- Higashijima, S.I.; Masino, M.A.; Mandel, G.; Fetcho, J.R. Engrailed-1 expression marks a primitive class of inhibitory spinal interneuron. J. Neurosci. 2004, 24, 5827–5839. [Google Scholar] [CrossRef] [PubMed]
- Choi, J.; Dong, L.; Ahn, J.; Dao, D.; Hammerschmidt, M.; Chen, J.N. FoxH1 negatively modulates flk1 gene expression and vascular formation in zebrafish. Dev. Biol. 2007, 304, 735–744. [Google Scholar] [CrossRef]
- Satou, C.; Kimura, Y.; Hirata, H.; Suster, M.L.; Kawakami, K.; Higashijima, S.I. Transgenic tools to characterize neuronal properties of discrete populations of zebrafish neurons. Development 2013, 140, 3927–3931. [Google Scholar] [CrossRef]
- Bernardos, R.L.; Raymond, P.A. GFAP transgenic zebrafish. Gene Expr. Patterns 2006, 6, 1007–1013. [Google Scholar] [CrossRef]
- McLean, D.L.; Fan, J.; Higashijima, S.I.; Hale, M.E.; Fetcho, J.R. A topographic map of recruitment in spinal cord. Nature 2007, 446, 71–75. [Google Scholar] [CrossRef]
- Higashijima, S.I.; Hotta, Y.; Okamoto, H. Visualization of cranial motor neurons in live transgenic zebrafish expressing green fluorescent protein under the control of the Islet-1 promoter/enhancer. J. Neurosci. 2000, 20, 206–218. [Google Scholar] [CrossRef]
- Heffer, A.; Marquart, G.D.; Aquilina-Beck, A.; Saleem, N.; Burgess, H.A.; Dawid, I.B. Generation and characterization of Kctd15 mutations in zebrafish. PLoS ONE 2017, 12. [Google Scholar] [CrossRef]
- Spiró, Z.; Koh, A.; Tay, S.; See, K.; Winkler, C. Transcriptional enhancement of Smn levels in motoneurons is crucial for proper axon morphology in zebrafish. Sci. Rep. 2016, 6. [Google Scholar] [CrossRef]
- Obholzer, N.; Wolfson, S.; Trapani, J.G.; Mo, W.; Nechiporuk, A.; Busch-Nentwich, E.; Seiler, C.; Sidi, S.; Söllner, C.; Duncan, R.N.; et al. Vesicular glutamate transporter 3 is required for synaptic transmission in zebrafish hair cells. J. Neurosci. 2008, 28, 2110–2118. [Google Scholar] [CrossRef] [PubMed]
- Blader, P.; Plessy, C.; Strähle, U. Multiple regulatory elements with spatially and temporally distinct activities control neurogenin1 expression in primary neurons of the zebrafish embryo. Mech. Dev. 2003, 120, 211–218. [Google Scholar] [CrossRef]
- Shin, J.; Park, H.C.; Topczewska, J.M.; Madwsley, D.J.; Appel, B. Neural cell fate analysis in zebrafish using olig2 BAC transgenics. Methods Cell Sci. 2003, 25, 7–14. [Google Scholar] [CrossRef]
- Lillesaar, C.; Stigloher, C.; Tannhäuser, B.; Wullimann, M.F.; Bally-Cuif, L. Axonal projections originating from raphe serotonergic neurons in the developing and adult Zebrafish, Danio Rerio, using transgenics to visualize Raphe-specific pet1 expression. J. Comp. Neurol. 2009, 512, 158–182. [Google Scholar] [CrossRef] [PubMed]
- Chen, A.; Chiu, C.N.; Mosser, E.A.; Kahn, S.; Spence, R.; Prober, D.A. QRFP and its receptors regulate locomotor activity and sleep in zebrafish. J. Neurosci. 2016, 36, 1823–1840. [Google Scholar] [CrossRef]
- Wada, N.; Javidan, Y.; Nelson, S.; Carney, T.J.; Kelsh, R.N.; Schilling, T.F. Hedgehog signaling is required for cranial neural crest morphogenesis and chondrogenesis at the midline in the zebrafish skull. Development 2005, 132, 3977–3988. [Google Scholar] [CrossRef]
- Takechi, M.; Hamaoka, T.; Kawamura, S. Fluorescence visualization of ultraviolet-sensitive cone photoreceptor development in living zebrafish. FEBS Lett. 2003, 553, 90–94. [Google Scholar] [CrossRef]
- Wen, L.; Wei, W.; Gu, W.; Huang, P.; Ren, X.; Zhang, Z.; Zhu, Z.; Lin, S.; Zhang, B. Visualization of monoaminergic neurons and neurotoxicity of MPTP in live transgenic zebrafish. Dev. Biol. 2008, 314, 84–92. [Google Scholar] [CrossRef]
