CYP6P9-Driven Signatures of Selective Sweep of Metabolic Resistance to Pyrethroids in the Malaria Vector Anopheles funestus Reveal Contemporary Barriers to Gene Flow

 ,
,  , ,
, , 
Abstract
:1. Introduction
2. Materials and Methods
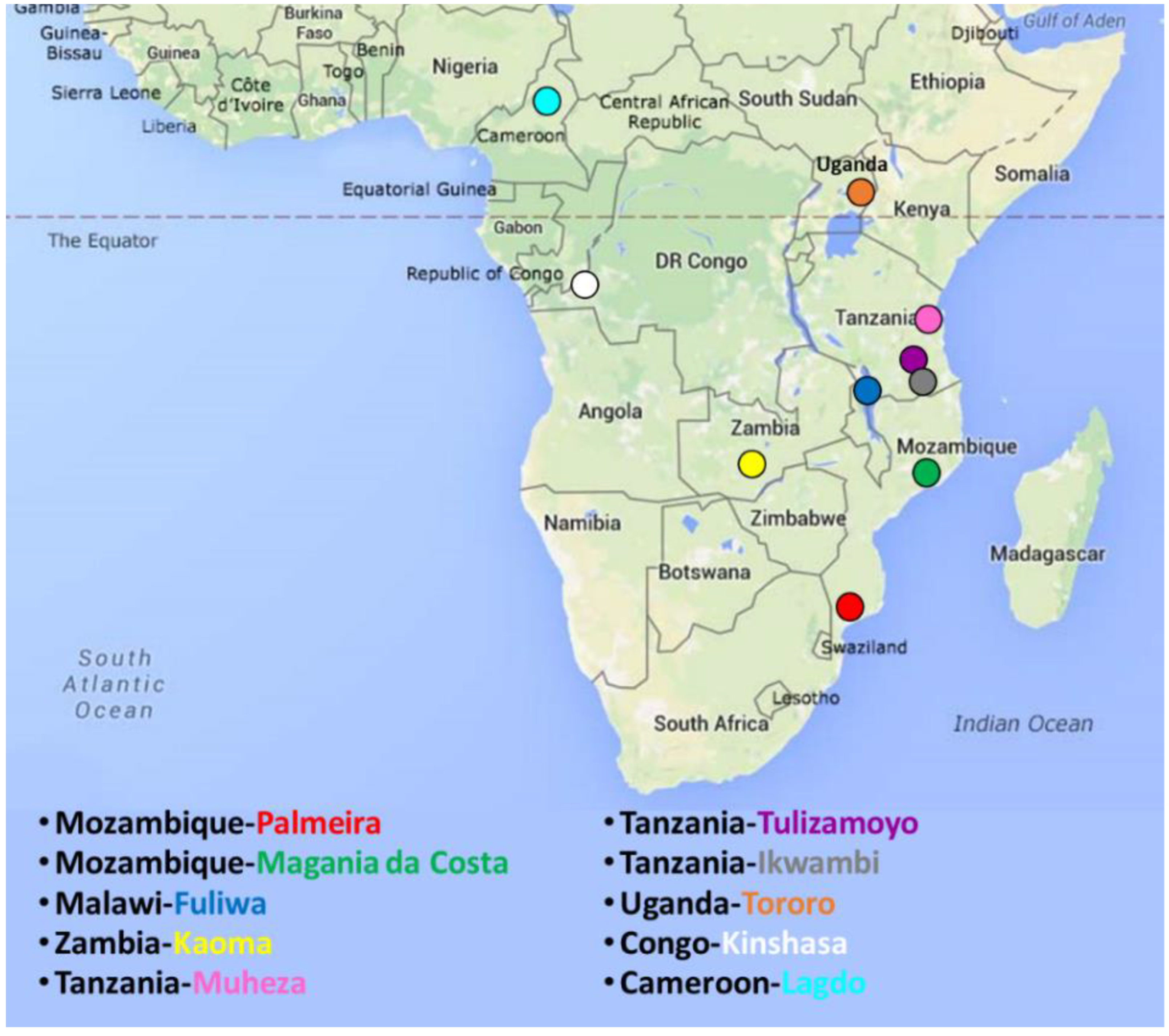
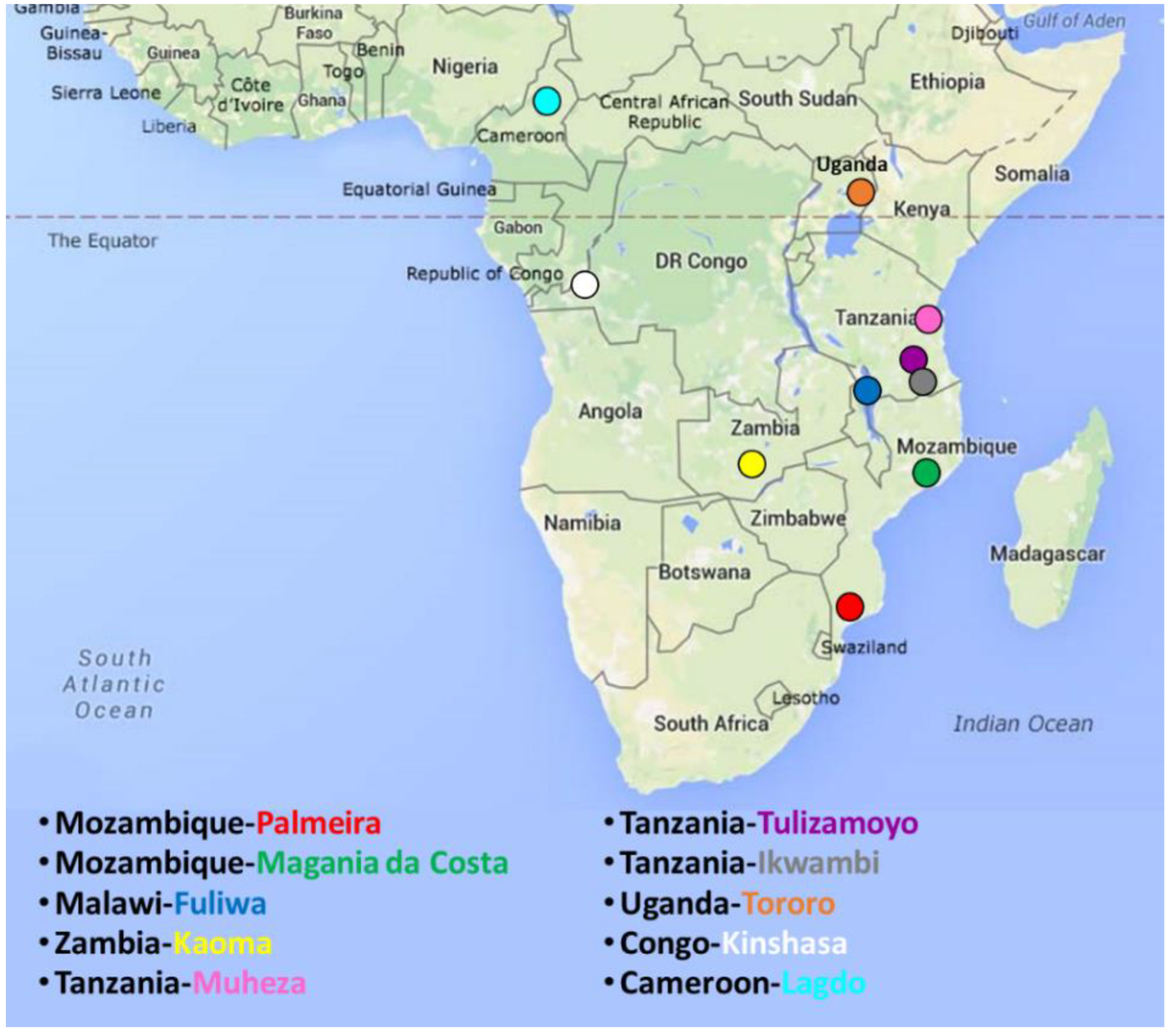
2.1. Sampling
2.2. PCR Amplification and Sequencing of CYP6P9a Gene and rp1 Fragments
2.3. Data Analysis
3. Results
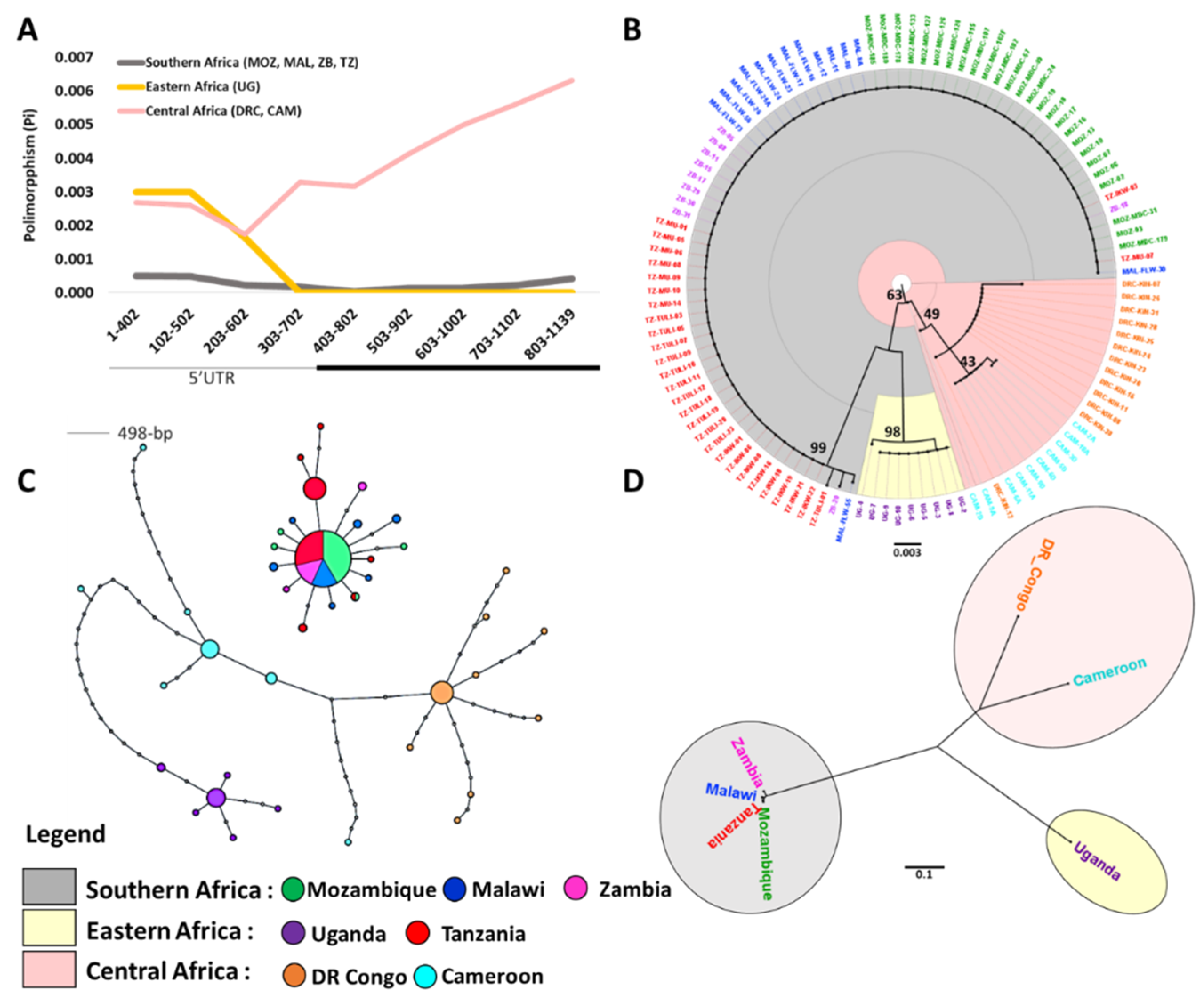
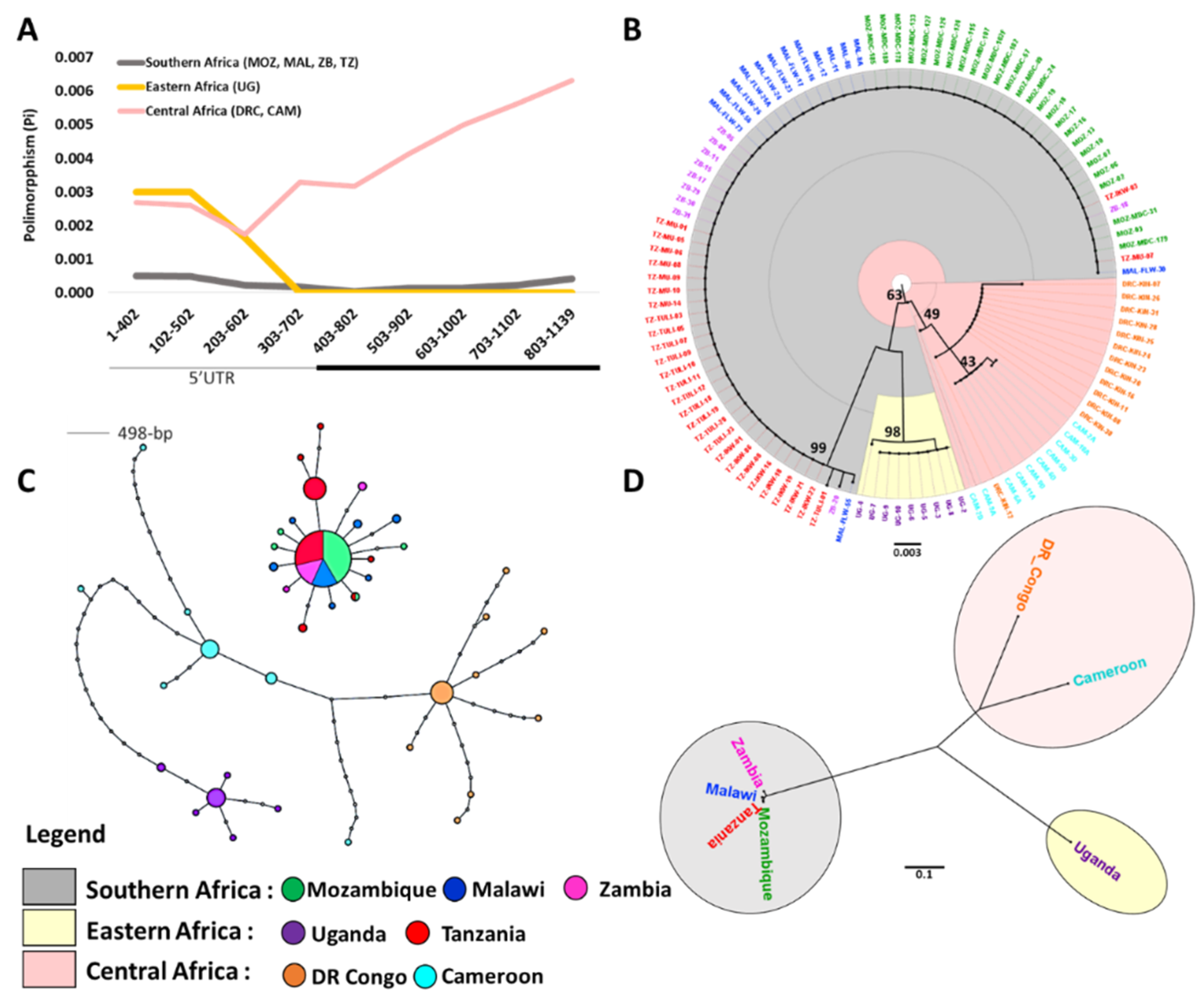
3.1. Sequence Diversity at CYP6P9a
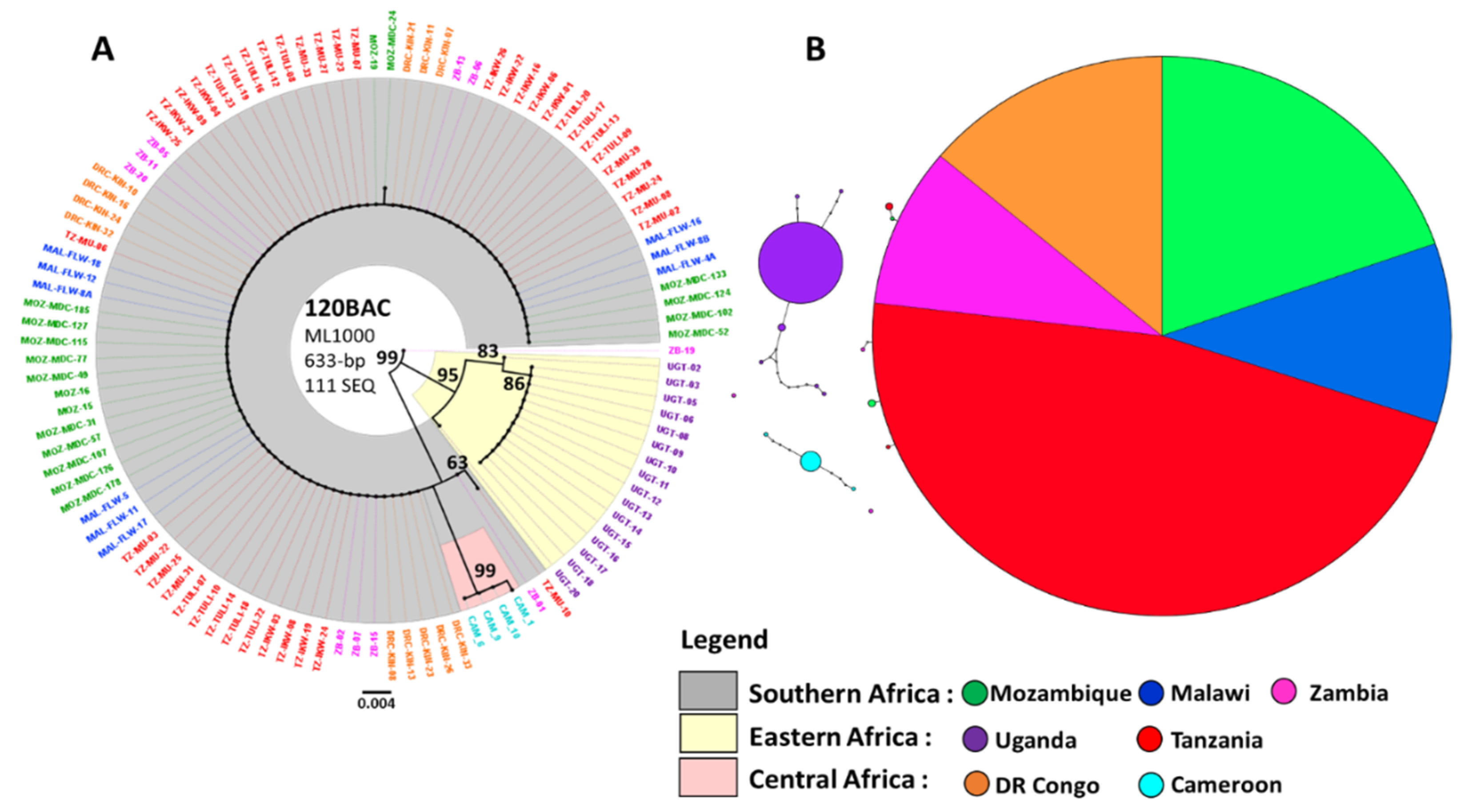
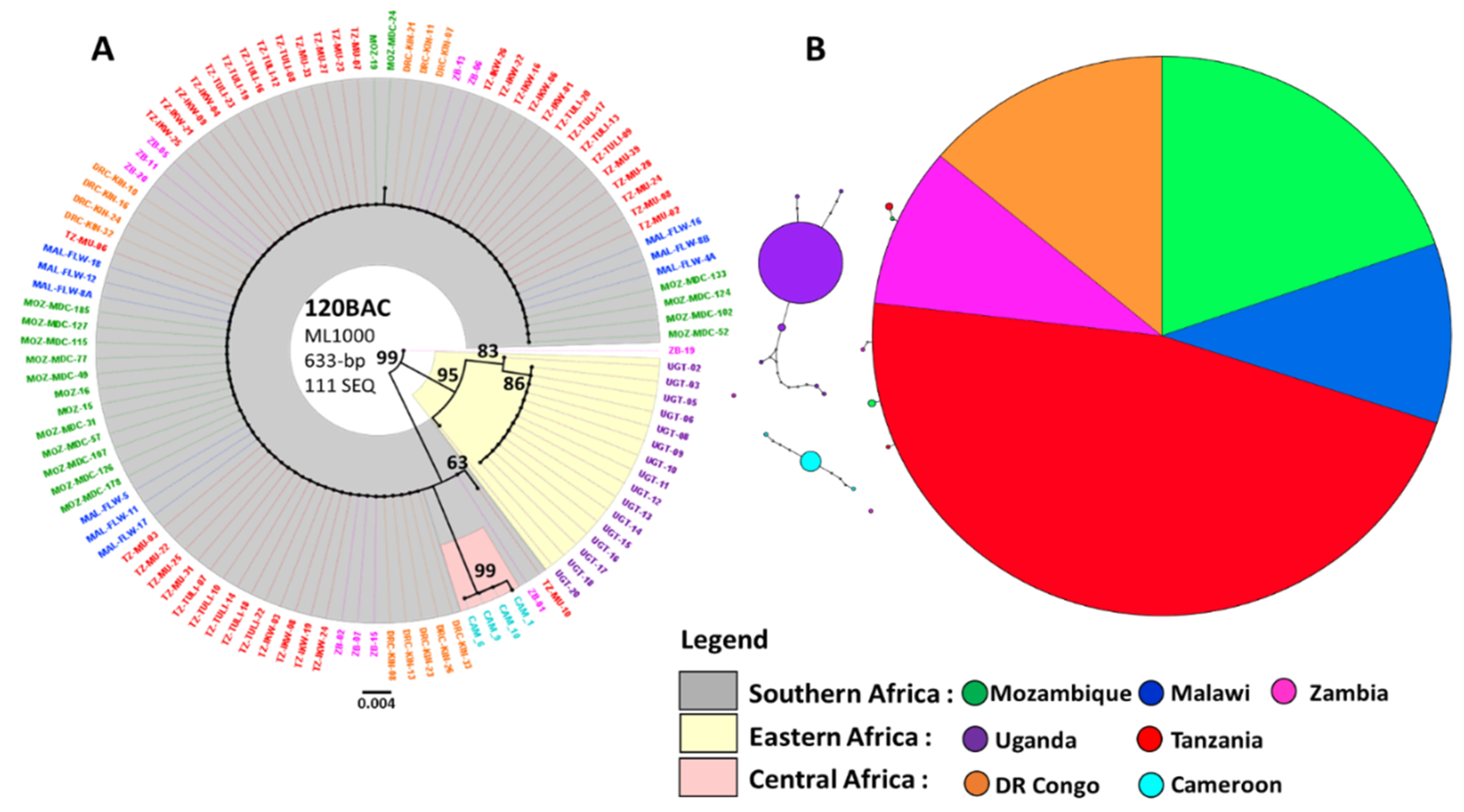
3.2. Maximum Likelihood Phylogenetic Tree and Haplotype Network for CYP6P9a
3.3. Genetic Differentiation of the CYP6P9a Gene
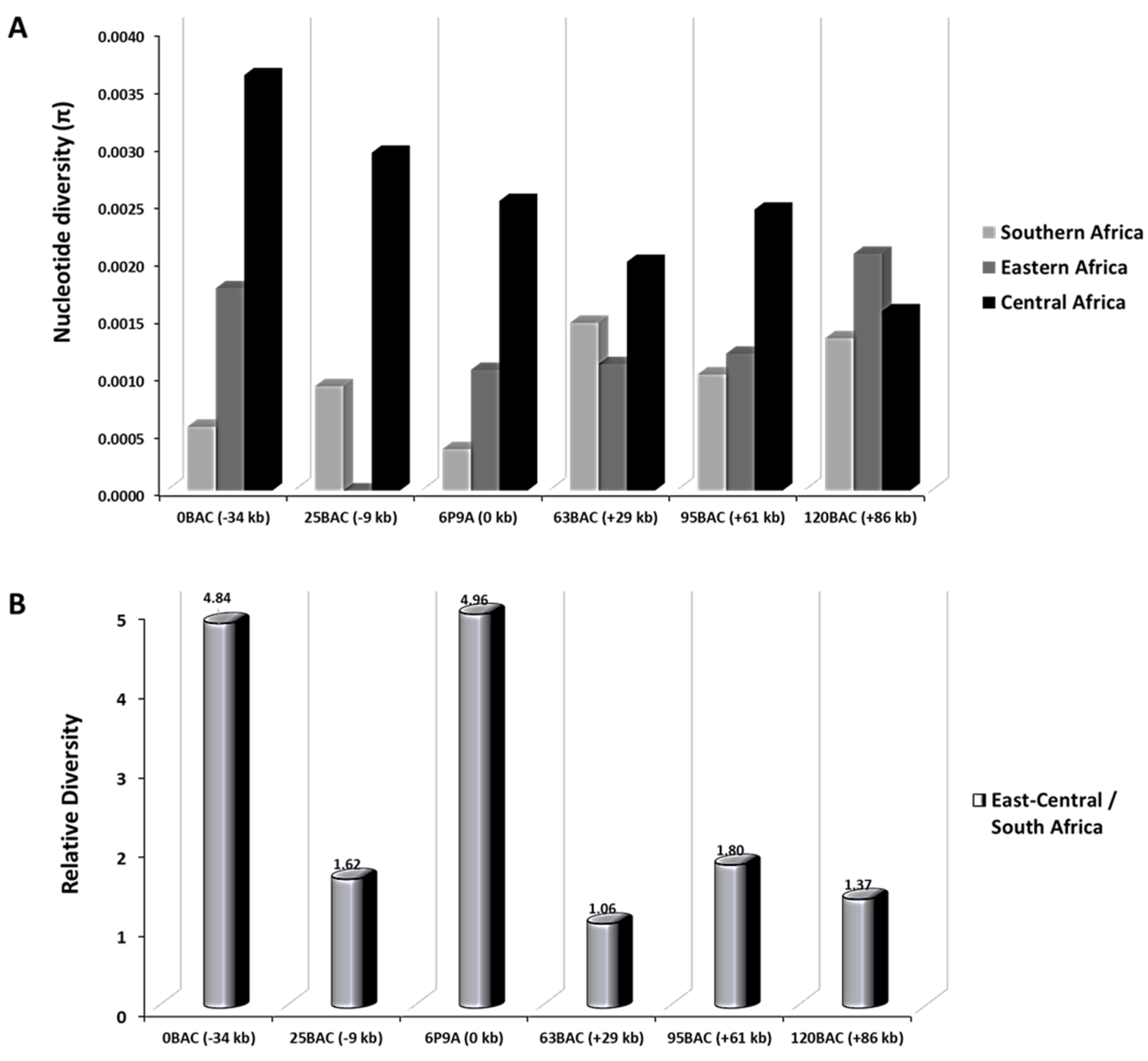
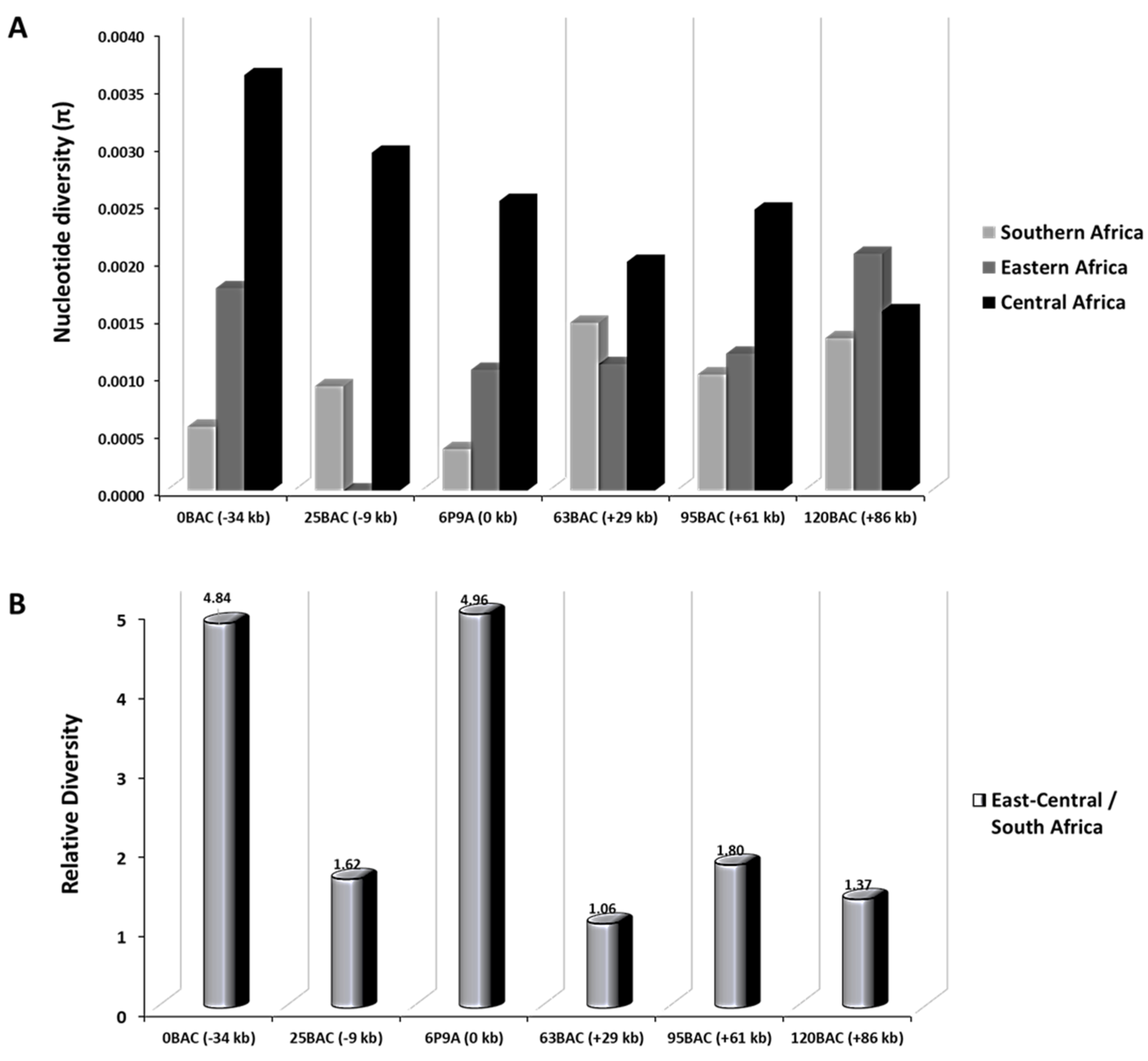
3.4. Selective Sweep Surrounding CYP6P9a
4. Discussion
4.1. Genetic Diversity and Selection on CYP6P9a
4.2. Spread of Resistance in An. Funestus Populations Across Africa and Barriers to Gene Flow
4.3. Pyrethroid Resistance in An. Funestus is Under Directional Selection
4.3.1. CYP6P9a Resistance Allele Has Moved Beyond Southern Africa into Eastern Africa
4.3.2. Reduced Polymorphism of the 120 kb rp1 Genomic Region Show a Selective Sweep in the Vicinity of CYP6P9a
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Wiebe, A.; Longbottom, J.; Gleave, K.; Shearer, F.M.; Sinka, M.E.; Massey, N.C.; Cameron, E.; Bhatt, S.; Gething, P.W.; Hemingway, J.; et al. Geographical distributions of African malaria vector sibling species and evidence for insecticide resistance. Malar J. 2017, 16, 85. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Collins, F.H.; Besansky, N.J. Vector biology and the control of malaria in Africa. Science 1994, 264, 1874–1875. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Manga, L.; Toto, J.C.; Le Goff, G.; Brunhes, J. The bionomics of Anopheles funestus and its role in malaria transmission in a forested area of southern Cameroon. Trans. R. Soc. Trop. Med. Hyg. 1997, 91, 387–388. [Google Scholar] [CrossRef]
- WHO. World Malaria Report 2019; WHO: Geneva, Switzerland, 2019; p. 232. [Google Scholar]
- Bhatt, S.; Weiss, D.J.; Cameron, E.; Bisanzio, D.; Mappin, B.; Dalrymple, U.; Battle, K.; Moyes, C.L.; Henry, A.; Eckhoff, P.A.; et al. The effect of malaria control on Plasmodium falciparum in Africa between 2000 and 2015. Nature 2015, 526, 207–211. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gething, P.W.; Casey, D.C.; Weiss, D.J.; Bisanzio, D.; Bhatt, S.; Cameron, E.; Battle, K.E.; Dalrymple, U.; Rozier, J.; Rao, P.C.; et al. Mapping Plasmodium falciparum Mortality in Africa between 1990 and 2015. N. Engl. J. Med. 2016, 375, 2435–2445. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wilson, A.L.; Courtenay, O.; Kelly-Hope, L.A.; Scott, T.W.; Takken, W.; Torr, S.J.; Lindsay, S.W. The importance of vector control for the control and elimination of vector-borne diseases. PLoS Negl. Trop. Dis. 2020, 14, e0007831. [Google Scholar] [CrossRef] [Green Version]
