Genetic Spectrum of Syndromic and Non-Syndromic Hearing Loss in Pakistani Families

 , , , ,
, , , , 
 , ,
, ,  , add
Show full author list
, add
Show full author list
Abstract
:1. Introduction
2. Materials and Methods
2.1. Clinical Evaluation
2.2. Autozygosity Mapping and Linkage Analysis
2.2.1. Genotyping and Quality Control
2.2.2. Linkage Analysis
2.3. Exome Sequencing
2.4. Variant Analysis and Prioritization
2.5. Variant Validation and Segregation Testing
3. Results
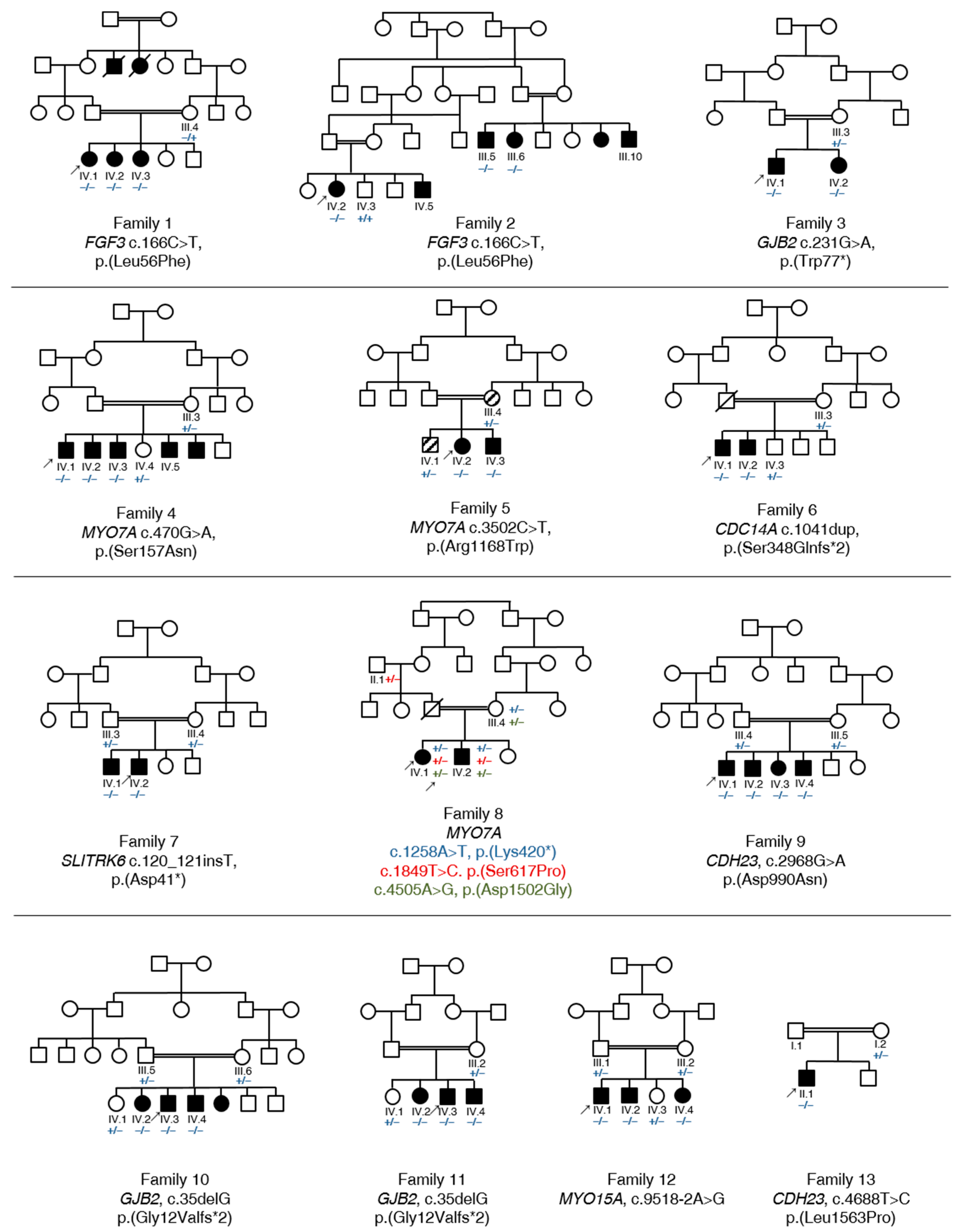
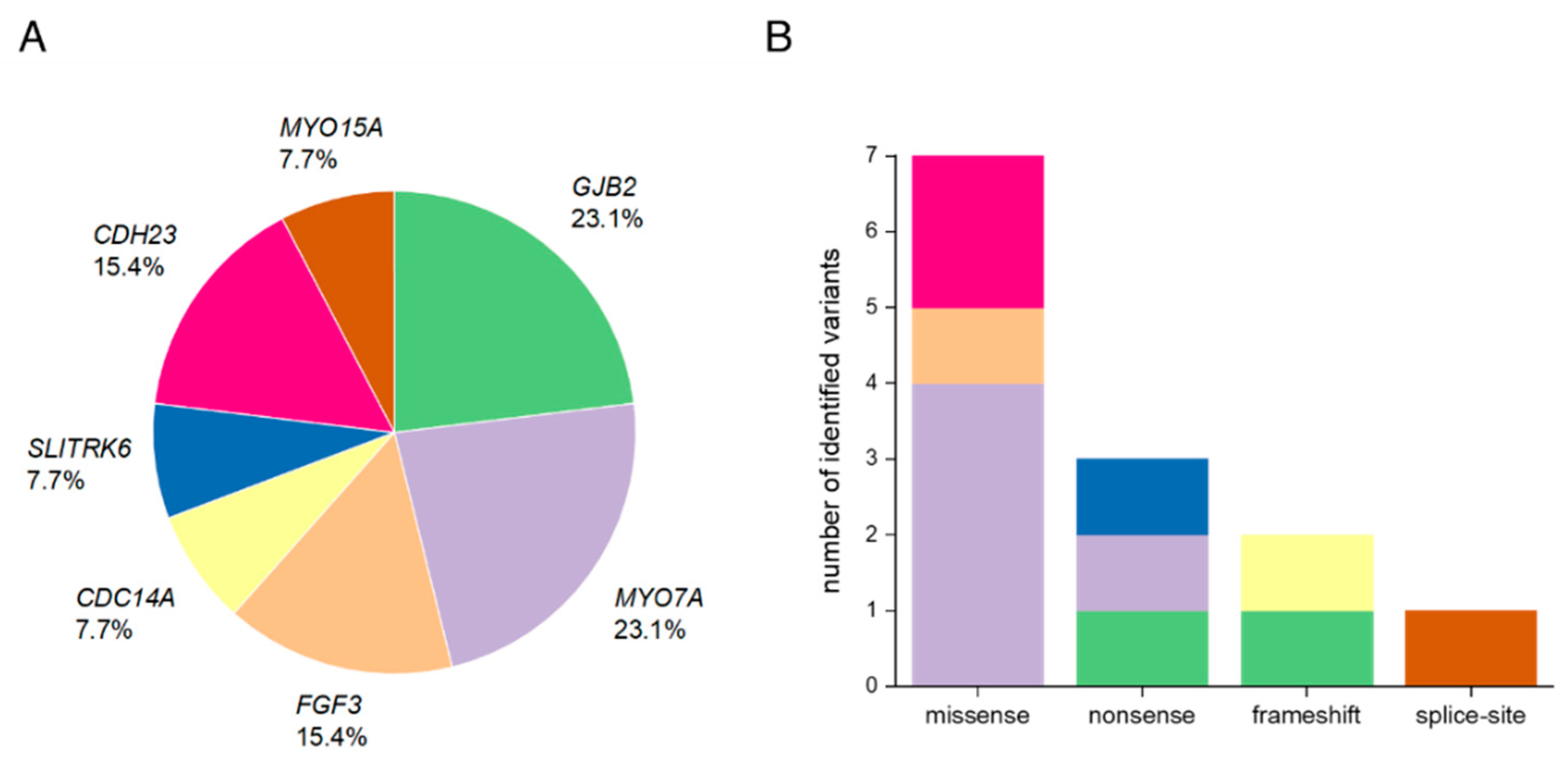
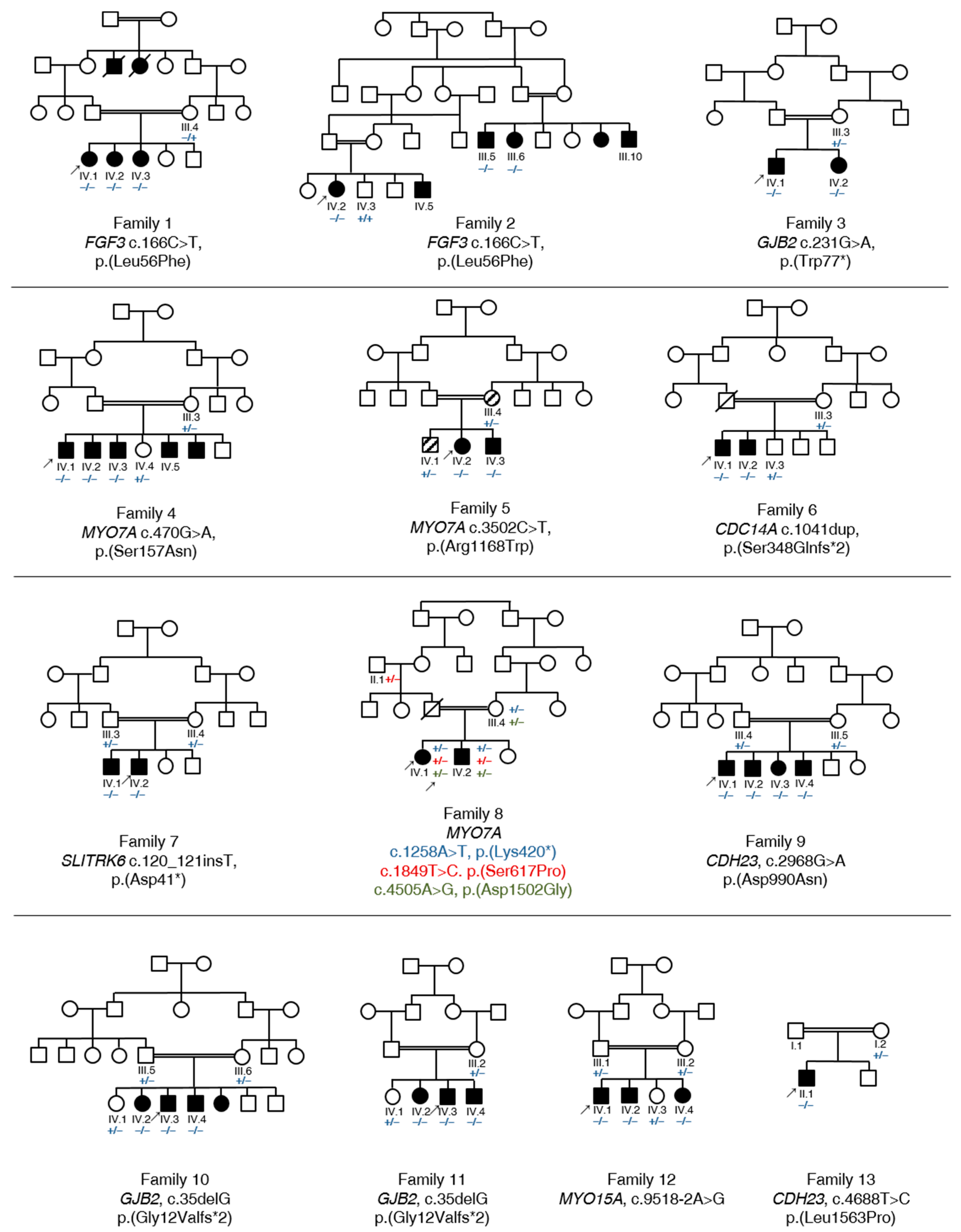
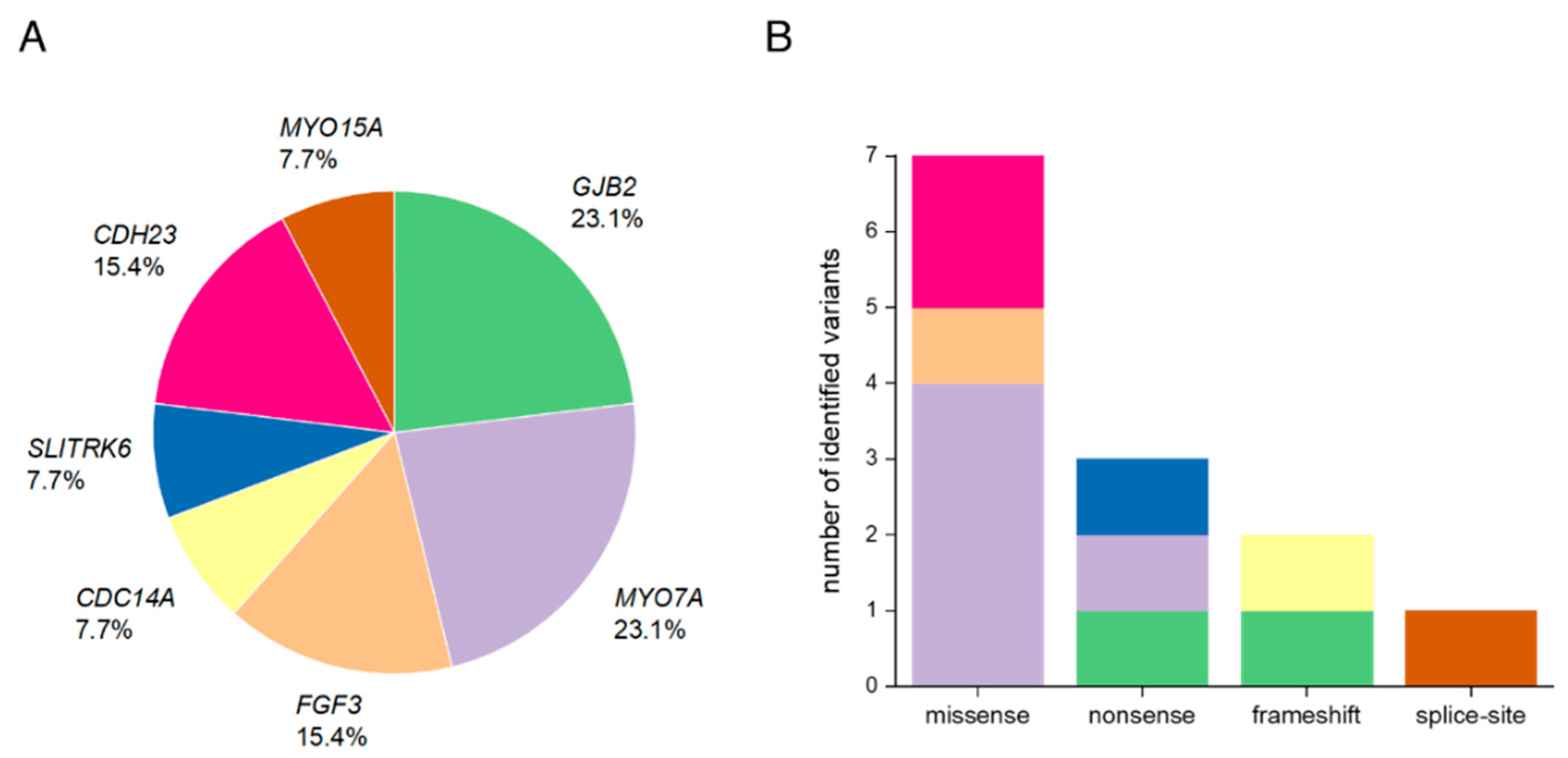
3.1. Summary of Affected Genes and Genetic Context
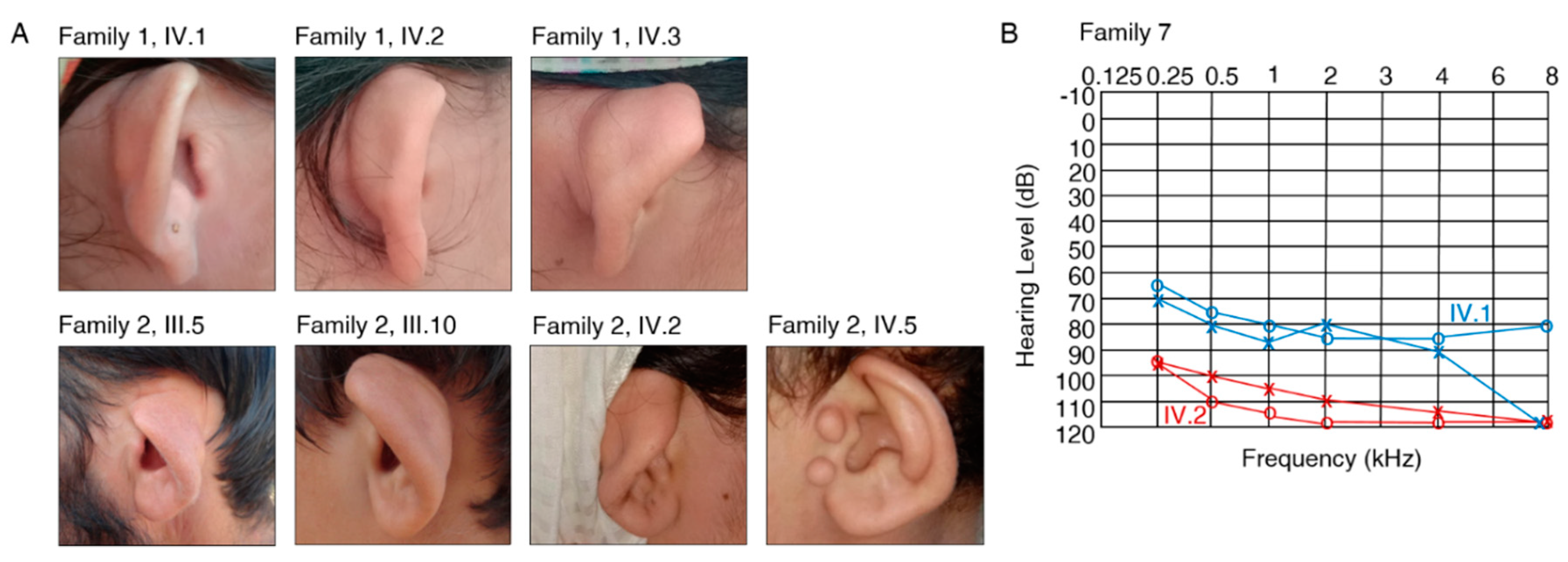
3.2. Clinical Features and Genetic Spectrum of Patients with Syndromic HL
FGF3, MYO7A and SLITRK6
3.3. Identification of Causative Variants in Patients with NSHL
3.3.1. GJB2
3.3.2. MYO7A
3.3.3. CDC14A, CDH23 and MYO15A
3.4. Autosomal Recessive HL Loci
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Hamamy, H. Consanguineous marriages: Preconception consultation in primary health care settings. J. Community Genet. 2012, 3, 185–192. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hussain, R.; Bittles, A.H. The prevalence and demographic characteristics of consanguineous marriages in Pakistan. J. Biosoc. Sci. 1998, 30, 261–275. [Google Scholar] [CrossRef] [PubMed]
- Zakzouk, S. Consanguinity and hearing impairment in developing countries: A custom to be discouraged. J. Laryngol. Otol. 2002, 116, 811–816. [Google Scholar] [CrossRef] [PubMed]
- Morton, C.C.; Nance, W.E. Newborn hearing screening--a silent revolution. N. Engl. J. Med. 2006, 354, 2151–2164. [Google Scholar] [CrossRef] [PubMed]
- Khan, A.; Han, S.; Wang, R.; Ansar, M.; Ahmad, W.; Zhang, X. Sequence variants in genes causing nonsyndromic hearing loss in a Pakistani cohort. Mol. Genet. Genom. Med. 2019, 7, e917. [Google Scholar] [CrossRef] [PubMed]
- Richard, E.M.; Santos-Cortez, R.L.P.; Faridi, R.; Rehman, A.U.; Lee, K.; Shahzad, M.; Acharya, A.; Khan, A.A.; Imtiaz, A.; Chakchouk, I.; et al. Global genetic insight contributed by consanguineous Pakistani families segregating hearing loss. Hum. Mutat. 2019, 40, 53–72. [Google Scholar] [CrossRef] [Green Version]
- Shafique, S.; Siddiqi, S.; Schraders, M.; Oostrik, J.; Ayub, H.; Bilal, A.; Ajmal, M.; Seco, C.Z.; Strom, T.M.; Mansoor, A.; et al. Genetic spectrum of autosomal recessive non-syndromic hearing loss in Pakistani families. PLoS ONE 2014, 9, e100146. [Google Scholar] [CrossRef] [Green Version]
- Parker, M.; Bitner-Glindzicz, M. Genetic investigations in childhood deafness. Arch. Dis. Child. 2015, 100, 271–278. [Google Scholar] [CrossRef]
- Bademci, G.; Cengiz, F.B.; Foster Ii, J.; Duman, D.; Sennaroglu, L.; Diaz-Horta, O.; Atik, T.; Kirazli, T.; Olgun, L.; Alper, H.; et al. Variations in Multiple Syndromic Deafness Genes Mimic Non-syndromic Hearing Loss. Sci. Rep. 2016, 6, 31622. [Google Scholar] [CrossRef]