- Ando, H.; Sato, T.; Ito, T.; Yamamoto, J.; Sakamoto, S.; Nitta, N.; Asatsuma-Okumura, T.; Shimizu, N.; Mizushima, R.; Aoki, I.; et al. Cereblon Control of Zebrafish Brain Size by Regulation of Neural Stem Cell Proliferation. iScience 2019, 15, 95–108. [Google Scholar] [CrossRef]
- Liu, T.; Shi, Y.; Chan, M.T.V.; Peng, G.; Zhang, Q.; Sun, X.; Zhu, Z.; Xie, Y.; Sham, K.W.Y.; Li, J.; et al. Developmental protein kinase C hyper-activation results in microcephaly and behavioral abnormalities in zebrafish. Transl. Psychiatry 2018, 8. [Google Scholar] [CrossRef]
- Pilorge, M.; Fassier, C.; Le Corronc, H.; Potey, A.; Bai, J.; De Gois, S.; Delaby, E.; Assouline, B.; Guinchat, V.; Devillard, F.; et al. Genetic and functional analyses demonstrate a role for abnormal glycinergic signaling in autism. Mol. Psychiatry 2016, 21, 936–945. [Google Scholar] [CrossRef] [PubMed]
- Baronio, D.; Puttonen, H.A.J.; Sundvik, M.; Semenova, S.; Lehtonen, E.; Panula, P. Embryonic exposure to valproic acid affects the histaminergic system and the social behaviour of adult zebrafish (Danio rerio). Br. J. Pharmacol. 2018, 175, 797–809. [Google Scholar] [CrossRef] [PubMed]
- James, D.M.; Kozol, R.A.; Kajiwara, Y.; Wahl, A.L.; Storrs, E.C.; Buxbaum, J.D.; Klein, M.; Moshiree, B.; Dallman, J.E. Intestinal dysmotility in a zebrafish (Danio rerio) shank3a;shank3b mutant model of autism. Mol. Autism 2019, 10. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.; Chun, H.S.; Lee, J.; Park, H.J.; Kim, K.T.; Kim, C.H.; Yoon, S.; Kim, W.K. Plausibility of the zebrafish embryos/larvae as an alternative animal model for autism: A comparison study of transcriptome changes. PLoS ONE 2018, 13. [Google Scholar] [CrossRef]
- Campbell, P.D.; Granato, M. Zebrafish as a tool to study schizophrenia-associated copy number variants. DMM Dis. Model. Mech. 2020, 13. [Google Scholar] [CrossRef]
- Brustein, E.; Saint-Amant, L.; Buss, R.R.; Chong, M.; McDearmid, J.R.; Drapeau, P. Steps during the development of the zebrafish locomotor network. J. Physiol. Paris 2003, 97, 77–86. [Google Scholar] [CrossRef]
- Valente, A.; Huang, K.-H.; Portugues, R.; Engert, F. Ontogeny of classical and operant learning behaviors in zebrafish. Learn. Mem. 2012, 19, 170–177. [Google Scholar] [CrossRef]
- Jain, R.A.; Wolman, M.A.; Marsden, K.C.; Nelson, J.C.; Shoenhard, H.; Echeverry, F.A.; Szi, C.; Bell, H.; Skinner, J.; Cobbs, E.N.; et al. A Forward Genetic Screen in Zebrafish Identifies the G-Protein-Coupled Receptor CaSR as a Modulator of Sensorimotor Decision Making. Curr. Biol. 2018, 28, 1357–1369.e5. [Google Scholar] [CrossRef]
- Gleason, M.R.; Nagiel, A.; Jamet, S.; Vologodskaia, M.; López-Schier, H.; Hudspeth, A.J. The transmembrane inner ear (Tmie) protein is essential for normal hearing and balance in the zebrafish. Proc. Natl. Acad. Sci. USA 2009, 106, 21347–21352. [Google Scholar] [CrossRef]
- Low, S.E.; Woods, I.G.; Lachance, M.; Ryan, J.; Schier, A.F.; Saint-Amant, L. Touch responsiveness in zebrafish requires voltage-gated calcium channel 2.1b. J. Neurophysiol. 2012, 108, 148–159. [Google Scholar] [CrossRef]
- Mathuru, A.S.; Kibat, C.; Cheong, W.F.; Shui, G.; Wenk, M.R.; Friedrich, R.W.; Jesuthasan, S. Chondroitin fragments are odorants that trigger fear behavior in fish. Curr. Biol. 2012, 22, 538–544. [Google Scholar] [CrossRef] [PubMed]
- Speedie, N.; Gerlai, R. Alarm substance induced behavioral responses in zebrafish (Danio rerio). Behav. Brain Res. 2008, 188, 168–177. [Google Scholar] [CrossRef] [PubMed]
- Mahabir, S.; Chatterjee, D.; Buske, C.; Gerlai, R. Maturation of shoaling in two zebrafish strains: A behavioral and neurochemical analysis. Behav. Brain Res. 2013, 247, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Qin, M.; Wong, A.; Seguin, D.; Gerlai, R. Induction of social behavior in zebrafish: Live versus computer animated fish as stimuli. Zebrafish 2014, 11, 185–197. [Google Scholar] [CrossRef]
