Plastome Characterization and Phylogenomics of East Asian Beeches with a Special Emphasis on Fagus multinervis on Ulleung Island, Korea



Abstract
:1. Introduction
2. Materials and Methods
2.1. Plant Sampling, DNA Isolation, and Plastome Sequencing and Annotation
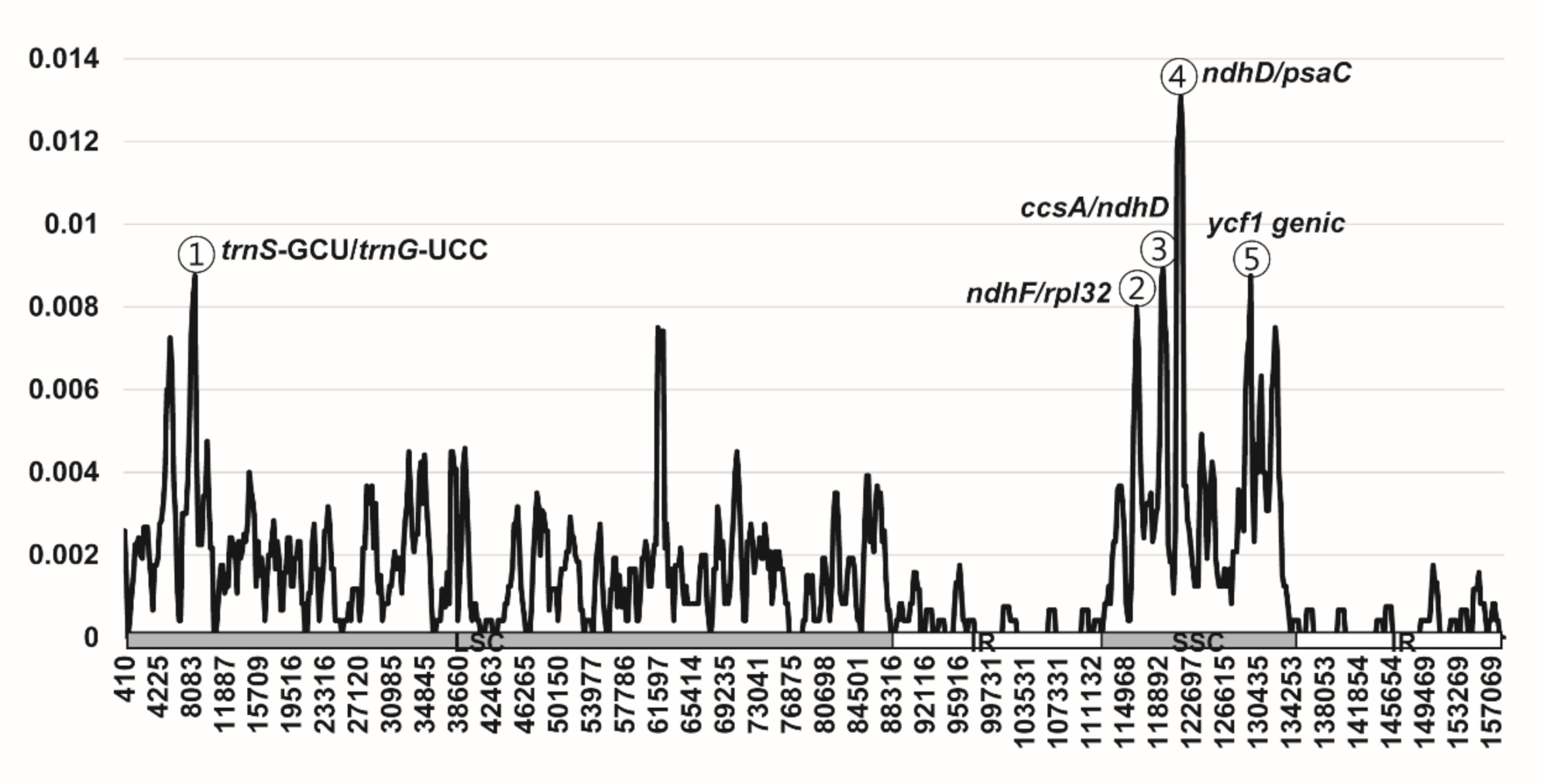
2.2. Comparative Plastome Analysis
2.3. Phylogenetic Analysis
3. Results
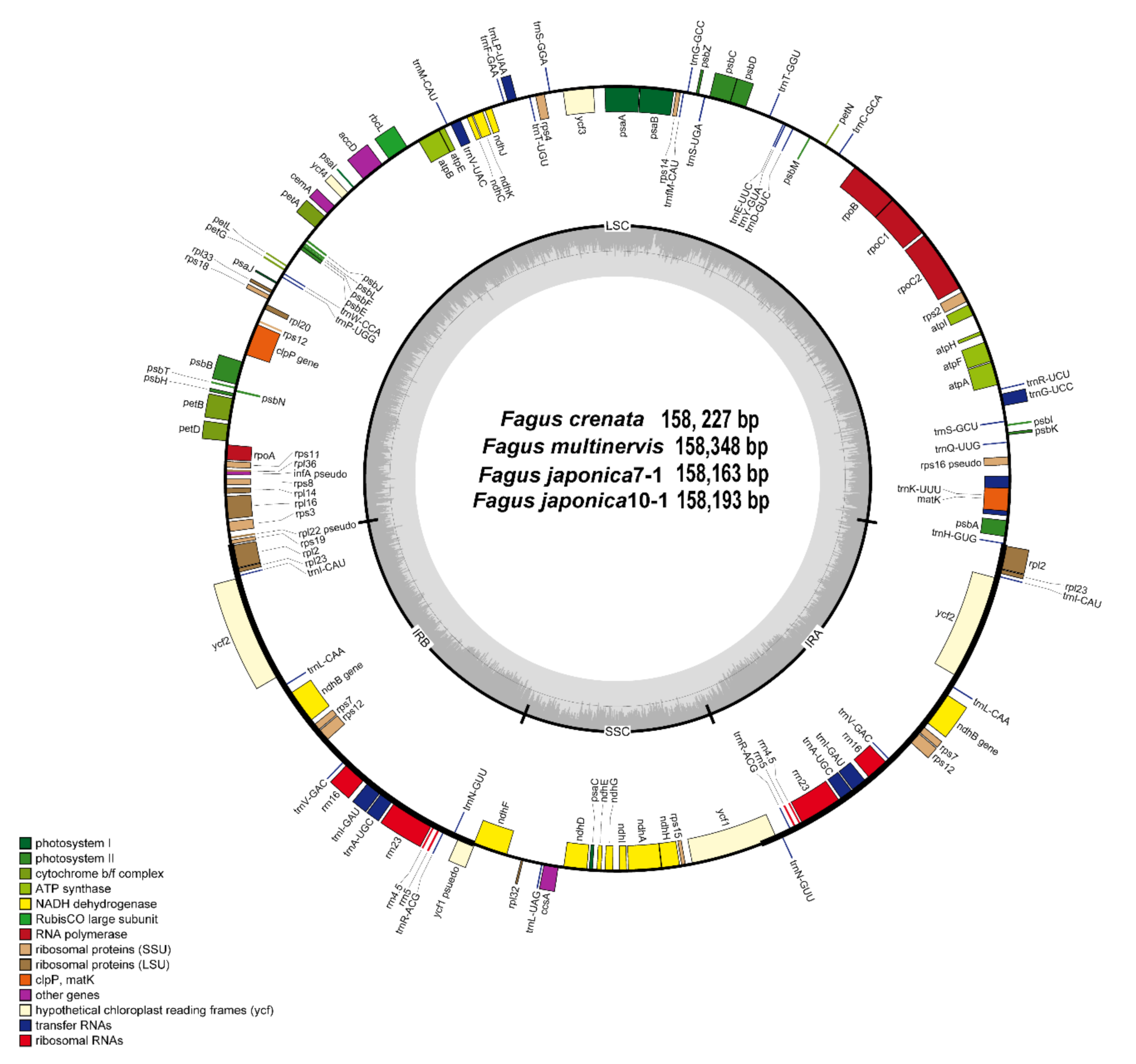
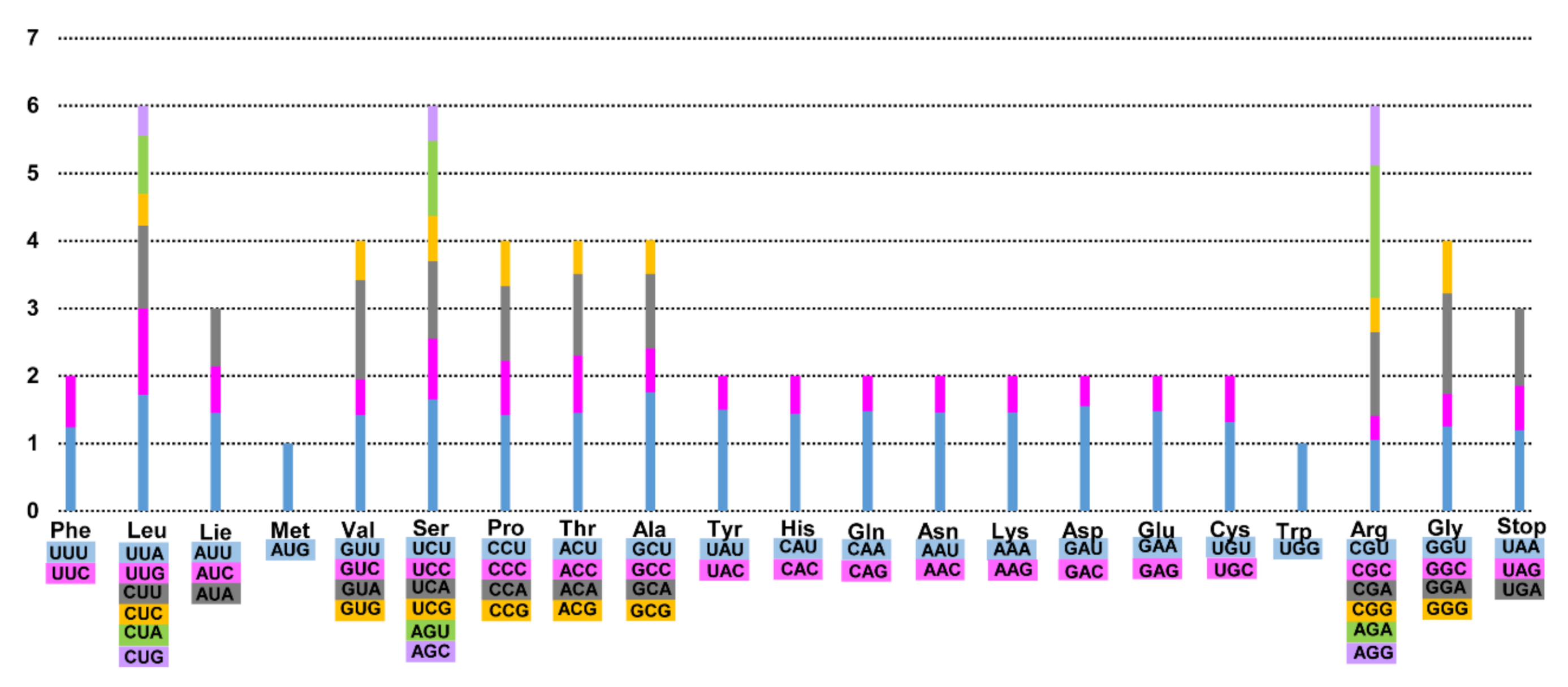
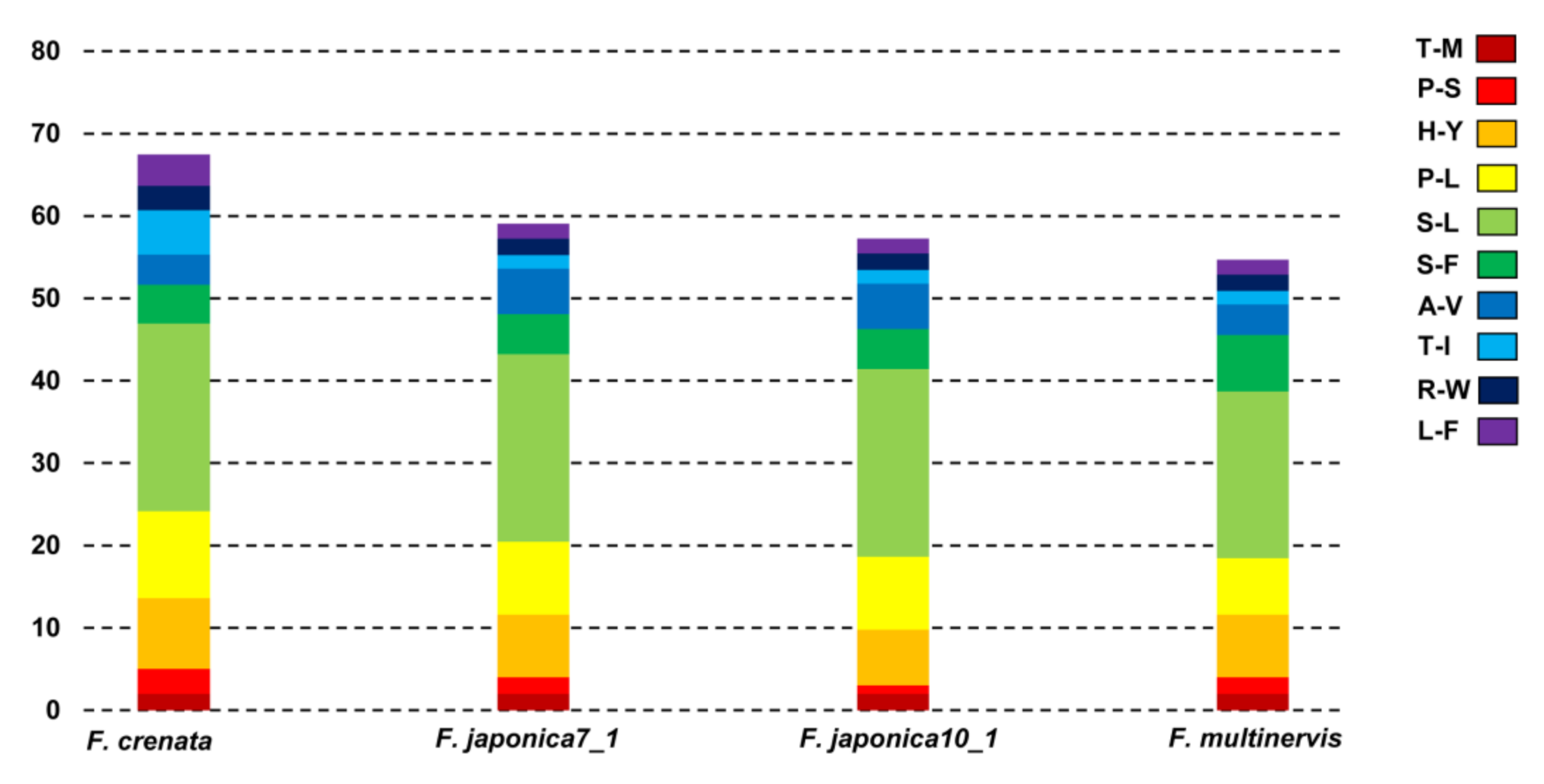
3.1. Genome Size and Characteristics
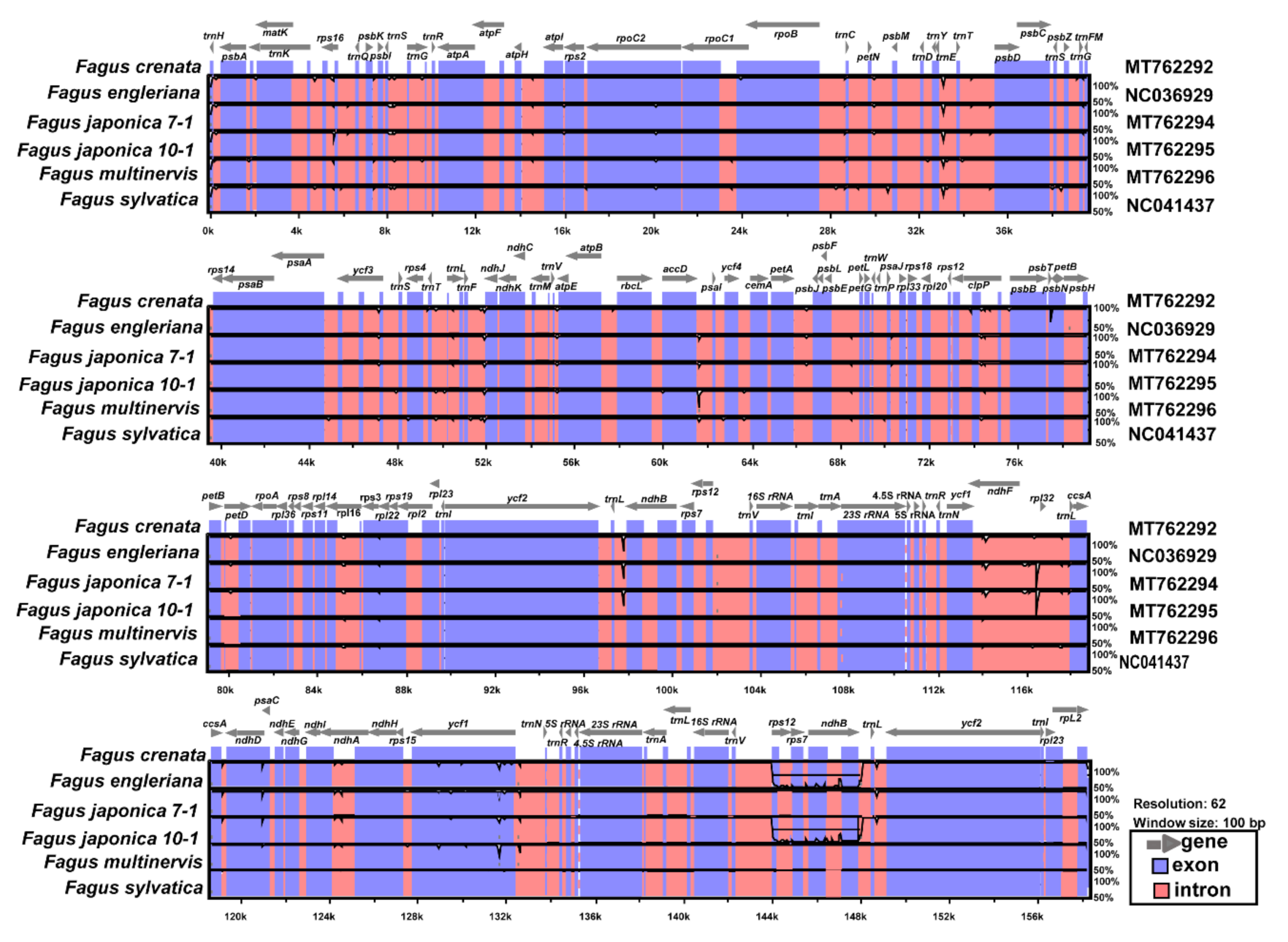
3.2. Comparative Analysis of Genome Structure
3.3. Phylogenetic Analysis
4. Discussion
4.1. Intraspecific Variation and Plastome Evolution in Fagus
4.2. Phylogenetic Position and Relationship of Fagus multinervis on Ulleung Island
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Denk, T. Phylogeny of Fagus L. (Fagaceae) based on morphological data. Plant. Syst. Evol. 2003, 240, 55–81. [Google Scholar] [CrossRef]
- Denk, T.; Grimm, G.W.; Hemleben, V. Patterns of molecular and morphological differentiation in Fagus (Fagaceae): Phylogenetic implications. Amer. J. Bot. 2005, 92, 1006–1016. [Google Scholar] [CrossRef] [PubMed]
- Zhou, G.; Li, X. Species composition and structure of Chinese beech forests. Nat. Hist. Res. 1994, 3, 21–26. [Google Scholar]
- Peters, R. Beech Forests; Kluwer Academic Publishers: Dordrecht, The Netherlands, 1997. [Google Scholar]
- Shen, C.-F. A monograph of the genus Fagus Tourn. ex L. (Fagaceae). Ph.D. Thesis, The City University of New York, New York, NY, USA, 1992. [Google Scholar]
- Renner, S.S.; Grimm, G.W.; Kapli, P.; Denk, T. Species relationships and divergence times in beeches: New insights from the inclusion of 53 young and old fossils in a birth–death clock model. Phil. Trans. R. Soc. B. 2016, 371, 20150135. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stanford, A.M. The Biogeography and Phylogeny of Castanea, Fagus, and Juglans Based on matK and ITS Sequence Data. Ph.D. Thesis, University of North Carolina, Chapel Hill, NC, USA, 1998. [Google Scholar]
- Manos, P.S.; Stanford, A.M. The historical biogeography of Fagaceae: Tracking the Tertiary history of temperate and subtropical forests of the Northern Hemisphere. Int. J. Plant. Sci. 2001, 162 (Suppl. S6), S77–S93. [Google Scholar] [CrossRef]
- Oh, S.-H.; Youn, J.-W.; Kim, Y.-I.; Kim, Y.-D. Phylogeny and evolution of endemic species on Ulleungdo Island, Korea: The case of Fagus multinervis (Fagaceae). Syst. Bot. 2016, 41, 617–625. [Google Scholar] [CrossRef]
- Lee, Y.N. Taxonomic studies on the Fagus multinervis, Fagus japonica and Fagus crenata. Bull. Korean Inst. Cult. Res. 1967, 10, 373–377. [Google Scholar]