- Killeen, G.F.; Ranson, H. Insecticide-resistant malaria vectors must be tackled. Lancet 2018, 391, 1551–1552. [Google Scholar] [CrossRef] [Green Version]
- WHO. Bulletin of the World Health Organization: Preserving Pyrethroids; WHO: Geneva, Switzerland, 2014; Volume 92, p. 159. [Google Scholar]
- Casimiro, S.; Coleman, M.; Mohloai, P.; Hemingway, J.; Sharp, B. Insecticide resistance in Anopheles funestus (Diptera: Culicidae) from Mozambique. J. Med. Entomol. 2006, 43, 267–275. [Google Scholar] [CrossRef] [Green Version]
- Hunt, R.; Edwardes, M.; Coetzee, M. Pyrethroid resistance in southern African Anopheles funestus extends to Likoma Island in Lake Malawi. Parasit Vectors 2010, 3, 122. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Matiya, D.J.; Philbert, A.B.; Kidima, W.; Matowo, J.J. Dynamics and monitoring of insecticide resistance in malaria vectors across mainland Tanzania from 1997 to 2017: A systematic review. Malar J. 2019, 18, 102. [Google Scholar] [CrossRef]
- Barnes, K.G.; Irving, H.; Chiumia, M.; Mzilahowa, T.; Coleman, M.; Hemingway, J.; Wondji, C.S. Restriction to gene flow is associated with changes in the molecular basis of pyrethroid resistance in the malaria vector Anopheles funestus. Proc. Natl. Acad. Sci. USA 2017, 114, 286–291. [Google Scholar] [CrossRef] [Green Version]
- Cuamba, N.; Morgan, J.C.; Irving, H.; Steven, A.; Wondji, C.S. High level of pyrethroid resistance in an Anopheles funestus population of the Chokwe District in Mozambique. PLoS ONE 2010, 5, e11010. [Google Scholar] [CrossRef]
- Riveron, J.M.; Chiumia, M.; Menze, B.D.; Barnes, K.G.; Irving, H.; Ibrahim, S.S.; Weedall, G.D.; Mzilahowa, T.; Wondji, C.S. Rise of multiple insecticide resistance in Anopheles funestus in Malawi: A major concern for malaria vector control. Malar J. 2015, 14, 344. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Riveron, J.M.; Irving, H.; Ndula, M.; Barnes, K.G.; Ibrahim, S.S.; Paine, M.J.; Wondji, C.S. Directionally selected cytochrome P450 alleles are driving the spread of pyrethroid resistance in the major malaria vector Anopheles funestus. Proc. Natl. Acad. Sci. USA 2013, 110, 252–257. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wondji, C.S.; Irving, H.; Morgan, J.; Lobo, N.F.; Collins, F.H.; Hunt, R.H.; Coetzee, M.; Hemingway, J.; Ranson, H. Two duplicated P450 genes are associated with pyrethroid resistance in Anopheles funestus, a major malaria vector. Genome Res. 2009, 19, 452–459. [Google Scholar] [CrossRef] [Green Version]
- Barnes, K.G.; Weedall, G.D.; Ndula, M.; Irving, H.; Mzihalowa, T.; Hemingway, J.; Wondji, C.S. Genomic Footprints of Selective Sweeps from Metabolic Resistance to Pyrethroids in African Malaria Vectors Are Driven by Scale up of Insecticide-Based Vector Control. PLoS Genet. 2017, 13, e1006539. [Google Scholar] [CrossRef]