- Babin, P.J.; Goizet, C.; Raldúa, D. Zebrafish models of human motor neuron diseases: Advantages and limitations. Prog. Neurobiol. 2014, 118, 36–58. [Google Scholar] [CrossRef]
- Cassar, S.; Adatto, I.; Freeman, J.L.; Gamse, J.T.; Iturria, I.; Lawrence, C.; Muriana, A.; Peterson, R.T.; Van Cruchten, S.; Zon, L.I. Use of Zebrafish in Drug Discovery Toxicology. Chem. Res. Toxicol. 2020, 33, 95–118. [Google Scholar] [CrossRef]
- Dwivedi, S.; Medishetti, R.; Rani, R.; Sevilimedu, A.; Kulkarni, P.; Yogeeswari, P. Larval zebrafish model for studying the effects of valproic acid on neurodevelopment: An approach towards modeling autism. J. Pharmacol. Toxicol. Methods 2019, 95, 56–65. [Google Scholar] [CrossRef]
- Lynch, M.; Force, A. The probability of duplicate gene preservation by subfunctionalization. Genetics 2000, 154, 459–473. [Google Scholar]
- Pereira, M.; Birtele, M.; Rylander Ottosson, D. Direct reprogramming into interneurons: Potential for brain repair. Cell. Mol. Life Sci. 2019, 76, 3953–3967. [Google Scholar] [CrossRef]
- Haapaniemi, E.; Botla, S.; Persson, J.; Schmierer, B.; Taipale, J. CRISPR–Cas9 genome editing induces a p53-mediated DNA damage response. Nat. Med. 2018, 24, 927–930. [Google Scholar] [CrossRef]
- Ihry, R.J.; Worringer, K.A.; Salick, M.R.; Frias, E.; Ho, D.; Theriault, K.; Kommineni, S.; Chen, J.; Sondey, M.; Ye, C.; et al. p53 inhibits CRISPR–Cas9 engineering in human pluripotent stem cells. Nat. Med. 2018, 24, 939–946. [Google Scholar] [CrossRef] [PubMed]
- Meijboom, F.L.B.; Kostrzewa, E.; Leenaars, C.H.C. Joining forces: The need to combine science and ethics to address problems of validity and translation in neuropsychiatry research using animal models. Philos. Ethics Humanit. Med. 2020, 15, 1. [Google Scholar] [CrossRef] [PubMed]
- DeGrazia, D.; Sebo, J. Necessary conditions for morally responsible animal research. Cambridge Q. Healthc. Ethics 2015, 24, 420–430. [Google Scholar] [CrossRef] [PubMed]
- Wong, C.H.; Siah, K.W.; Lo, A.W. Estimation of clinical trial success rates and related parameters. Biostatistics 2019, 20, 273–286. [Google Scholar] [CrossRef]
- Freires, I.A.; de Sardi, J.C.O.; de Castro, R.D.; Rosalen, P.L. Alternative animal and non-animal models for drug discovery and development: Bonus or burden? Pharm. Res. 2017, 34, 681–686. [Google Scholar] [CrossRef]
- Bovenkerk, B.; Kaldewaij, F. The use of animal models in behavioural neuroscience research. Curr. Top. Behav. Neurosci. 2015, 19, 17–46. [Google Scholar]
- Kilkenny, C.; Browne, W.J.; Cuthill, I.C.; Emerson, M.; Altman, D.G. Improving bioscience research reporting: The ARRIVE guidelines for reporting animal research. PLoS Biol. 2010, 8, e1000412. [Google Scholar] [CrossRef]
- Smith, A.J.; Clutton, R.E.; Lilley, E.; Hansen, K.E.A.; Brattelid, T. PREPARE: Guidelines for planning animal research and testing. Lab. Anim. 2018, 52, 135–141. [Google Scholar] [CrossRef]





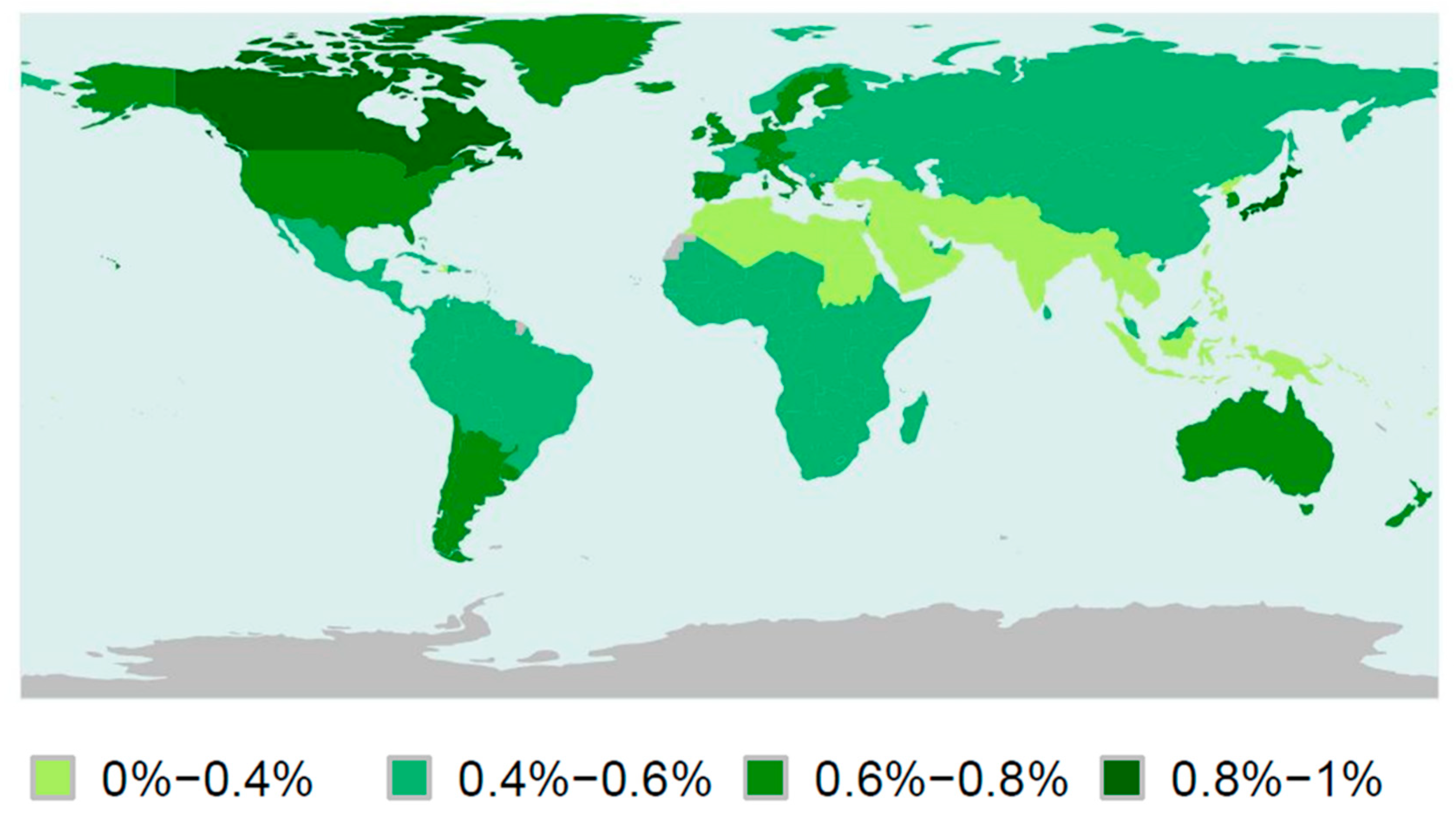{kind=link}
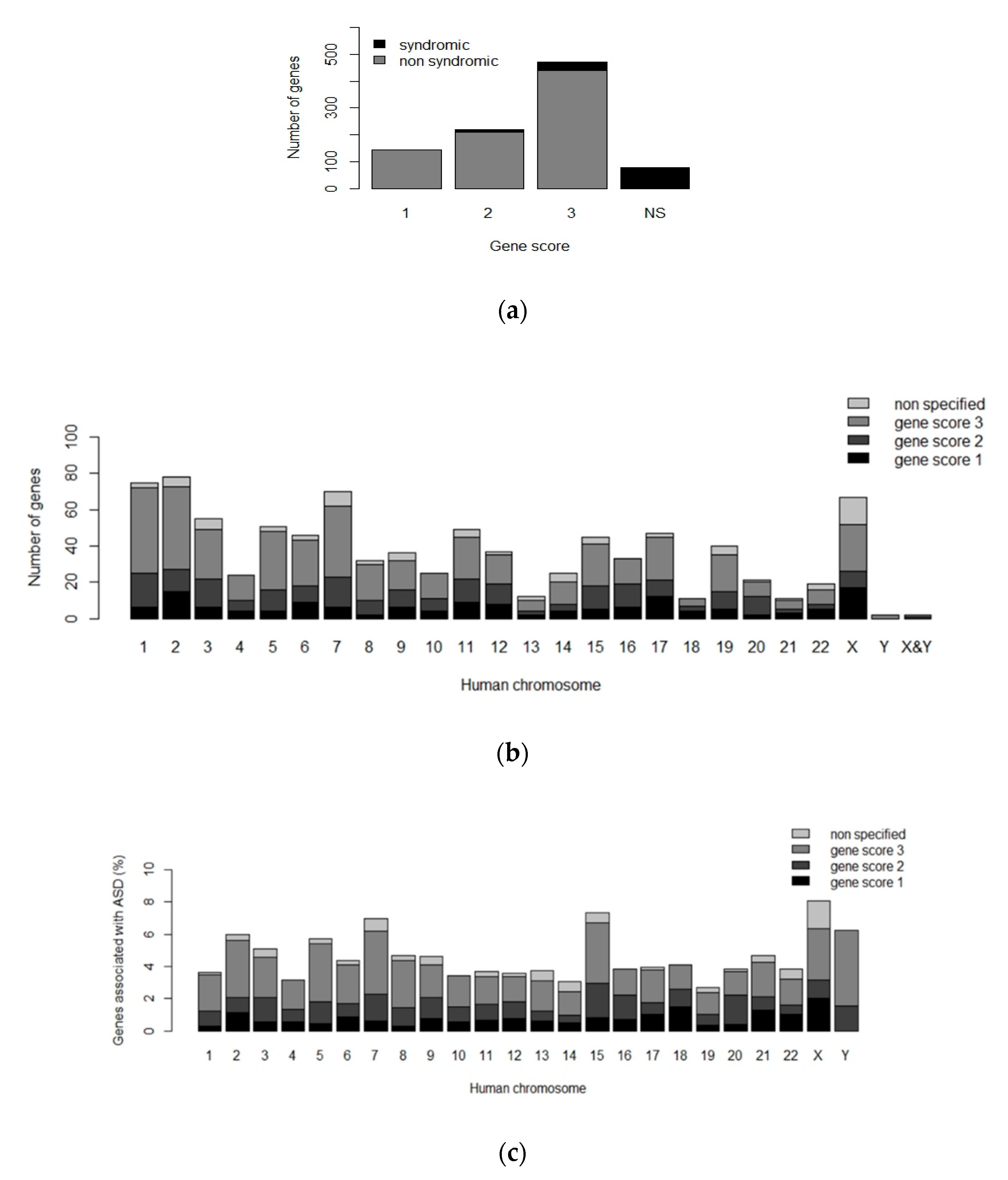{kind=link}
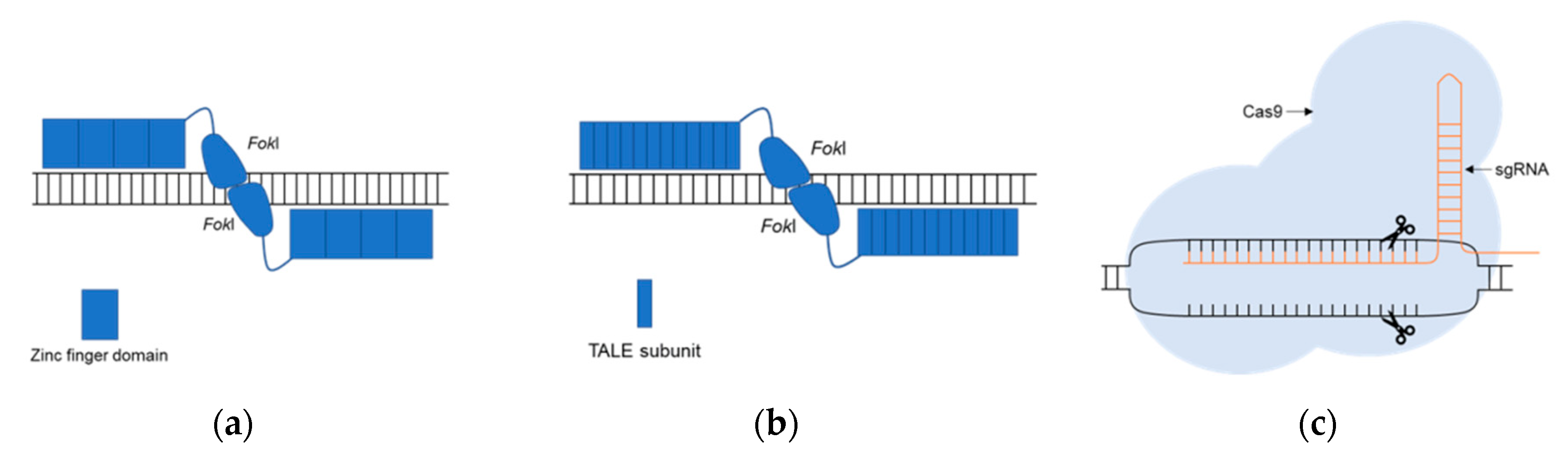{kind=link}
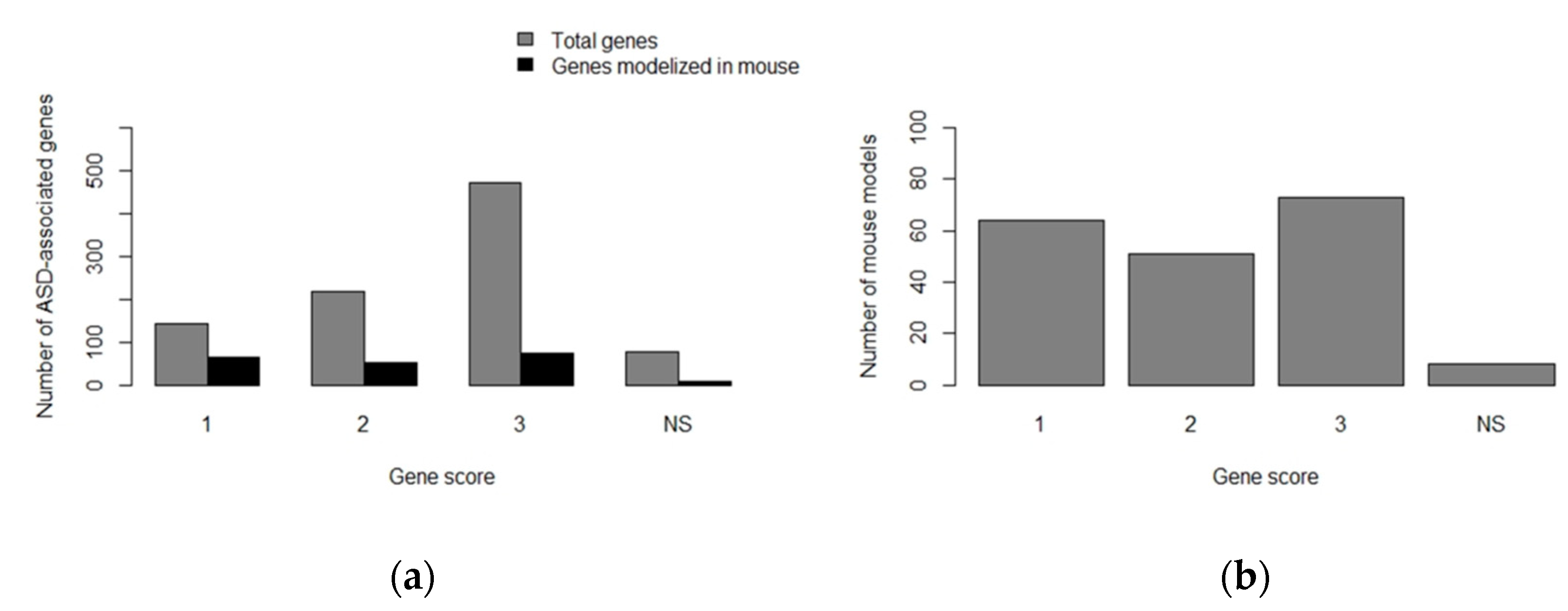{kind=link}
| Clinical Diagnosis Criteria for ASD |
|---|
| Deficits in social communication and interaction |
| Restricted and repetitive patterns of behavior, interests, or activities Symptoms present during early development Presence of impairments in important areas of an individual’s functioning Symptoms are not better explained by other mental disorder |
| Type of Therapy | Therapy | Procedure | Areas with Improvement | Side Effects |