- Chang, C.-S.; Fagus, L. The Genera of Vascular Plants of Korea; Park, C.W., Ed.; Academy Publishing Co.: Seoul, Korea, 2007; pp. 268–269. [Google Scholar]
- Sun, B.-Y.; Shin, H.; Hyun, J.-O.; Kim, Y.-D.; Oh, S.-H. Vascular Plants of Dokdo and Ulleungdo Islands in Korea; National Institute of Biological Resources: Incheon, Korea, 2014. [Google Scholar]
- Chung, H.G.; Chung, J.M.; Chung, M.G. Allozyme variation in six flowering plant species characterizing Ullung Island, Korea. J. Jpn Bot. 1998, 73, 241–247. [Google Scholar]
- Ohkawa, T.; Kitamura, K.; Takasu, H.; Kawano, S. Genetic variation in Fagus multinervis Nakai (Fagaceae), a beech species endemic to Ullung Island, South Korea. Plant. Spec. Biol. 2006, 21, 135–145. [Google Scholar] [CrossRef]
- Hukusima, T.; Matsui, T.; Nishio, T.; Pignatti, S.; Yang, L.; Lu, S.Y.; Kim, M.-H.; Yoshikawa, M.; Honma, H.; Wang, Y. Phytosociology of the beech (Fagus) forest in East Asia. In Syntaxonomy of the East Asiatic Fagus Forests; Pedrotii, F., Ed.; Springer: Berlin/Heidelberg, Germany, 2013; pp. 9–47. [Google Scholar]
- Daniell, H.; Lin, C.-S.; Yu, M.; Chang, W.-J. Chloroplast genomes: Diversity, evolution, and applications in genetic engineering. Genome Biol. 2016, 17, 134. [Google Scholar] [CrossRef] [Green Version]
- Jansen, R.K.; Cai, Z.; Raubeson, L.A.; Daniell, H.; DePamphilis, C.W.; Leebens-Mack, J.; Muller, K.F.; Guisinger-Bellian, M.; Haberle, R.C.; Hansen, A.K.; et al. Analysis of 81 genes from 64 plastid genomes resolves relationships in angiosperms and identifies genome-scale evolutionary patterns. Proc. Natl. Acad. Sci. USA 2007, 104, 19369–19374. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moore, M.J.; Bell, C.D.; Soltis, P.S.; Soltis, D.E. Using plastid genome scale data to resolve enigmatic relationships among basal angiosperms. Proc. Natl. Acad. Sci. USA 2007, 104, 19363–19368. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barrett, C.F.; Davis, J.I.; Leebens-Mack, J.; Conran, J.G.; Stevenson, D.W. Plastid genomes and deep relationships among the commelinid monocot angiosperms. Cladistics 2013, 29, 65–87. [Google Scholar] [CrossRef]
- Barrett, C.F.; Baker, W.J.; Comer, J.R.; Conran, J.G.; Lahmeyer, S.C.; Leebens-Mack, J.H.; Li, J.; Lim, G.S.; Mayfield-Jones, D.R.; Perez, L.; et al. Plastid genomes reveal support for deep phylogenetic relationships and extensive rate variation among palms and other commelinid monocots. New Phytol. 2016, 209, 855–870. [Google Scholar] [CrossRef] [PubMed]