- Weedall, G.D.; Mugenzi, L.M.J.; Menze, B.D.; Tchouakui, M.; Ibrahim, S.S.; Amvongo-Adjia, N.; Irving, H.; Wondji, M.J.; Tchoupo, M.; Djouaka, R.; et al. A cytochrome P450 allele confers pyrethroid resistance on a major African malaria vector, reducing insecticide-treated bednet efficacy. Sci. Transl. Med. 2019, 11, eaat7386. [Google Scholar] [CrossRef] [Green Version]
- Riveron, J.M.; Huijben, S.; Tchapga, W.; Tchouakui, M.; Wondji, M.J.; Tchoupo, M.; Irving, H.; Cuamba, N.; Maquina, M.; Paaijmans, K.; et al. Escalation of Pyrethroid Resistance in the Malaria Vector Anopheles funestus Induces a Loss of Efficacy of Piperonyl Butoxide-Based Insecticide-Treated Nets in Mozambique. J. Infect. Dis. 2019, 220, 467–475. [Google Scholar] [CrossRef]
- Weedall, G.D.; Riveron, J.M.; Hearn, J.; Irving, H.; Kamdem, C.; Fouet, C.; White, B.J.; Wondji, C.S. An Africa-wide genomic evolution of insecticide resistance in the malaria vector Anopheles funestus involves selective sweeps, copy number variations, gene conversion and transposons. PLoS Genet. 2020, 16, e1008822. [Google Scholar] [CrossRef] [PubMed]
- Michel, A.P.; Ingrasci, M.J.; Schemerhorn, B.J.; Kern, M.; Le Goff, G.; Coetzee, M.; Elissa, N.; Fontenille, D.; Vulule, J.; Lehmann, T.; et al. Rangewide population genetic structure of the African malaria vector Anopheles funestus. Mol. Ecol. 2005, 14, 4235–4248. [Google Scholar] [CrossRef]
- Kaindoa, E.W.; Ngowo, H.S.; Limwagu, A.J.; Tchouakui, M.; Hape, E.; Abbasi, S.; Kihonda, J.; Mmbando, A.S.; Njalambaha, R.M.; Mkandawile, G.; et al. Swarms of the malaria vector Anopheles funestus in Tanzania. Malar J. 2019, 18, 29. [Google Scholar] [CrossRef]
- Mulamba, C.; Riveron, J.M.; Ibrahim, S.S.; Irving, H.; Barnes, K.G.; Mukwaya, L.G.; Birungi, J.; Wondji, C.S. Widespread pyrethroid and DDT resistance in the major malaria vector Anopheles funestus in East Africa is driven by metabolic resistance mechanisms. PLoS ONE 2014, 9, e110058. [Google Scholar] [CrossRef] [Green Version]
- Riveron, J.M.; Watsenga, F.; Irving, H.; Irish, S.R.; Wondji, C.S. High Plasmodium Infection Rate and Reduced Bed Net Efficacy in Multiple Insecticide-Resistant Malaria Vectors in Kinshasa, Democratic Republic of Congo. J. Infect. Dis. 2018, 217, 320–328. [Google Scholar] [CrossRef] [Green Version]
- Livak, K.J. Organization and mapping of a sequence on the Drosophila melanogaster X and Y chromosomes that is transcribed during spermatogenesis. Genetics 1984, 107, 611–634. [Google Scholar]
- Koekemoer, L.L.; Kamau, L.; Hunt, R.H.; Coetzee, M. A cocktail polymerase chain reaction assay to identify members of the Anopheles funestus (Diptera: Culicidae) group. Am. J. Trop. Med. Hyg. 2002, 66, 804–811. [Google Scholar] [CrossRef] [Green Version]
- Kumar, S.; Stecher, G.; Li, M.; Knyaz, C.; Tamura, K. MEGA X: Molecular Evolutionary Genetics Analysis across Computing Platforms. Mol. Biol. Evol. 2018, 35, 1547–1549. [Google Scholar] [CrossRef]