|---|---|---|---|---|
| Psychosocial therapies | Applied behavior analysis (ABA) | Repetition of learning trials (positive reinforcement) | Intellectual functioning, language, daily living skills and socialization | Long-term and costly therapy, need patient’s cooperation and motivation |
| Pivotal Response Treatment (PRT) | Targets specific skills and motivations | Improve communication skills and less disruptive behaviors compared to ABA | No significant side effects | |
| Parent-mediated early interventions | Interventions that can be applied at home by parents | Socialization and communication | No significant side effects | |
| Social skills interventions | Interventions to improve social skills | Emotional regulation, communication and socialization | No significant side effects | |
| Pharmacology | Risperidone | Atypical Antipsychotics | Irritability, socialization and communication | Weight gain, increased appetite and somnolence |
| Aripiprazole | Atypical Antipsychotics | Irritability | Weight gain and somnolence | |
| Olanzapine | Atypical Antipsychotics | Irritability | Weight gain | |
| Ziprasidone | Atypical Antipsychotics | Irritability | Cardiovascular alterations and somnolence | |
| Paliperidone | Atypical Antipsychotics | Irritability | Weight gain and extrapyramidal symptoms | |
| Haloperidol | Typical Antipsychotics | Hyperactivity, stereotypical behaviors and learning on discrimination tasks | Somnolence, irritability and dystonic reactions | |
| Antidepressants: venlafaxine | Typical Antipsychotics | Repetitive behaviors, socialization and communication | Hyperactivity, inattention, nausea and polyuria | |
| Antidepressants: clomipramine | Typical Antipsychotics | Stereotypical behavior and anger management | No significant side effects | |
| Divalproex sodium | Mood stabilizers | Irritability and repetitive behaviors | No significant side effects | |
| Methylphenidate | Stimulants/atomoxetine/α-2 agonists | Hyperactivity | Appetite decrease, insomnia, irritability and emotional outbursts | |
| Atomoxetine | Stimulants/atomoxetine/α-2 agonists | Hyperactivity and impulsivity | No significant side effects | |
| α-2 agonists: clonidine and guanfacine | Stimulants/atomoxetine/α-2 agonists | Hyperactivity | Somnolence | |
| Naltrexone | Other medications | Hyperactivity and impulsivity | No significant side effects | |
| Complementary alternative medicine | Melatonin | Sleep disturbances | No significant side effects |
| Cell Lines Derived from hiPSCs | ASD-Associated Gene | Alterations Due to the Lack of Expression of ASD-Associated Gene | References |
|---|---|---|---|
| Cortical neurons | EHMT1 | Reduced neurite length and complexity Altered neuronal activity Increased expression of proliferation genes Decreased expression of maturation and migration genes | [64] |
| MECP2 | Increased synaptogenesis and dendritic complexity Altered neuronal network synchronization | [65] | |
| NRXN1 | Altered ion transport and calcium signaling | [66] | |
| PTCHD1 | Decreased frequency of miniature excitatory postsynaptic currents N-methyl-D-aspartate receptor (NMDARs) hypofunction | [61] | |
| PTCHD1-AS | Decreased frequency of miniature excitatory postsynaptic currents | [61] | |
| SHANK2 | Increased number of synapses, dendritic length and complexity Increased frequency of spontaneous excitatory postsynaptic currents Altered expression of genes associated to neuronal morphogenesis, plasticity and synapse | [67] | |
| SHANK3 | Synaptic alteration and decreased dendritic spines | [68,69] | |
| TSC2 | Mitochondria disorganization and altered mitophagy Increased soma size and neurite number mTORC1 signaling pathway hyperactivation Increased neuronal activity and upregulation of cell adhesion genes | [70,71] | |