- Ma, P.F.; Zhang, Y.X.; Zeng, C.X.; Guo, Z.H.; Li, D.Z. Chloroplast phylogenomic analysis resolve deep-level relationships of an intractable bamboo tribe Arundinarieae (Poaceae). Syst. Biol. 2014, 63, 933–950. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Parks, M.; Cronn, R.; Liston, A. Increasing phylogenetic resolution at low taxonomic levels using massively parallel sequencing of chloroplast genomes. BMC Biol. 2009, 7, 84. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nikiforova, S.V.; Cavalieri, D.; Velasco, R.; Goremykin, V. Phylogenetic analysis of 47 chloroplast genomes clarifies the contribution of wild species to the domesticated apple maternal line. Mol. Biol. Evol. 2013, 30, 1751–1760. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carbonell-Caballero, J.; Alonso, R.; Ibañez, V.; Terol, J.; Talon, M.; Dopazo, J. A phylogenetic analysis of 34 chloroplast genomes elucidates the relationships between wild and domestic species within the genus Citrus. Mol. Biol. Evol. 2015, 32, 2015–2035. [Google Scholar] [CrossRef] [Green Version]
- Viljeon, E.; Odeny, D.A.; Coetzee, M.P.A.; Berger, D.K.; Rees, D.J.G. Application of chloroplast phylogenomics to resolve species relationships within the plant genus Amaranthus. J. Mol. Evol. 2018, 86, 216–239. [Google Scholar] [CrossRef]
- Cho, M.-S.; Kim, J.H.; Kim, C.-S.; Mejias, J.A.; Kim, S.-C. Sow thistle chloroplast genomes: Insights into the plastome evolution and relationship of two weedy species, Sonchus asper and Sonchus oleraceus (Asteraceae). Genes 2019, 10, 881. [Google Scholar] [CrossRef] [Green Version]
- Jeon, J.-H.; Kim, S.-C. Comparative analysis of the complete chloroplast genome sequences of three closely related east-Asian wild roses (Rosa sect. Synstylae; Rosaceae). Genes 2019, 10, 23. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, L.-S.; Herrando-Moraira, S.; Susanna, A.; Galbany-Casals, M.; Chen, Y.-S. Phylogeny, origin and dispersal of Saussurea (Asteraceae) based on chloroplast genome data. Mol. Phylogenet. Evol. 2019, 141, 106613. [Google Scholar] [CrossRef] [PubMed]
- Kyalo, C.M.; Lim, Z.-Z.; Mkala, E.M.; Malombe, I.; Hu, G.-W.; Wang, Q.-F. The first glimpse of Streptocarpus ionanthus (Gesneriaceae) phylogenomics: Analysis of five subspecies’ chloroplast genomes. Plants 2020, 9, 456. [Google Scholar] [CrossRef] [Green Version]
- Mader, M.; Liesebach, H.; Liesebach, M.; Kersten, B. The complete chloroplast genome sequence of Fagus sylvatica L. (Fagaceae). Mitochondrial DNA B 2019, 4, 1818–1819. [Google Scholar] [CrossRef]
- Park, J.-S.; Jin, D.-P.; Park, J.-W.; Choi, B.-H. Complete chloroplast genome of Fagus multinervis, a beech species endemic to Ulleung Island in South Korea. Mitochondrial DNA B 2019, 4, 1698–1699. [Google Scholar] [CrossRef] [Green Version]
- Worth, J.R.P.; Liu, L.; Wei, F.-J.; Tomaru, N. The complete chloroplast genome of Fagus crenata (subgenus Fagus) and comparison with F. engleriana (subgenus Engleriana). PeerJ 2019, 7, e7026. [Google Scholar] [CrossRef]