- Rozas, J.; Ferrer-Mata, A.; Sanchez-DelBarrio, J.C.; Guirao-Rico, S.; Librado, P.; Ramos-Onsins, S.E.; Sanchez-Gracia, A. DnaSP 6: DNA Sequence Polymorphism Analysis of Large Data Sets. Mol. Biol. Evol. 2017, 34, 3299–3302. [Google Scholar] [CrossRef]
- Clement, M.; Posada, D.; Crandall, K.A. TCS: A computer program to estimate gene genealogies. Mol. Ecol. 2000, 9, 1657–1659. [Google Scholar] [CrossRef] [Green Version]
- Murias dos Santos, A.; Cabezas, M.P.; Tavares, A.I.; Xavier, R.; Branco, M. tcsBU: A tool to extend TCS network layout and visualization. Bioinformatics 2016, 32, 627–628. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Crow, J.; Kimura, M. An Introduction to Population Genetics Theory; Harper and Row: New York, NY, USA, 1970. [Google Scholar]
- Jost, L.; Archer, F.; Flanagan, S.; Gaggiotti, O.; Hoban, S.; Latch, E. Differentiation measures for conservation genetics. Evol. Appl. 2018, 11, 1139–1148. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weir, B.S.; Cockerham, C.C. Estimating F-Statistics for the Analysis of Population Structure. Evolution 1984, 38, 1358–1370. [Google Scholar] [PubMed]
- Nei, M. Analysis of gene diversity in subdivided populations. Proc. Natl. Acad. Sci. USA 1973, 70, 3321–3323. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hudson, R.R.; Boos, D.D.; Kaplan, N.L. A statistical test for detecting geographic subdivision. Mol. Biol. Evol. 1992, 9, 138–151. [Google Scholar]
- Grivet, D.; Petit, R.J. Phylogeography of the common ivy (Hedera sp.) in Europe: Genetic differentiation through space and time. Mol. Ecol. 2002, 11, 1351–1362. [Google Scholar] [CrossRef]
- Mouhamadou, C.S.; de Souza, S.S.; Fodjo, B.K.; Zoh, M.G.; Bli, N.K.; Koudou, B.G. Evidence of insecticide resistance selection in wild Anopheles coluzzii mosquitoes due to agricultural pesticide use. Infect. Dis. Poverty 2019, 8, 64. [Google Scholar] [CrossRef]
- Mugenzi, L.M.J.; Menze, B.D.; Tchouakui, M.; Wondji, M.J.; Irving, H.; Tchoupo, M.; Hearn, J.; Weedall, G.D.; Riveron, J.M.; Wondji, C.S. Cis-regulatory CYP6P9b P450 variants associated with loss of insecticide-treated bed net efficacy against Anopheles funestus. Nat. Commun. 2019, 10, 4652. [Google Scholar] [CrossRef] [Green Version]
- Riveron, J.M.; Ibrahim, S.S.; Chanda, E.; Mzilahowa, T.; Cuamba, N.; Irving, H.; Barnes, K.G.; Ndula, M.; Wondji, C.S. The highly polymorphic CYP6M7 cytochrome P450 gene partners with the directionally selected CYP6P9a and CYP6P9b genes to expand the pyrethroid resistance front in the malaria vector Anopheles funestus in Africa. BMC Genom. 2014, 15, 817. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Riveron, J.M.; Ibrahim, S.S.; Mulamba, C.; Djouaka, R.; Irving, H.; Wondji, M.J.; Ishak, I.H.; Wondji, C.S. Genome-Wide Transcription and Functional Analyses Reveal Heterogeneous Molecular Mechanisms Driving Pyrethroids Resistance in the Major Malaria Vector Anopheles funestus Across Africa. G3 (Bethesda) 2017, 7, 1819–1832. [Google Scholar] [CrossRef] [Green Version]