| Dopaminergic neurons | RELN | Altered neuronal migration | [72] |
| Glutamatergic neurons | AFF2 | Alteration in genes associated with neuronal development Decreased synaptic activity: reduced spontaneous excitatory postsynaptic currents | [73] |
| ASTN2 | Alteration in genes associated with neuronal development Decreased synaptic activity: reduced spontaneous excitatory postsynaptic currents | [73] | |
| ATRX | Alteration in genes associated with neuronal development Decreased synaptic activity: reduced spontaneous excitatory postsynaptic currents | [73] | |
| CNTN5 | Increased neuronal activity | [74] | |
| KCNQ2 | Decreased synaptic activity: reduced spontaneous excitatory postsynaptic currents | [73] | |
| SCN2A | Alteration in genes associated with morphogenesis Decreased synaptic activity: reduced spontaneous excitatory postsynaptic currents | [73] | |
| Neuron-like cells | ARHGEF9 | Altered mTORC1 signaling pathway | [75] |
| CACNA1C | Altered calcium signaling Altered differentiation of neurons from cortical layers Increased production of norepinephrine and dopamine Altered expression of tyrosine hydrolase | [76,77] | |
| CDKL5 | Alterations in neuronal activity | [78] | |
| CHD8 | Altered expression of genes associated with neural development, β-catenin/Wnt signaling, extracellular matrix and skeletal system development | [79] | |
| COSMOC | Impaired redox homeostasis Altered PTBP2 splicing | [62] | |
| FMR1 | Altered DNA methylation patterns Altered expression of genes associated with neuronal development, migration and maturation Altered neurite formation and neuronal differentiation | [80,81,82] | |
| SHANK3 | Alterations in the soma and neurites, as well as alterations in synaptic transmission Altered expression of genes associated to motility and neurogenesis | [83,84] | |
| TRPC6 | Reduce neurite length and complexity Altered glutamatergic synapse formation and reduced sodium influx | [85] | |
| Neural organoids | CHD8 | Alterations in the expression of gens associated with neurogenesis, β-catenin/Wnt signaling, neuronal differentiation and axonal guidance | [86] |
| Neural progenitor cells | NRXN1 RELN | Alterations in neuronal adhesion and differentiation Overactivation of mTORC1 pathway | [87,88] [89] |
| TRPC6 | Altered calcium signaling and expression of genes involved in cell adhesion and neurite formation | [85] | |
| ZNF804A | Altered expression of pathways mediated by interferon-α 2 | [90] | |
| Olfactory placodal neurons | SHANK3 | Decreased number of synapses Alterations during neural development in the soma and neurites | [91] |
| Purkinje cells | TSC2 | Hypoexcitability and synaptic dysfunction mTORC1 pathway hyperactivation Altered neuronal differentiation | [92] |
| Areas of Interest | Behavioral Assays in Rodents | Behavioral Assays in Zebrafish |
|---|---|---|
| Socialization |
|
|
| Non-social patterns of behavior |
|
|
| Communication |
|
|
| ASD-Associated Gene/Mus musculus | Gene Modification Technique | Main Phenotypical Observations | Reference |
|---|---|---|---|
| ADNP/Adnp | KO by homologous recombination | Embryonic lethality (KO) Developmental delay Decreased neuronal survival Social and memory impairments | [101,102,103] |
| ARID1B/Arid1b | KO by CRISPR/Cas9 Conditional heterozygous KO by Cas9 (floxed allele) | Increased lethality Abnormal brain and heart development Decreased neuronal precursor proliferation and cortical thickness Anxiety and social interaction alterations Decreased cognitive flexibility | [104,105] |