- Park, J.S.; Oh, S.-H. A second complete chloroplast genome sequence of Fagus multinervis Nakai (Fagaceae): Intraspecific variations on chloroplast genome. Mitochondrial DNA B 2020, 5, 1868–1869. [Google Scholar] [CrossRef] [Green Version]
- Yang, Y.; Zhu, J.; Feng, L.; Zhou, T.; Bai, G.; Yang, J.; Zhao, G. Plastid Genome Comparative and Phylogenetic Analyses of the Key Genera in Fagaceae: Highlighting the Effect of Codon Composition Bias in Phylogenetic Inference. Front. Plant. Sci. 2018, 9, 82. [Google Scholar] [CrossRef]
- Zerbino, D.R.; Birney, E. Velvet: Algorithms for de novo short read assembly using de Bruijn graphs. Genome Res. 2008, 18, 821–829. [Google Scholar] [CrossRef] [Green Version]
- Lowe, T.M.; Eddy, S.R. tRNAscan-SE: A program for improved detection of transfer RNA genes in genomic sequence. Nucleic Acids Res. 1997, 25, 955–964. [Google Scholar] [CrossRef]
- Kearse, M.; Moir, R.; Wilson, A.; Stones-Havas, S.; Cheung, M.; Sturrock, S.; Buxton, S.; Cooper, A.; Markowitz, S.; Duran, C.; et al. Geneious Basic: An integrated and extendable desktop software platform for the organization and analysis of sequence data. Bioinformatics 2012, 28, 1647–1649. [Google Scholar] [CrossRef] [PubMed]
- Lohse, M.; Drechsel, O.; Bock, R. Organellar genome DRAW (OGDRAW): A tool for the easy generation of high-quality custom graphical maps of plastid and mitochondrial genomes. Curr. Genet. 2009, 25, 1451–1452. [Google Scholar]
- Brundo, M.; Malde, S.; Poliakov, A.; Do, C.B.; Couronne, O.; Dubchak, I.; Batzoglou, S. Global Alignment: Finding rearrangements during alignment. Bioinformatics 2003, 19 (Suppl. S1), i54–i62. [Google Scholar]
- Frazer, K.A.; Pachter, L.; Poliakov, A.; Rubin, E.M.; Dubchak, I. VISTA: Computational tools for comparative genomics. Nucleic Acids Res. 2004, 32, W273–W279. [Google Scholar] [CrossRef] [PubMed]
- Katoh, K.; Standley, D.M. MAFFT multiple sequence alignment software v7: Improvements in performance and usability. Mol. Biol. Evol. 2013, 30, 772–780. [Google Scholar] [CrossRef] [Green Version]
- Rozas, J.; Ferrer-Mata, A.; Sánchez-DelBarrio, J.C.; Guirao-Rico, S.; Librado, P.; Ramos-Onsins, S.E.; Sánchez-Gracia, A. DnaSP v6: DNA sequence polymorphism analysis of large datasets. Mol. Biol. Evol. 2017, 34, 3299–3302. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.; Stecher, G.; Tamura, K. MEGA7: Molecular evolutionary genetics analysis version 7.0 for bigger datasets. Mol. Biol. Evol. 2016, 33, 1870–1874. [Google Scholar] [CrossRef] [Green Version]