- Sajjad, M.; Khattak, A.A.; Bunn, J.E.; Mackenzie, I. Causes of childhood deafness in Pukhtoonkhwa Province of Pakistan and the role of consanguinity. J. Laryngol. Otol. 2008, 122, 1057–1063. [Google Scholar] [CrossRef] [Green Version]
- Vona, B.; Nanda, I.; Hofrichter, M.A.; Shehata-Dieler, W.; Haaf, T. Non-syndromic hearing loss gene identification: A brief history and glimpse into the future. Mol. Cell Probes 2015, 29, 260–270. [Google Scholar] [CrossRef] [PubMed]
- Mazzoli, M.; Van Camp, G.; Newton, V.; Giarbini, N.; Declau, F.; Parving, A. Recommendations for the description of genetic and audiological data for families with nonsyndromic hereditary hearing impairment. Audiol. Med. 2003, 1, 148–150. [Google Scholar]
- Ruschendorf, F.; Nurnberg, P. ALOHOMORA: A tool for linkage analysis using 10K SNP array data. Bioinformatics 2005, 21, 2123–2125. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abecasis, G.R.; Cherny, S.S.; Cookson, W.O.; Cardon, L.R. GRR: Graphical representation of relationship errors. Bioinformatics 2001, 17, 742–743. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- O’Connell, J.R.; Weeks, D.E. PedCheck: A program for identification of genotype incompatibilities in linkage analysis. Am. J. Hum. Genet. 1998, 63, 259–266. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abecasis, G.R.; Cherny, S.S.; Cookson, W.O.; Cardon, L.R. Merlin-rapid analysis of dense genetic maps using sparse gene flow trees. Nat. Genet. 2002, 30, 97–101. [Google Scholar] [CrossRef]
- Sambrook, J.; Russell, D.W. Purification of nucleic acids by extraction with phenol: Chloroform. CSH Protoc. 2006, 2006. [Google Scholar] [CrossRef]
- Green, M.R.; Sambrook, J. Precipitation of DNA with Ethanol. Cold Spring Harb. Protoc. 2016, 2016. [Google Scholar] [CrossRef]
- Li, H.; Durbin, R. Fast and accurate long-read alignment with Burrows-Wheeler transform. Bioinformatics 2010, 26, 589–595. [Google Scholar] [CrossRef] [Green Version]
- DePristo, M.A.; Banks, E.; Poplin, R.; Garimella, K.V.; Maguire, J.R.; Hartl, C.; Philippakis, A.A.; del Angel, G.; Rivas, M.A.; Hanna, M.; et al. A framework for variation discovery and genotyping using next-generation DNA sequencing data. Nat. Genet. 2011, 43, 491–498. [Google Scholar] [CrossRef]
- McKenna, A.; Hanna, M.; Banks, E.; Sivachenko, A.; Cibulskis, K.; Kernytsky, A.; Garimella, K.; Altshuler, D.; Gabriel, S.; Daly, M.; et al. The Genome Analysis Toolkit: A MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 2010, 20, 1297–1303. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Adzhubei, I.A.; Schmidt, S.; Peshkin, L.; Ramensky, V.E.; Gerasimova, A.; Bork, P.; Kondrashov, A.S.; Sunyaev, S.R. A method and server for predicting damaging missense mutations. Nat. Methods 2010, 7, 248–249. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ng, P.C.; Henikoff, S. Predicting the effects of amino acid substitutions on protein function. Annu. Rev. Genom. Hum. Genet. 2006, 7, 61–80. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schwarz, J.M.; Cooper, D.N.; Schuelke, M.; Seelow, D. MutationTaster2: Mutation prediction for the deep-sequencing age. Nat. Methods 2014, 11, 361–362. [Google Scholar] [CrossRef] [PubMed]