- Lehmann, T.; Hawley, W.A.; Grebert, H.; Danga, M.; Atieli, F.; Collins, F.H. The Rift Valley complex as a barrier to gene flow for Anopheles gambiae in Kenya. J. Hered. 1999, 90, 613–621. [Google Scholar] [CrossRef] [Green Version]
- Lehmann, T.; Licht, M.; Elissa, N.; Maega, B.T.; Chimumbwa, J.M.; Watsenga, F.T.; Wondji, C.S.; Simard, F.; Hawley, W.A. Population Structure of Anopheles gambiae in Africa. J. Hered. 2003, 94, 133–147. [Google Scholar] [CrossRef]
- Kamau, L.; Munyekenye, G.O.; Koekemoer, L.L.; Hunt, R.H.; Coetzee, M. A survey of the Anopheles funestus (Diptera: Culicidae) group of mosquitoes from 10 sites in Kenya with special emphasis on population genetic structure based on chromosomal inversion karyotypes. J. Med. Entomol. 2003, 40, 664–671. [Google Scholar] [CrossRef] [Green Version]
- Amenya, D.A.; Naguran, R.; Lo, T.C.; Ranson, H.; Spillings, B.L.; Wood, O.R.; Brooke, B.D.; Coetzee, M.; Koekemoer, L.L. Over expression of a cytochrome P450 (CYP6P9) in a major African malaria vector, Anopheles Funestus, resistant to pyrethroids. Insect Mol. Biol. 2008, 17, 19–25. [Google Scholar] [CrossRef]
- Wondji, C.S.; Dabire, R.K.; Tukur, Z.; Irving, H.; Djouaka, R.; Morgan, J.C. Identification and distribution of a GABA receptor mutation conferring dieldrin resistance in the malaria vector Anopheles funestus in Africa. Insect Biochem. Mol. Biol. 2011, 41, 484–491. [Google Scholar] [CrossRef] [Green Version]
- Kaddumukasa, M.A.; Wright, J.; Muleba, M.; Stevenson, J.C.; Norris, D.E.; Coetzee, M. Genetic differentiation and population structure of Anopheles funestus from Uganda and the southern African countries of Malawi, Mozambique, Zambia and Zimbabwe. Parasites Vectors 2020, 13, 87. [Google Scholar] [CrossRef] [Green Version]
- Muller, P.; Warr, E.; Stevenson, B.J.; Pignatelli, P.M.; Morgan, J.C.; Steven, A.; Yawson, A.E.; Mitchell, S.N.; Ranson, H.; Hemingway, J.; et al. Field-caught permethrin-resistant Anopheles gambiae overexpress CYP6P3, a P450 that metabolises pyrethroids. PLoS Genet. 2008, 4, e1000286. [Google Scholar] [CrossRef] [Green Version]
- Ishak, I.H.; Riveron, J.M.; Ibrahim, S.S.; Stott, R.; Longbottom, J.; Irving, H.; Wondji, C.S. The Cytochrome P450 gene CYP6P12 confers pyrethroid resistance in kdr-free Malaysian populations of the dengue vector Aedes albopictus. Sci. Rep. 2016, 6, 24707. [Google Scholar] [CrossRef] [Green Version]
- Ibrahim, S.S.; Riveron, J.M.; Bibby, J.; Irving, H.; Yunta, C.; Paine, M.J.; Wondji, C.S. Allelic Variation of Cytochrome P450s Drives Resistance to Bednet Insecticides in a Major Malaria Vector. PLoS Genet. 2015, 11, e1005618. [Google Scholar] [CrossRef] [Green Version]