| ASH1L/Ash1l | KO with gene trap vector, piggyBac or CRISPR/Cas9 | Increased lethality and infertility Delayed eye development Reduced adiposity Altered immune response Reduced chromatin modification | [106,107,108] |
| CHD2/Chd2 | Targeted KO with cassette Cre-flox Conditional LOF in interneurons | Growth delay and increased mortality Abnormal synaptic transmission Reduced number of neural precursors and interneurons Altered hippocampal morphology Decreased object recognition memory Decreased spatial working memory | [109,110] |
| CHD8/Chd8 | Knockdown (shRNAs) KO by CRISPR/Cas9 or Cre-LoxP | Altered brain development, corticogenesis and differentiation of neural precursors Reduced density of the dendritic tree Decreased myelination Increased anxiety and altered sociability Increased repetitive behaviors Altered memory patterns | [111,112,113,114,115,116,117,118] |
| CIC/Cic | Conditional LOF in the neocortex, hippocampus and pallium | Altered hippocampal and cortical morphology Reduced number of postmitotic excitatory neurons of the forebrain Reduced dendritic complexity Reduced social interactions | [119] |
| CNTNAP2/Cntnap2 | Targeted KO by gene replacement | Delayed growth Cortical disorganization in the brain Decreased levels of neuroreceptors Repetitive behaviors and seizures Impairments in social interactions | [120,121,122,123,124,125] |
| GABRB3/Gabrb3 | Conditional LOF in endothelial cells Targeted KO | Altered brain morphology Reduced number of interneurons Reduced neuronal migration Decreased levels of GABA neurotransmitter Increased seizures, anxiety and depression Reduced social and tactile memory | [126,127,128,129,130] |
| PTEN/Pten | Conditional LOF in: forebrain gabaergic and dopaminergic neurons; secondary progenitors in the subpallium SVZ; Purkinje cells; dentate gyrus, hippocampus, cortex or ventricular zone of the MGE | Increased lethality Altered brain morphology Reduced number of interneurons Increased neuronal size and connectivity Impaired neuronal differentiation Altered synaptic function Increased apoptosis in brain cells Increased thickness in the cerebellum Decreased number of Purkinje cells Reduced coordination Reduced social memory | [131,132,133,134,135,136,137] |
| RELN/Reln | Spontaneous mutation | Altered morphology of the brain, cerebellum, cortex and olfactory bulb Reduced number of Purkinje cells Altered neuronal migration patterns Altered metabolism of neurotransmitters Impaired coordination Increased anxiety response levels | [138,139,140] |
| SCN2A/Scn2a | Targeted KO by gene interruption Conditional LOF in dorsal telencephalic excitatory neurons | Increased apoptosis and mortality Seizures and hyperactivity Increased rearing Reduced anxiety responses | [141,142,143] |
| SHANK2/Shank2 | Conditional LOF in Purkinje cells Targeted KO | Altered synaptic currents Increased anxiety and hyperactivity Reduced coordination Increased repetitive behaviors Reduced social approach Decreased spatial learning and memory | [144,145,146,147,148] |
| TAOK2/Taok2 | Targeted KO by Cre-LoxP | Abnormal brain morphology and spine density Reduced dendritic length and complexity Reduced cortical lamination and thickness Impaired memory of context | [149] |
| TBR1/Tbr1 | Conditional LOF in neurons of cortical layer 6 and subplate Targeted KO by homologous recombination | Altered brain morphology Reduced neuronal connectivity Reduced number of interneurons Altered differentiation of brain cells Altered cortical organization Altered synaptic currents Increased anxiety aggressiveness Increased aggressive | [146,150,151,152,153] |
| UPF3B/Upf3b | Targeted KO by gene trap | Reduced spine density Altered morphology of cortical neurons Poor differentiation of neural progenitors Impaired sensorimotor gating Abnormal clasping reflex Abnormal sleep pattern Impaired startle response to acoustic stimuli | [154] |