- Shihab, H.A.; Gough, J.; Cooper, D.N.; Stenson, P.D.; Barker, G.L.; Edwards, K.J.; Day, I.N.; Gaunt, T.R. Predicting the functional, molecular, and phenotypic consequences of amino acid substitutions using hidden Markov models. Hum. Mutat. 2013, 34, 57–65. [Google Scholar] [CrossRef]
- Chun, S.; Fay, J.C. Identification of deleterious mutations within three human genomes. Genome Res. 2009, 19, 1553–1561. [Google Scholar] [CrossRef] [Green Version]
- Cooper, G.M.; Stone, E.A.; Asimenos, G.; Green, E.D.; Batzoglou, S.; Sidow, A. Distribution and intensity of constraint in mammalian genomic sequence. Genome Res. 2005, 15, 901–913. [Google Scholar] [CrossRef] [Green Version]
- Shearer, A.E.; Eppsteiner, R.W.; Booth, K.T.; Ephraim, S.S.; Gurrola, J., II; Simpson, A.; Black-Ziegelbein, E.A.; Joshi, S.; Ravi, H.; Giuffre, A.C.; et al. Utilizing ethnic-specific differences in minor allele frequency to recategorize reported pathogenic deafness variants. Am. J. Hum. Genet. 2014, 95, 445–453. [Google Scholar] [CrossRef] [Green Version]
- Scott, E.M.; Halees, A.; Itan, Y.; Spencer, E.G.; He, Y.; Azab, M.A.; Gabriel, S.B.; Belkadi, A.; Boisson, B.; Abel, L.; et al. Characterization of Greater Middle Eastern genetic variation for enhanced disease gene discovery. Nat. Genet. 2016, 48, 1071–1076. [Google Scholar] [CrossRef]
- Karczewski, K.J.; Francioli, L.C.; Tiao, G.; Cummings, B.B.; Alföldi, J.; Wang, Q.; Collins, R.L.; Laricchia, K.M.; Ganna, A.; Birnbaum, D.P.; et al. Variation across 141,456 human exomes and genomes reveals the spectrum of loss-of-function intolerance across human protein-coding genes. bioRXiv 2019. [Google Scholar] [CrossRef]
- Fromer, M.; Purcell, S.M. Using XHMM Software to Detect Copy Number Variation in Whole-Exome Sequencing Data. Curr. Protoc. Hum. Genet. 2014, 81, 7–23. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Untergasser, A.; Cutcutache, I.; Koressaar, T.; Ye, J.; Faircloth, B.C.; Remm, M.; Rozen, S.G. Primer3-new capabilities and interfaces. Nucleic Acids Res. 2012, 40, e115. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kelsell, D.P.; Dunlop, J.; Stevens, H.P.; Lench, N.J.; Liang, J.N.; Parry, G.; Mueller, R.F.; Leigh, I.M. Connexin 26 mutations in hereditary non-syndromic sensorineural deafness. Nature 1997, 387, 80–83. [Google Scholar] [CrossRef] [PubMed]
- Jaijo, T.; Aller, E.; Oltra, S.; Beneyto, M.; Najera, C.; Ayuso, C.; Baiget, M.; Carballo, M.; Antinolo, G.; Valverde, D.; et al. Mutation profile of the MYO7A gene in Spanish patients with Usher syndrome type I. Hum. Mutat. 2006, 27, 290–291. [Google Scholar] [CrossRef] [PubMed]