- Riveron, J.M.; Yunta, C.; Ibrahim, S.S.; Djouaka, R.; Irving, H.; Menze, B.D.; Ismail, H.M.; Hemingway, J.; Ranson, H.; Albert, A.; et al. A single mutation in the GSTe2 gene allows tracking of metabolically based insecticide resistance in a major malaria vector. Genome Biol. 2014, 15, R27. [Google Scholar] [CrossRef] [Green Version]
- Thomsen, E.K.; Strode, C.; Hemmings, K.; Hughes, A.J.; Chanda, E.; Musapa, M.; Kamuliwo, M.; Phiri, F.N.; Muzia, L.; Chanda, J.; et al. Underpinning sustainable vector control through informed insecticide resistance management. PLoS ONE 2014, 9, e99822. [Google Scholar] [CrossRef] [Green Version]
- Wondji, C.S.; Coleman, M.; Kleinschmidt, I.; Mzilahowa, T.; Irving, H.; Ndula, M.; Rehman, A.; Morgan, J.; Barnes, K.G.; Hemingway, J. Impact of pyrethroid resistance on operational malaria control in Malawi. Proc. Natl. Acad. Sci. USA 2012, 109, 19063–19070. [Google Scholar] [CrossRef] [Green Version]
- Ranson, H.; N’Guessan, R.; Lines, J.; Moiroux, N.; Nkuni, Z.; Corbel, V. Pyrethroid resistance in African anopheline mosquitoes: What are the implications for malaria control? Trends Parasitol. 2011, 27, 91–98. [Google Scholar] [CrossRef]
- Ishak, I.H.; Jaal, Z.; Ranson, H.; Wondji, C.S. Contrasting patterns of insecticide resistance and knockdown resistance (kdr) in the dengue vectors Aedes aegypti and Aedes albopictus from Malaysia. Parasit. Vectors 2015, 8, 181. [Google Scholar] [CrossRef] [Green Version]
- Biswas, S.; Akey, J.M. Genomic insights into positive selection. Trends Genet. 2006, 22, 437–446. [Google Scholar] [CrossRef]
- Yik, J.H.; Chen, R.; Nishimura, R.; Jennings, J.L.; Link, A.J.; Zhou, Q. Inhibition of P-TEFb (CDK9/Cyclin T) kinase and RNA polymerase II transcription by the coordinated actions of HEXIM1 and 7SK snRNA. Mol. Cell 2003, 12, 971–982. [Google Scholar] [CrossRef]
- Yik, J.H.; Chen, R.; Pezda, A.C.; Zhou, Q. Compensatory contributions of HEXIM1 and HEXIM2 in maintaining the balance of active and inactive positive transcription elongation factor b complexes for control of transcription. J. Biol. Chem. 2005, 280, 16368–16376. [Google Scholar] [CrossRef] [Green Version]
- Ghurye, J.; Koren, S.; Small, S.T.; Redmond, S.; Howell, P.; Phillippy, A.M.; Besansky, N.J. A chromosome-scale assembly of the major African malaria vector Anopheles funestus. Gigascience 2019, 1, 8. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| 2n | S | h | hd | π | D | D * | FS | |
|---|---|---|---|---|---|---|---|---|
| Complete sequence fragment (1139 bp) | ||||||||
| Mozambique_Palmeira | 20 | 1 | 2 | 0.10 | 0.00009 | −1.16 | −1.54 | −0.88 |
| Mozambique_Magania | 32 | 3 | 3 | 0.12 | 0.00016 | −1.73 | −2.73 * | −1.71 |
| Mozambique | 52 | 4 | 4 | 0.11 | 0.00014 | −1.86 * | −3.43 ** | −3.63 * |
| Malawi | 28 | 7 | 8 | 0.59 | 0.00061 | −1.84 * | −1.46 | −5.83 ** |
| Zambia | 20 | 3 | 3 | 0.28 | 0.00042 | −1.16 | −0.12 | −0.29 |
| Tanzania_Muheza | 16 | 3 | 3 | 0.24 | 0.00033 | −1.70 | −2.20 | −0.90 |