- Sharp, P.M.; Li, W.H. The codon adaptation index–a measure of directional synonymous codon usage bias, and its potential applications. Nucleic Acids Res. 1987, 15, 1281–1295. [Google Scholar] [CrossRef] [Green Version]
- Kozak, M. Comparison of initiation of protein synthesis in procaryotes, eucaryotes, and organelles. Microbiol. Rev. 1983, 47, 1–45. [Google Scholar] [CrossRef] [Green Version]
- Mower, J.P. The PREP suite: Predictive RNA editors for plant mitochondrial genes, chloroplast genes and user-defined alignments. Nucleic Acids Res. 2009, 37, W253–W259. [Google Scholar] [CrossRef]
- Nguyen, L.-T.; Schmidt, H.A.; von Haeseler, A.; Minh, B.Q. IQ-TREE: A fast and effective stochastic Algorithm for estimating maximum-likelihood phylogenies. Mol. Biol. Evol. 2015, 32, 268–274. [Google Scholar] [CrossRef] [PubMed]
- Gao, F.; Chen, C.; Arab, D.A.; Du, Z.; He, Y.; Ho, S.Y.W. EasyCodeML: A visual tool for analysis of selection using CodeML. Ecol. Evolut. 2019, 9, 3891–3898. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, Z. PAML: A program package for phylogenetic analysis by maximum likelihood. Bioinformatics 1997, 13, 555–556. [Google Scholar] [CrossRef] [PubMed]
- Fujii, N.; Tomaru, N.; Okuyama, K.; Koike, T.; Mikami, T.; Ueda, K. Chloroplast DNA phylogeography of Fagus crenata (Fagaceae) in Japan. Plant. Syst. Evol. 2002, 232, 21–33. [Google Scholar] [CrossRef]
- Oh, S.-H. Sea, wind, or bird: Origin of Fagus multinervis (Fagaceae) inferred from chloroplast DNA sequences. Korean J. Plant Taxon 2015, 45, 213–220. [Google Scholar] [CrossRef] [Green Version]
- Yang, Y.; Zhou, T.; Duan, D.; Yang, J.; Feng, L.; Zhao, G. Comparative analysis of the complete chloroplast genomes of five Quercus species. Front. Plant Sci. 2016, 7, 959. [Google Scholar] [CrossRef] [Green Version]
- Ravi, V.; Khurana, J.P.; Tyagi, A.K.; Khurana, P. An update chloroplast genomes. Plant Syst. Evol. 2008, 271, 101–122. [Google Scholar] [CrossRef]
- Morton, B.R. Selection on the codon bias of chloroplast and cyanelle genes in different plant and algal lineages. J. Mol. Evol. 1998, 46, 449–459. [Google Scholar] [CrossRef]
- Rabah, S.O.; Lee, C.; Hajrah, N.H.; Makki, R.M.; Alharby, H.F.; Alhebshi, A.M.; Sabir, J.S.M.; Jansen, R.K.; Ruhlman, T.A. Plastome sequencing of ten nonmodel crop species uncovers a large insertion of mitochondrial DNA in cashew. Plant Genome 2017, 10, 1–14. [Google Scholar] [CrossRef] [Green Version]
- Pinard, D.; Myburg, A.A.; Mizrachi, E. The plastid and mitochondrial genomes of Eucalyptus grandis. BMC Genom. 2019, 20, 1–14. [Google Scholar] [CrossRef]