- Le Guedard-Mereuze, S.; Vache, C.; Baux, D.; Faugere, V.; Larrieu, L.; Abadie, C.; Janecke, A.; Claustres, M.; Roux, A.F.; Tuffery-Giraud, S. Ex vivo splicing assays of mutations at noncanonical positions of splice sites in USHER genes. Hum. Mutat. 2010, 31, 347–355. [Google Scholar] [CrossRef] [PubMed]
- Doll, J.; Kolb, S.; Schnapp, L.; Rad, A.; Ruschendorf, F.; Khan, I.; Adli, A.; Hasanzadeh, A.; Liedtke, D.; Knaup, S.; et al. Novel Loss-of-Function Variants in CDC14A are Associated with Recessive Sensorineural Hearing Loss in Iranian and Pakistani Patients. Int. J. Mol. Sci. 2020, 21, 311. [Google Scholar] [CrossRef] [Green Version]
- Cremers, F.P.; Kimberling, W.J.; Kulm, M.; de Brouwer, A.P.; van Wijk, E.; te Brinke, H.; Cremers, C.W.; Hoefsloot, L.H.; Banfi, S.; Simonelli, F.; et al. Development of a genotyping microarray for Usher syndrome. J. Med. Genet. 2007, 44, 153–160. [Google Scholar] [CrossRef] [Green Version]
- Carss, K.J.; Arno, G.; Erwood, M.; Stephens, J.; Sanchis-Juan, A.; Hull, S.; Megy, K.; Grozeva, D.; Dewhurst, E.; Malka, S.; et al. Comprehensive Rare Variant Analysis via Whole-Genome Sequencing to Determine the Molecular Pathology of Inherited Retinal Disease. Am. J. Hum. Genet. 2017, 100, 75–90. [Google Scholar] [CrossRef] [Green Version]
- Bork, J.M.; Peters, L.M.; Riazuddin, S.; Bernstein, S.L.; Ahmed, Z.M.; Ness, S.L.; Polomeno, R.; Ramesh, A.; Schloss, M.; Srisailpathy, C.R.; et al. Usher syndrome 1D and nonsyndromic autosomal recessive deafness DFNB12 are caused by allelic mutations of the novel cadherin-like gene CDH23. Am. J. Hum. Genet. 2001, 68, 26–37. [Google Scholar] [CrossRef] [Green Version]
- Zelante, L.; Gasparini, P.; Estivill, X.; Melchionda, S.; D’Agruma, L.; Govea, N.; Mila, M.; Monica, M.D.; Lutfi, J.; Shohat, M.; et al. Connexin26 mutations associated with the most common form of non-syndromic neurosensory autosomal recessive deafness (DFNB1) in Mediterraneans. Hum. Mol. Genet. 1997, 6, 1605–1609. [Google Scholar] [CrossRef] [Green Version]
- Schultz, J.M.; Bhatti, R.; Madeo, A.C.; Turriff, A.; Muskett, J.A.; Zalewski, C.K.; King, K.A.; Ahmed, Z.M.; Riazuddin, S.; Ahmad, N.; et al. Allelic hierarchy of CDH23 mutations causing non-syndromic deafness DFNB12 or Usher syndrome USH1D in compound heterozygotes. J. Med. Genet. 2011, 48, 767–775. [Google Scholar] [CrossRef] [PubMed]
- Oza, A.M.; DiStefano, M.T.; Hemphill, S.E.; Cushman, B.J.; Grant, A.R.; Siegert, R.K.; Shen, J.; Chapin, A.; Boczek, N.J.; Schimmenti, L.A.; et al. Expert specification of the ACMG/AMP variant interpretation guidelines for genetic hearing loss. Hum. Mutat. 2018, 39, 1593–1613. [Google Scholar] [CrossRef] [PubMed]