| ASD-Associated Gene/Rattus norvegicus | Gene Modification Technique | Main Phenotypical Observations | Reference |
|---|---|---|---|
| BCKDK/Bckdk | KO by spontaneous mutation | Neuronal alterations Reduced protein phosphorylation Infertility Altered development | [155] |
| CACNA1C/Cacna1c | KO by ZFN | Altered social behavior and reduced USVs Increased perseverative behaviors | [156,157] |
| CNTNAP2/Cntnap2 | KO by ZFN | Seizures Hyperactivity Altered audition and sleep routines | [158,159] |
| CYFIP1/Cyfip1 | KO by CRISPR/Cas9 | Neuronal alterations Altered behavioral flexibility in learning tasks | [160] |
| FMR1/Fmr1 | KO by ZFN | Increased repetitive behaviors and social alterations. Altered sensorimotor gating Memory difficulties Neuronal alterations Altered auditory responses | [161,162,163] |
| MECP2/Mecp2 | KO by ZFN | High mortality Malocclusion Neuronal alterations Hypoactivity Altered social interaction and speech responses. Memory alterations Decreased grip strength | [164,165] |
| NLGN2/Nlgn2 | Overexpression in the hippocampus | Decreased response to new stimuli and aggressive behavior | [166] |
| NLGN3/Nlgn3 | KO by ZFN | Increased repetitive behaviors Hyperactivity and altered sleep routines Decreased body weight Altered juvenile play behavior and startle response Altered sensorimotor gating | [162,167] |
| NRXN1/Nrxn1 | KO by biallelic deletion | Hyperactivity Altered startle response Memory alterations | [168] |
| PTEN/Pten | Heterozygous KO by ZFN | Neuronal alterations | [169] |
| SCN1A/Scn1a | KO by ENU mutagenesis | Increased repetitive behaviors Hyperactivity and anxiety Learning and memory difficulties Motor alterations Reduced dopamine levels | [170] |
| SHANK2/Shank2 | KO by ZFN | Alterations in social behavior Hyperactivity and increased repetitive behavior Memory alterations Neuronal alterations | [171] |
| SHANK3/Shank3 | KO by ZFN | Alterations in social behavior Neuronal alterations | [172] |
| TCF4/Tcf4 | KO by CRISPR/Cas9 and knockdown by shRNA in the prefrontal cortex | Altered electrophysiological properties in neurons | [173] |
| TSC2/Tsc2 | KO by spontaneous mutation | Enhanced episodic-like memory Enhanced seizure-induced plasticity Increased induction of phospho-p42-MAPK in the hippocampus Increased basal oxygen consumption in the brain | [174,175] |
| UBE3A/Ube3a | KO by CRISPR/Cas9 | Motor, learning and memory difficulties | [176] |
| ASD-Linked Gene/Danio rerio | Gene Modification Technique | Main Phenotypical Observations | Reference |
|---|---|---|---|
| ARID1B/arid1b | Knockdown by MOs | Reduced body length Altered expression of chondrogenic/osteogenic genes | [219] |
| ARX/arxa | Knockdown by MOs | Altered brain development Neuronal alterations | [220] |
| AUTS2/auts2a and auts2b | Knockdown by MOs | Microcephaly Altered jaw development Motor alterations Neuronal alterations | [221] |
| CACNA1C/cacna1c | Knockdown by MOs | Cardiac alterations Altered jaw development | [222] |
| CEP41/cep41 | Knockdown by MOs | Neuronal alterations Social behavior alterations | [223] |
| CHD2/chd2 | Knockdown by MOs | Altered development Microcephaly, abnormal body curvature Swim bladder absence Motor difficulties | [224] |
| CHD8/chd8 | Knockout by CRISPR/Cas9 and knockdown by MOs | Macrocephaly Reduction in post-mitotic enteric neurons | [225,226] |
| CNTNAP2/cntnap2a and cntap2b | Knockout by ZFN | Altered development Microcephaly Neuronal alterations Motor alterations | [36] |
| CTNND2/ctnnd2b | Knockdown by MOs | Reduced body length Notochord alterations | [227] |