- Kim, S.-H.; Yang, J.Y.; Park, J.S.; Yamada, T.; Maki, M.; Kim, S.-C. Comparison of whole plastome sequences between thermogenic skunk cabbage Symplocarpus renifolius and nonthermogenic S. nipponicus (Orontioideae; Araceae) in East Asia. Int. J. Mol. Sci. 2019, 20, 4678. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Menezes, A.P.A.; Resende-Moreira, L.C.; Buzatti, R.S.O.; Nazareno, A.G.; Carlsen, M.; Lobo, F.P.; Kalapothakis, E.; Lovato, M.B. Chloroplast genomes of Byrsonima species (Malpighiaceae): Comparative analysis and screening of high divergence sequences. Sci. Rep. 2018, 8, 2210. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Millen, R.S.; Olmstead, R.G.; Adams, K.L.; Palmer, J.D.; Lao, N.T.; Heggie, L.; Kavanagh, T.A.; Hibberd, J.M.; Gray, J.C.; Morden, C.M.; et al. Many parallel losses of infA from chloroplast DNA during angiosperm evolution with multiple independent transfers to the nucleus. Plant Cell. 2001, 13, 645–658. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ueda, M.; Nishikawa, T.; Fujimoto, M.; Takanashi, H.; Arimura, S.-I.; Tsutsumi, N.; Kadowaki, K.-I. Substitution of the gene for chloroplast rps16 was assisted by generation of a dual targeting signal. Mol. Biol. Evol. 2008, 25, 1566–1575. [Google Scholar] [CrossRef] [Green Version]
- Park, S.J.; Jansen, R.K.; Park, S.J. Complete plastome sequence of Thalictrum coreanum (Ranunculadeae) and transfer of the rpl32 gene to the nucleus in the ancestor of the subfamily Thalictroideae. BMC Plant Biol. 2015, 15, 40. [Google Scholar] [CrossRef] [Green Version]
- Stegemann, S.; Hartmann, S.; Ruf, S.; Bock, R. High-frequency gene transfer from the chloroplast genome to the nucleus. Proc. Natl. Acad. Sci. USA 2003, 100, 882–8833. [Google Scholar] [CrossRef] [Green Version]
- Timmis, J.N.; Ayliffe, M.A.; Huang, C.Y.; Martin, W. Endosymbiotic gene transfer: Organelle genomes forge eukaryotic chromosomes. Nature Rev. Genet. 2004, 5, 123–135. [Google Scholar] [CrossRef]
- Noutsos, C.; Richly, E.; Leister, D. Generation and evolutionary fate of insertions of organelle DNA in the nuclear genomes of flowering plants. Genome Res. 2005, 15, 616–628. [Google Scholar] [CrossRef] [Green Version]
- Sloan, D.B.; Triant, D.A.; Forrester, N.J.; Bergner, L.M.; Wu, M.; Taylor, D.R. A recurring syndrome of accelerated plastid genome evolution in the angiosperm tribe Sileneae (Caryophyllaceae). Mol. Phylogenet. Evol. 2014, 72, 82–89. [Google Scholar] [CrossRef]
- Cheng, H.; Li, J.; Zhang, H.; Cai, B.; Gao, Z.; Qiao, Y.; Mi, L. The complete chloroplast genome sequence of strawberry (Fragaria×ananassa Duch.) and comparison with related species of Rosaceae. PeerJ 2017, 5, e3919. [Google Scholar] [CrossRef] [Green Version]
- Jiang, P.; Shi, F.X.; Li, M.R.; Liu, B.; Wen, J.; Xiao, H.X.; Li, L.F. Positive selection driving cytoplasmic genome evolution of the medicinally important ginseng plant genus Panax. Front. Plant Sci. 2018, 9, 359. [Google Scholar] [CrossRef] [PubMed]