- Tekin, M.; Hismi, B.O.; Fitoz, S.; Ozdag, H.; Cengiz, F.B.; Sirmaci, A.; Aslan, I.; Inceoglu, B.; Yuksel-Konuk, E.B.; Yilmaz, S.T.; et al. Homozygous mutations in fibroblast growth factor 3 are associated with a new form of syndromic deafness characterized by inner ear agenesis, microtia, and microdontia. Am. J. Hum. Genet. 2007, 80, 338–344. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aparisi, M.J.; Garcia-Garcia, G.; Aller, E.; Sequedo, M.D.; de la Camara, C.M.-F.; Rodrigo, R.; Armengot, M.; Cortijo, J.; Milara, J.; Diaz, L.M.; et al. Study of USH1 splicing variants through minigenes and transcript analysis from nasal epithelial cells. PLoS ONE 2013, 8, e57506. [Google Scholar] [CrossRef] [PubMed]
- Tekin, M.; Chioza, B.A.; Matsumoto, Y.; Diaz-Horta, O.; Cross, H.E.; Duman, D.; Kokotas, H.; Moore-Barton, H.L.; Sakoori, K.; Ota, M.; et al. SLITRK6 mutations cause myopia and deafness in humans and mice. J. Clin. Investig. 2013, 123, 2094–2102. [Google Scholar] [CrossRef] [Green Version]
- Yan, D.; Kannan-Sundhari, A.; Vishwanath, S.; Qing, J.; Mittal, R.; Kameswaran, M.; Liu, X.Z. The Genetic Basis of Nonsyndromic Hearing Loss in Indian and Pakistani Populations. Genet. Test. Mol. Biomark. 2015, 19, 512–527. [Google Scholar] [CrossRef] [Green Version]
- Heutink, P.; Oostra, B.A. Gene finding in genetically isolated populations. Hum. Mol. Genet. 2002, 11, 2507–2515. [Google Scholar] [CrossRef] [Green Version]
- Friedman, T.B.; Griffith, A.J. Human nonsyndromic sensorineural deafness. Annu. Rev. Genom. Hum. Genet. 2003, 4, 341–402. [Google Scholar] [CrossRef]
- Sloan-Heggen, C.M.; Babanejad, M.; Beheshtian, M.; Simpson, A.C.; Booth, K.T.; Ardalani, F.; Frees, K.L.; Mohseni, M.; Mozafari, R.; Mehrjoo, Z.; et al. Characterising the spectrum of autosomal recessive hereditary hearing loss in Iran. J. Med. Genet. 2015, 52, 823–829. [Google Scholar] [CrossRef]
- Azaiez, H.; Booth, K.T.; Ephraim, S.S.; Crone, B.; Black-Ziegelbein, E.A.; Marini, R.J.; Shearer, A.E.; Sloan-Heggen, C.M.; Kolbe, D.; Casavant, T.; et al. Genomic Landscape and Mutational Signatures of Deafness-Associated Genes. Am. J. Hum. Genet. 2018, 103, 484–497. [Google Scholar] [CrossRef] [Green Version]
- Anwar, S.; Riazuddin, S.; Ahmed, Z.M.; Tasneem, S.; Ateequl, J.; Khan, S.Y.; Griffith, A.J.; Friedman, T.B.; Riazuddin, S. SLC26A4 mutation spectrum associated with DFNB4 deafness and Pendred’s syndrome in Pakistanis. J. Hum. Genet. 2009, 54, 266–270. [Google Scholar] [CrossRef] [PubMed]
- Hilgert, N.; Smith, R.J.; Van Camp, G. Forty-six genes causing nonsyndromic hearing impairment: Which ones should be analyzed in DNA diagnostics? Mutat. Res. 2009, 681, 189–196. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schultz, J.M.; Khan, S.N.; Ahmed, Z.M.; Riazuddin, S.; Waryah, A.M.; Chhatre, D.; Starost, M.F.; Ploplis, B.; Buckley, S.; Velasquez, D.; et al. Noncoding mutations of HGF are associated with nonsyndromic hearing loss, DFNB39. Am. J. Hum. Genet. 2009, 85, 25–39. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Anjum, S.; Azhar, A.; Tariq, M.; Baig, S.M.; Bolz, H.J.; Qayyum, M.; Naqvi, S.M.S.; Raja, G.K. GJB2 Gene Mutations Causing Hearing Loss in Pakistani Families. Pak. J. Life Soc. Sci. 2014, 12, 126–131. [Google Scholar]