| Tanzania_Tulizamoyo | 24 | 3 | 3 | 0.24 | 0.00035 | −1.26 | −0.19 | −0.50 |
| Tanzania_Ikwambi | 18 | 1 | 2 | 0.11 | 0.00010 | −1.16 | −1.50 | −0.79 |
| Tanzania | 58 | 7 | 6 | 0.20 | 0.00027 | −2.05 * | −2.77 * | −4.89 * |
| Uganda | 18 | 9 | 6 | 0.56 | 0.00105 | −1.92 * | −2.11 | −1.91 |
| DR Congo | 26 | 24 | 9 | 0.53 | 0.00200 | −2.33 ** | −2.90 ** | −2.04 |
| Cameroon | 20 | 23 | 7 | 0.68 | 0.00304 | −1.79 | −1.93 | 0.44 |
| Overall | 222 | 84 | 39 | 0.61 | 0.01095 | −0.42 | −3.96 ** | −0.53 |
| Coding Region (642 bp) | ||||||||
| Mozambique_Palmeira | 20 | 1 | 2 | 0.10 | 0.00016 | −1.16 | −1.54 | −0.88 |
| Mozambique_Magania | 32 | 3 | 3 | 0.12 | 0.00029 | −1.73 | −2.73 * | −1.71 |
| Mozambique | 52 | 4 | 4 | 0.11 | 0.00024 | −1.86 * | −3.43 * | −3.63 * |
| Malawi | 28 | 2 | 3 | 0.27 | 0.00043 | −0.97 | 0.82 | −1.09 |
| Zambia | 20 | 0 | 1 | 0.00 | 0.00000 | - | - | - |
| Tanzania_Muheza | 16 | 3 | 3 | 0.24 | 0.00058 | −1.70 | −2.20 | −0.90 |
| Tanzania_Tulizamoyo | 24 | 0 | 1 | 0.00 | 0.00000 | - | - | - |
| Tanzania_Ikwambi | 18 | 1 | 2 | 0.11 | 0.00017 | −1.16 | −1.50 | −0.79 |
| Tanzania | 58 | 4 | 4 | 0.10 | 0.00021 | −1.85 * | −3.51 ** | -3.84 * |
| Uganda | 18 | 0 | 1 | 0.00 | 0.00000 | - | - | - |
| DR Congo | 26 | 6 | 4 | 0.22 | 0.00083 | −1.96 * | −2.52 * | −1.19 |
| Cameroon | 20 | 16 | 6 | 0.62 | 0.00403 | −1.58 | −1.37 | 0.47 |
| Overall | 222 | 41 | 20 | 0.54 | 0.01328 | 0.54 | −2.42 * | 4.08 * |
| Non-Coding Region (497 bp) | ||||||||
| Mozambique_Palmeira | 20 | 0 | 1 | 0.00 | 0.00000 | - | - | - |
| Mozambique_Magania | 32 | 0 | 1 | 0.00 | 0.00000 | - | - | - |
| Mozambique | 52 | 0 | 1 | 0.00 | 0.00000 | - | - | - |
| Malawi | 28 | 5 | 6 | 0.39 | 0.00085 | −1.86 * | −2.30 | −4.66 ** |
| Zambia | 20 | 3 | 3 | 0.28 | 0.00096 | −1.16 | −0.12 | −0.29 |
| Tanzania_Muheza | 16 | 0 | 1 | 0.00 | 0.00000 | - | - | - |
| Tanzania_Tulizamoyo | 24 | 3 | 3 | 0.24 | 0.00081 | −1.26 | −0.19 | -0.50 |
| Tanzania_Ikwambi | 18 | 0 | 1 | 0.00 | 0.00000 | - | - | - |
| Tanzania | 58 | 3 | 3 | 0.10 | 0.00034 | −1.47 | −0.45 | −1.60 |
| Uganda | 18 | 9 | 6 | 0.56 | 0.00242 | −1.92 * | −2.11 | −2.11 |
| DR Congo | 26 | 18 | 8 | 0.47 | 0.00353 | −2.24 ** | −2.61 * | −2.07 |
| Cameroon | 20 | 7 | 6 | 0.45 | 0.00175 | −1.84 * | −2.46 * | −2.70 * |
| Overall | 222 | 43 | 28 | 0.54 | 0.00790 | −1.34 | −4.28 ** | −6.63 ** |
| Synonymous Sites | Synonymous Substitution | Synonymous Polymorphism Per Site (pS) | Non-Synonymous Sites | Non-Synonymous Substitution | Non-Synonymous Polymorphism Per Site (pN) | Polymorphism Rate (pN/pS) | |
|---|---|---|---|---|---|---|---|
| Mozambique | 153.47 | 1 | 0.007 | 488.53 | 3 | 0.006 | 0.94 |
| Malawi | 153.43 | 0 | 0.000 | 488.57 | 2 | 0.004 | - |
| Tanzania | 153.50 | 0 | 0.000 | 488.50 | 0 | 0.000 | - |
| Zambia | 153.53 | 2 | 0.013 | 488.47 | 2 | 0.004 | 0.31 |
| Uganda | 155.17 | 0 | 0.000 | 486.83 | 0 | 0.000 | - |