- Li, P.; Lou, G.; Cai, X.; Zhang, B.; Cheng, Y.; Wang, H. Comparison of the complete plastomes and the phylogenetic analysis of Paulownia species. Sci. Rep. 2020, 10, 2225. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, L.Q.; Zhang, H.; Jiang, M.; Chen, H.; Huang, L.; Liu, C. Complete plastome sequence of Iodes cirrhosa Turcz., the first in the Icacinaceae, comparative genomic analyses and possible split of Idoes species in response to climate changes. PeerJ 2019, 7, e6663. [Google Scholar] [CrossRef] [PubMed]
- Caspermeyer, J. Most comprehensive study to date reveals evolutionary history of Citrus. Mol. Biol. Evol. 2015, 32, 2217–2218. [Google Scholar] [CrossRef] [Green Version]
- Fan, X.; Zhang, J.; Li, W.; Peng, L. The ndhV subunit is required to stabilize the chloroplast NADH dehydrogenase-like complex in Arabidopsis. Plant J. 2015, 82, 221–231. [Google Scholar] [CrossRef] [Green Version]
- Horvath, E.M.; Peter, S.O.; Joët, T.; Rumeau, D.; Cournac, L.; Horváth, G.V.; Kavanagh, T.A.; Schafer, C.; Peltier, G.; Medgyesy, P. Targeted inactivation of the plastid ndhB gene in tobacco results in an enhanced sensitivity of photosynthesis to moderate stomatal closure. Plant Physiol. 2000, 123, 1337–1350. [Google Scholar] [CrossRef] [Green Version]
- Burri, R.; Salamin, N.; Studer, R.A.; Roulin, A.; Fumagalli, L. Adaptive divergence of ancient gene duplicates in the avian MHC class II β. Mol. Biol. Evol. 2010, 27, 2360–2374. [Google Scholar] [CrossRef] [Green Version]
- Piot, A.; Hackel, J.; Christin, P.A.; Besnard, G. One-third of the plastid genes evolved under positive selection in PACMAD grasses. Planta 2018, 247, 255–266. [Google Scholar] [CrossRef]
- Okaura, T.; Harada, K. Phylogeographical structure revealed by chloroplast DNA variation in Japanese Beech (Fagus crenata Blume). Heredity 2002, 88, 322–329. [Google Scholar] [CrossRef] [Green Version]
- Cannon, C.H.; Manos, P.S. Phylogeography of the Southeast Asian stone oaks (Lithocarpus). J. Biogeogr. 2003, 30, 211–226. [Google Scholar] [CrossRef]
- Paik, I.-S.; Kim, H.-J.; Kim, K.; Jeong, E.-K.; Kang, H.-C.; Lee, H.-I.; Uemura, K. Leaf beds in the Early Miocene lacustrine deposits of the Geumgwangdong Formation, Korea: Occurrence, plant-insect interaction records, taphonomy and paleoenviromental implications. Rev. Palaeobot. Palynol. 2012, 170, 1–14. [Google Scholar] [CrossRef]
- Nakai, T. Notulae ad plantas Japoniae et Koreae XVII. Bot. Mag. 1918, 32, 103–110. [Google Scholar] [CrossRef] [Green Version]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Taxa | F. crenata | F. japonica7-1 | F. japonica10-1 | F. multinervis |
|---|---|---|---|---|
| Total cpDNA size (bp) | 158,227 | 158,163 | 158,193 | 158,348 |