- Liu, X.Z.; Walsh, J.; Mburu, P.; Kendrick-Jones, J.; Cope, M.J.; Steel, K.P.; Brown, S.D. Mutations in the myosin VIIA gene cause non-syndromic recessive deafness. Nat. Genet. 1997, 16, 188–190. [Google Scholar] [CrossRef] [PubMed]
- Weil, D.; Blanchard, S.; Kaplan, J.; Guilford, P.; Gibson, F.; Walsh, J.; Mburu, P.; Varela, A.; Levilliers, J.; Weston, M.D.; et al. Defective myosin VIIA gene responsible for Usher syndrome type 1B. Nature 1995, 374, 60–61. [Google Scholar] [CrossRef]
- Vanniya, S.P.; Chandru, J.; Pavithra, A.; Jeffrey, J.M.; Kalaimathi, M.; Ramakrishnan, R.; Karthikeyen, N.P.; Srikumari, C.R.S. Recurrence of reported CDH23 mutations causing DFNB12 in a special cohort of South Indian hearing impaired assortative mating families—An evaluation. Ann. Hum. Genet. 2018, 82, 119–126. [Google Scholar] [CrossRef]
- Riazuddin, S.; Ahmed, Z.M.; Hegde, R.S.; Khan, S.N.; Nasir, I.; Shaukat, U.; Riazuddin, S.; Butman, J.A.; Griffith, A.J.; Friedman, T.B.; et al. Variable expressivity of FGF3 mutations associated with deafness and LAMM syndrome. BMC Med. Genet. 2011, 12, 21. [Google Scholar] [CrossRef] [Green Version]
- Maquat, L.E. Nonsense-mediated mRNA decay: Splicing, translation and mRNP dynamics. Nat. Rev. Mol. Cell Biol. 2004, 5, 89–99. [Google Scholar] [CrossRef]
- Salime, S.; Riahi, Z.; Elrharchi, S.; Elkhattabi, L.; Charoute, H.; Nahili, H.; Rouba, H.; Kabine, M.; Bonnet, C.; Petit, C.; et al. A novel mutation in SLITRK6 causes deafness and myopia in a Moroccan family. Gene 2018, 659, 89–92. [Google Scholar] [CrossRef]
- Van Beeck Calkoen, E.A.; Engel, M.S.D.; van de Kamp, J.M.; Yntema, H.G.; Goverts, S.T.; Mulder, M.F.; Merkus, P.; Hensen, E.F. The etiological evaluation of sensorineural hearing loss in children. Eur. J. Pediatr. 2019, 178, 1195–1205. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ordonez, J.L.; Tekin, M. Deafness and Myopia Syndrome. In GeneReviews((R)); Adam, M.P., Ardinger, H.H., Pagon, R.A., Wallace, S.E., Bean, L.J.H., Stephens, K., Amemiya, A., Eds.; University of Washington: Seattle, WA, USA, 1993. [Google Scholar]
- Anna, A.; Monika, G. Splicing mutations in human genetic disorders: Examples, detection, and confirmation. J. Appl. Genet. 2018, 59, 253–268. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sheppard, S.; Biswas, S.; Li, M.H.; Jayaraman, V.; Slack, I.; Romasko, E.J.; Sasson, A.; Brunton, J.; Rajagopalan, R.; Sarmady, M.; et al. Utility and limitations of exome sequencing as a genetic diagnostic tool for children with hearing loss. Genet. Med. 2018, 20, 1663–1676. [Google Scholar] [CrossRef] [PubMed] [Green Version]