| DR Congo | 157.09 | 1 | 0.006 | 484.91 | 5 | 0.010 | 1.62 |
| Cameroon | 156.63 | 0 | 0.000 | 485.38 | 16 | 0.033 | - |
| Position (kb) | Length (bp) | Mozambique | Malawi | Zambia | Tanzania | Uganda | DR Congo | Cameroon | Pi Ratio * | |||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| hd | π*10E3 | D | hd | π*10E3 | D | hd | π*10E3 | D | hd | π*10E3 | D | hd | π*10E3 | D | hd | π*10E3 | D | hd | π*10E3 | D | ||||
| 0BAC | −34 | 626 | 0.04 | 0.06 | −1.10 | 0.31 | 0.72 | −1.43 | 0.22 | 0.36 | −1.22 | 0.36 | 1.08 | −2.18 ** | 0.36 | 1.76 | −1.61 | 0.15 | 0.74 | −2.09 * | 0.91 | 6.49 | −0.80 | 4.84 |
| 25BAC | −9 | 754 | 0.04 | 0.14 | −1.68 | 0.38 | 1.27 | −1.24 | 0.21 | 0.94 | −2.20 ** | 0.23 | 1.28 | −1.92 * | n.a | 0.00 | n.a | 0.13 | 0.35 | −1.89 * | 0.86 | 5.53 | −0.54 | 1.62 |
| CYP6P9a | 0 | 1139 | 0.11 | 0.14 | −1.86 * | 0.59 | 0.61 | −1.84 * | 0.28 | 0.42 | −1.16 | 0.20 | 0.27 | −2.05 * | 0.56 | 1.05 | −1.92 * | 0.53 | 2.00 | −2.33 ** | 0.68 | 3.04 | −1.79 | 1.41 |
| 63BAC | +29 | 614 | 0.43 | 1.11 | −0.68 | - | - | - | - | - | - | 0.64 | 1.81 | 1.05 | 0.60 | 1.10 | 0.18 | 0.46 | 1.99 | -1.68 * | - | - | - | 1.06 |
| 95BAC | +61 | 671 | 0.41 | 1.24 | −1.38 | 0.20 | 0.30 | −1.51 | 0.79 | 1.76 | −1.03 | 0.26 | 0.74 | −1.85 * | 0.33 | 1.19 | −1.93 * | 0.24 | 0.62 | −1.70 | 0.85 | 4.27 | −1.66 | 1.80 |
| 120BAC | +86 | 633 | 0.11 | 0.17 | −0.81 | n.a | 0.00 | n.a | 0.07 | 0.19 | −1.49 | 0.37 | 4.94 | −2.09 * | 0.39 | 2.06 | −1.83 * | n.a | 0.00 | n.a | 0.46 | 3.12 | −1.76 * | 1.37 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Djuicy, D.D.; Hearn, J.; Tchouakui, M.; Wondji, M.J.; Irving, H.; Okumu, F.O.; Wondji, C.S. CYP6P9-Driven Signatures of Selective Sweep of Metabolic Resistance to Pyrethroids in the Malaria Vector Anopheles funestus Reveal Contemporary Barriers to Gene Flow. Genes 2020, 11, 1314. https://doi.org/10.3390/genes11111314
Djuicy DD, Hearn J, Tchouakui M, Wondji MJ, Irving H, Okumu FO, Wondji CS. CYP6P9-Driven Signatures of Selective Sweep of Metabolic Resistance to Pyrethroids in the Malaria Vector Anopheles funestus Reveal Contemporary Barriers to Gene Flow. Genes. 2020; 11(11):1314. https://doi.org/10.3390/genes11111314
Chicago/Turabian StyleDjuicy, Delia Doreen, Jack Hearn, Magellan Tchouakui, Murielle J. Wondji, Helen Irving, Fredros O. Okumu, and Charles S. Wondji. 2020. "CYP6P9-Driven Signatures of Selective Sweep of Metabolic Resistance to Pyrethroids in the Malaria Vector Anopheles funestus Reveal Contemporary Barriers to Gene Flow" Genes 11, no. 11: 1314. https://doi.org/10.3390/genes11111314
APA StyleDjuicy, D. D., Hearn, J., Tchouakui, M., Wondji, M. J., Irving, H., Okumu, F. O., & Wondji, C. S. (2020). CYP6P9-Driven Signatures of Selective Sweep of Metabolic Resistance to Pyrethroids in the Malaria Vector Anopheles funestus Reveal Contemporary Barriers to Gene Flow. Genes, 11(11), 1314. https://doi.org/10.3390/genes11111314