| GC content (%) | 37.1% | 37.1% | 37.1% | 37.1% |
| LSC size (bp)/GC content (%) | 87,552/35.1% | 87,590/35.1% | 87,620/35.1% | 87,659/35.0% |
| IR size (bp)/GC content (%) | 25,873/42.7% | 25,894/42.7% | 25,893/42.7% | 25,893/42.7% |
| SSC size (bp)/GC content (%) | 18,929/31.1% | 18,785/31.2% | 18,787/31.2% | 18,903/31.1% |
| Number of genes | 131 | 131 | 131 | 131 |
| Number of protein-coding genes | 82 | 82 | 82 | 82 |
| Number of tRNA genes | 37 | 37 | 37 | 37 |
| Number of rRNA genes | 8 | 8 | 8 | 8 |
| Number of duplicated genes | 17 | 17 | 17 | 17 |
| Accession Number | MT762292 | MT762294 | MT762295 | MT762296 |
| Gene Name | Models | np | ln L | Likelihood Ratio Test p-Value | Positively Selected Sites |
|---|---|---|---|---|---|
| ndhD | M8 | 15 | −2087.368596 | 0.000000130 | 385 T 0.951 * |
| M7 | 13 | −2103.224804 | |||
| ndhJ | M8 | 15 | −670.195417 | 0.000000009 | 151 F 0.995 ** |
| M7 | 13 | −688.698395 | |||
| rpoB | M8 | 15 | −4347.768637 | 0.000000009 | 248 Q 0.959 * |
| M7 | 13 | −4372.590740 | |||
| rpoC2 | M8 | 15 | −5635.415430 | 0.000000000 | 748 Y 0.984 *,1338 I 0.983 * |
| M7 | 13 | −5656.937844 | |||
| rps16 | M8 | 15 | −242.458940 | 0.000000720 | 1 M 0.995 ** |
| M7 | 13 | −256.602491 | |||
| ycf1 | M8 | 15 | −7160.045295 | 0.000000000 | 372 K 1.000 **, 373 T 1.000 **, 842 F 0.991 **, 905 P 0.991 **, 1066 R 0.992 **, 1142 F 0.991 **, 1284 N 1.000 **, 1286 D 0.991 **, 1359 F 0.999 ** |
| M7 | 13 | −7218.177588 | |||
| ycf2 | M8 | 15 | −9220.322295 | 0.000000000 | 388 T 0.982 * |
| M7 | 13 | −9248.972947 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yang, J.; Takayama, K.; Youn, J.-S.; Pak, J.-H.; Kim, S.-C. Plastome Characterization and Phylogenomics of East Asian Beeches with a Special Emphasis on Fagus multinervis on Ulleung Island, Korea. Genes 2020, 11, 1338. https://doi.org/10.3390/genes11111338
Yang J, Takayama K, Youn J-S, Pak J-H, Kim S-C. Plastome Characterization and Phylogenomics of East Asian Beeches with a Special Emphasis on Fagus multinervis on Ulleung Island, Korea. Genes. 2020; 11(11):1338. https://doi.org/10.3390/genes11111338
Chicago/Turabian StyleYang, JiYoung, Koji Takayama, Jin-Suk Youn, Jae-Hong Pak, and Seung-Chul Kim. 2020. "Plastome Characterization and Phylogenomics of East Asian Beeches with a Special Emphasis on Fagus multinervis on Ulleung Island, Korea" Genes 11, no. 11: 1338. https://doi.org/10.3390/genes11111338
APA StyleYang, J., Takayama, K., Youn, J.-S., Pak, J.-H., & Kim, S.-C. (2020). Plastome Characterization and Phylogenomics of East Asian Beeches with a Special Emphasis on Fagus multinervis on Ulleung Island, Korea. Genes, 11(11), 1338. https://doi.org/10.3390/genes11111338