{kind=link}
{kind=link}
{kind=link}
| ID | Gene | DFN locus | Transcript | Nucleotide | Protein | Zygosity | MT | PP | SIFT | GERP | LRT | DVD |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Family 1 | FGF3 | -- | NM_005247.2 | c.166C>T | p.(Leu56Phe) | 1/1 | DC | PrD | D | C | U | U |
| Family 2 | FGF3 | -- | NM_005247.2 | c.166C>T | p.(Leu56Phe) | 1/1 | DC | PrD | D | C | U | U |
| Family 3 | GJB2 | DFNB1 | NM_004004.5 | c.231G>A | p.(Trp77*) | 1/1 | DC | -- | -- | C | D | P [33] |
| Family 4 | MYO7A | DFNB2 | NM_000260.3 | c.470G>A | p.(Ser157Asn) | 1/1 | DC | PrD | D | C | D | P [34] |
| Family 5 | MYO7A | DFNB2 | NM_000260.3 | c.3502C>T | p.(Arg1168Trp) | 1/1 | DC | PrD | D | C | D | LP [35] |
| Family 6 | CDC14A | DFNB32 | NM_033312.2 | c.1041dup [36] | p.(Ser348Glnfs*2) [36] | 1/1 | -- | -- | -- | -- | -- | -- |
| Family 7 | SLITRK6 | -- | NM_032229.2 | c.120_121insT | p.(Asp41*) | 1/1 | -- | -- | -- | -- | -- | -- |
| Family 8 | MYO7A | DFNB2 | NM_000260.3 | c.1258A>T | p.(Lys420*) | 0/1 | DC | -- | -- | C | D | P [37] |
| c.1849T>C | p.(Ser617Pro) | 0/1 | DC | PrD | D | C | D | U [38] | ||||
| c.4505A>G | p.(Asp1502Gly) | 0/1 | DC | PrD | D | C | D | U [6] | ||||
| Family 9 | CDH23 | DFNB12 | NM_022124.5 | c.2968G>A | p.(Asp990Asn) | 1/1 | DC | PrD | D | C | D | P [39] |
| Family 10 | GJB2 | DFNB1 | NM_004004.5 | c.35delG | p.(Gly12Valfs*2) | 1/1 | -- | -- | -- | -- | -- | P [40] |
| Family 11 | GJB2 | DFNB1 | NM_004004.5 | c.35delG | p.(Gly12Valfs*2) | 1/1 | -- | -- | -- | -- | -- | P [40] |
| Family 12 | MYO15A | DFNB3 | NM_016239.3 | c.9518-2A>G | 1/1 | DC | -- | -- | C | -- | U [5] | |
| Family 13 | CDH23 | DFNB12 | NM_022124.5 | c.4688T>C | p.(Leu1563Pro) | 1/1 | DC | PrD | D | C | D | P [41] |
| ID | Phenotype | Affected Family Members | Unaffected Family Members |
|---|---|---|---|
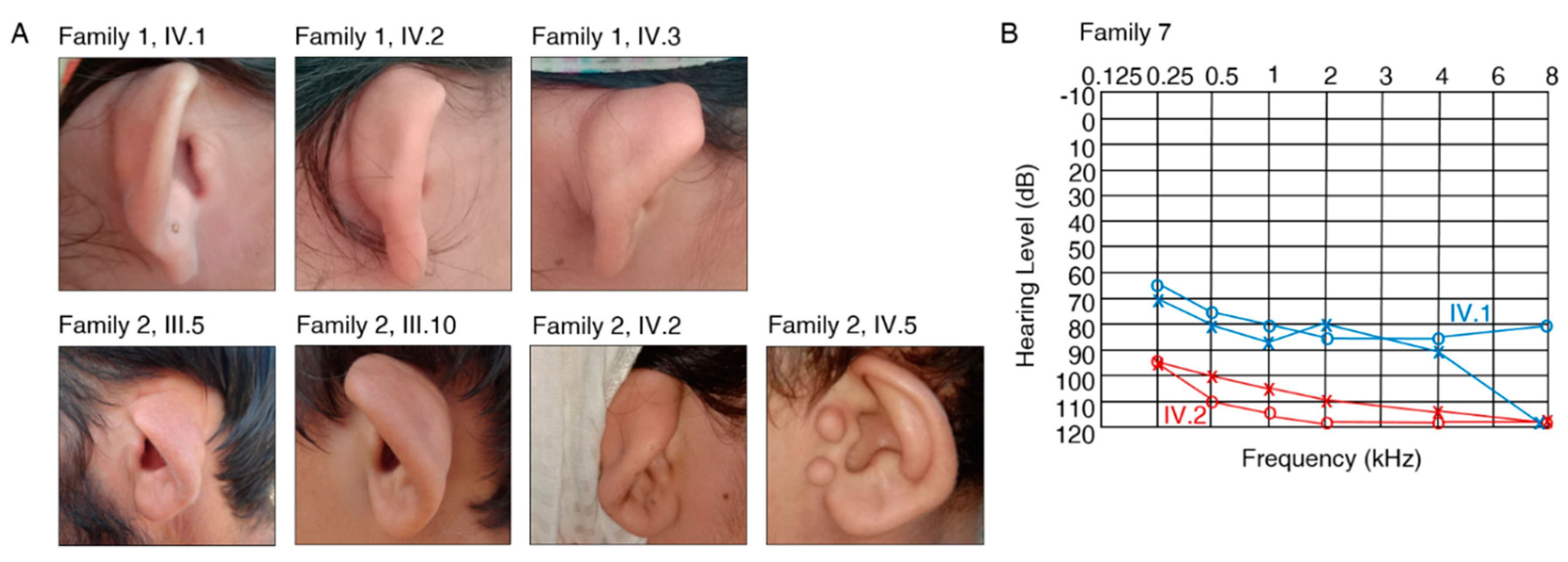
| Family 1 | HL, cupped ears | IV.1, IV.2, IV.3 | III.4 |
| Family 2 | HL, cupped ears | III.5, III.6, III.10 1, IV.2, IV.5 1 | IV.3 |
| Family 3 | HL | IV.1 (25 y/o), IV.2 (10 y/o) | III.3 |
| Family 4 | Usher syndrome | IV.1 (35 y/o), IV.2 (33 y/o), IV.3 (32 y/o), IV.5 (33 y/o) 1 | III.3, IV.4 |
| Family 5 | Usher syndrome 2, bone disorder 2 | IV.2 (30 y/o), IV.3 (18 y/o); Usher syndrome | III.4, IV.1; bone disorder |
| Family 6 | severe-to-profound HL, compound myopic astigmatism | IV.1 (30 y/o), IV.2 (28 y/o) | III.3, IV.3 |
| Family 7 | severe-to-profound HL, compound myopic astigmatism, glaucoma | IV.1 (26 y/o), IV.2 (23 y/o) | III.3, III.4 |
| Family 8 | HL | IV.1 (13 y/o), IV.2 (12 y/o) | II.1, III.4 |
| Family 9 | HL | IV.1 (33 y/o), IV.2 (32 y/o), IV.3 (20 y/o), IV.4 (18 y/o) | III.4, III.5 |
| Family 10 | HL | IV.2 (14 y/o), IV.3 (13 y/o), IV.4 (12 y/o) | III.5, III.6, IV.1 |
| Family 11 | HL | IV.2 (16 y/o), IV.3 (14 y/o), IV.4 (12 y/o) | III.2, IV.1 |