| DYRK1A/dyrk1a | Knockout by TALENs | Altered response to social stimuli | [228] |
| FMR/fmr1 | Knockout by ENU-mutagenesis and CRISPR/Cas9 | Altered cephalic development Hyperactivity Increased anxiety Altered social behavior Learning difficulties | [229,230,231] |
| KCNJ10/kcnj10 | Knockdown by MOs | Motor alterations Altered development | [232] |
| KDM6A/kdm6a | Knockdown by MOs | Reduced body length Altered development Notochord alterations Neuronal alterations | [233,234] |
| MECP2/mecp2 | Knockout by ENU-mutagenesis and knockdown by MOs | Altered immune response Neuronal alterations | [235,236,237] |
| MET/met | Knockdown by MOs | High mortality Neuronal alterations | [238] |
| MYT1L/mytl1a and mytl1b | Knockdown by MOs | Reduced levels of oxytocin | [239] |
| NBEA/nbea | Knockout by ENU-mutagenesis and TALENs | Neuronal alterations Altered response to startle stimuli | [240] |
| NR3C2/nr3c2 | Knockout by CRISPR/Cas9 | Altered social behavior Altered sleep routines | [241] |
| OXTR/oxtr | Knockout by TALENs | Altered oxytocin signaling pathway Memory alterations in social and non-social recognition | [242] |
| RELN/reln | Knockout by TALENs | Altered social behavior Altered serotonin signaling pathway | [243] |
| RERE/rerea and rereb | Knockout by ENU-mutagenesis | Altered startle response to stimuli Vision and hearing difficulties | [244] |
| SHANK3/shank3a and shankb | Knockout by CRISPR/Cas9 | Altered development Neuronal alterations Reduced social behavior, hypoactivity | [245,246] |
| SYNGAP1/syngap1a and syngap1b | Knockdown by MOs | Delayed development High mortality Neuronal alterations Motor difficulties | [245] |
| Transgenic Line | Expression Pattern | Reference |
|---|---|---|
| ath5:GFP | Retinal ganglion cells | [247] |
| brn3c:GFP | Retinal ganglion cells | [248] |
| dat:EGFP | Dopaminergic neurons | [249] |
| elavl3:lynTagRFP | Post-mitotic neurons | [250] |
| En-1:GFP | Circumferential ascending interneurons | [251] |
| flk1:GFP | Endothelial cells | [252] |
| gad1b:RFP | Gabaergic neurons | [253] |
| gfap:GFP | Radial glial cells | [254] |
| glyt2:GFP | Glycinergic neurons | [255] |
| gsx1:GFP | Gabaergic neurons | [253] |
| isl1:GFP | Cranial motor neurons | [256] |
| kctd15a:GFP | Torus lateralis | [257] |
| mnx1:GFP | Motor neurons | [258] |
| neurod:EGFP | Immature neurons | [259] |
| neurog1:GFP | Primary neurons | [260] |
| olig2:EGFP | Oligodendrocytes | [261] |
| pet1:GFP | Serotonergic neurons | [262] |
| qrfp:GFP | Rostral hipothalamus | [263] |
| sox10:GFP | Neural crest cells/Neurocranium cartilague | [264] |
| tbx2b:EGFP | Cone photoreceptor cells | [265] |
| Vglut2a:GFP | Glutamatergic neurons | [253] |
| vmat2:GFP | Monoaminergic neurons | [266] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pensado-López, A.; Veiga-Rúa, S.; Carracedo, Á.; Allegue, C.; Sánchez, L. Experimental Models to Study Autism Spectrum Disorders: hiPSCs, Rodents and Zebrafish. Genes 2020, 11, 1376. https://doi.org/10.3390/genes11111376
Pensado-López A, Veiga-Rúa S, Carracedo Á, Allegue C, Sánchez L. Experimental Models to Study Autism Spectrum Disorders: hiPSCs, Rodents and Zebrafish. Genes. 2020; 11(11):1376. https://doi.org/10.3390/genes11111376
Chicago/Turabian StylePensado-López, Alba, Sara Veiga-Rúa, Ángel Carracedo, Catarina Allegue, and Laura Sánchez. 2020. "Experimental Models to Study Autism Spectrum Disorders: hiPSCs, Rodents and Zebrafish" Genes 11, no. 11: 1376. https://doi.org/10.3390/genes11111376
APA StylePensado-López, A., Veiga-Rúa, S., Carracedo, Á., Allegue, C., & Sánchez, L. (2020). Experimental Models to Study Autism Spectrum Disorders: hiPSCs, Rodents and Zebrafish. Genes, 11(11), 1376. https://doi.org/10.3390/genes11111376