| Family 12 | HL | IV.1 (15 y/o), IV.2 (15 y/o), IV.4 (10 y/o) | III.1, III.2, IV.3 |
| Family 13 | HL | II.1 (11 y/o) | I.2 |
| Family ID | Chromosomal Band | Region of Autozygosity Identified by Linkage Analysis (hg19) | Length (Mb) | LOD | Causal Gene in Locus | Gene Coordinates (hg19) |
|---|---|---|---|---|---|---|
| Family 1 | 11p12-q13.4 | 39,536,493–73,025,971 | 33.5 | 2.529 | FGF3 | chr11:69,624,736–69,634,192 |
| Family 2 | 11q13.1-q13.3 | 63,870,810–69,964,525 | 6.1 | 3.73 | FGF3 | chr11:69,624,736–69,634,192 |
| Family 3 * | 13q12.11-q14.11 | 22,661,666–41,063,028 | 18.4 | 1.927 | ||
| Family 4 | 11q13.3-q14.3 | 69,063,393–88,489,081 | 19.4 | 2.529 | MYO7A | chr11:76,839,310–76,926,284 |
| Family 5 | 11q13.5-q14.1 | 76,792,431–80,457,784 | 3.7 | 1.927 | MYO7A | chr11:76,839,310–76,926,284 |
| Family 6 | 1p22.2-p21.2 | 88,430,037–102,069,696 | 13.6 | 1.927 | CDC14A | chr1:100,810,598–100,985,833 |
| Family 7 | 13q22.1-q31.3 | 74,995,660–90,925,494 | 15.9 | 1.2 | SLITRK6 | chr13:86,366,925–86,373,554 |
| Family 8 | No interval close to MYO7A | |||||
| Family 9 | 10q21.2-q22.3 | 64,059,261–78,005,230 | 13.9 | 3.006 | CDH23 | chr10:73,156,691–73,575,702 |
| Family 10 | 13q11-q12.11 | 19,121,950–22,395,049 | 3.3 | 2.529 | GJB2 | chr13:20,761,609–20,767,037 |
| Family 11 | 13q11-q12.12 | 19,121,950–23,534,670 | 4.4 | 2.529 | GJB2 | chr13:20,761,609–20,767,037 |
| Family 12 | 17p12-p11.2 | 13,801,016–21,539,613 | 7.7 | 2.529 | MYO15A | chr17:18,012,020–18,083,116 |
| Family 13 | 10q21.2-q23.31 | 61,998,060–91,002,927 | 29.0 | 1.2 | CDH23 | chr10:73,156,691–73,575,702 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Doll, J.; Vona, B.; Schnapp, L.; Rüschendorf, F.; Khan, I.; Khan, S.; Muhammad, N.; Alam Khan, S.; Nawaz, H.; Khan, A.; et al. Genetic Spectrum of Syndromic and Non-Syndromic Hearing Loss in Pakistani Families. Genes 2020, 11, 1329. https://doi.org/10.3390/genes11111329
Doll J, Vona B, Schnapp L, Rüschendorf F, Khan I, Khan S, Muhammad N, Alam Khan S, Nawaz H, Khan A, et al. Genetic Spectrum of Syndromic and Non-Syndromic Hearing Loss in Pakistani Families. Genes. 2020; 11(11):1329. https://doi.org/10.3390/genes11111329
Chicago/Turabian StyleDoll, Julia, Barbara Vona, Linda Schnapp, Franz Rüschendorf, Imran Khan, Saadullah Khan, Noor Muhammad, Sher Alam Khan, Hamed Nawaz, Ajmal Khan, and et al. 2020. "Genetic Spectrum of Syndromic and Non-Syndromic Hearing Loss in Pakistani Families" Genes 11, no. 11: 1329. https://doi.org/10.3390/genes11111329
APA StyleDoll, J., Vona, B., Schnapp, L., Rüschendorf, F., Khan, I., Khan, S., Muhammad, N., Alam Khan, S., Nawaz, H., Khan, A., Ahmad, N., Kolb, S. M., Kühlewein, L., Labonne, J. D. J., Layman, L. C., Hofrichter, M. A. H., Röder, T., Dittrich, M., Müller, T., ... Haaf, T. (2020). Genetic Spectrum of Syndromic and Non-Syndromic Hearing Loss in Pakistani Families. Genes, 11(11), 1329. https://doi.org/10.3390/genes11111329