Assessment of Imputation from Low-Pass Sequencing to Predict Merit of Beef Steers

 , ,
, ,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Data Source
2.2. SNP Array Genotypes
2.3. Genetic Prediction
2.4. Low-Pass Sequence and Imputation
- Aligned the reads to the ARS-UCD1.2 genome using bwa mem v0.7.17 [28]
- Sorted the reads using samtools v1.10 [29]
- Marked duplicate reads using GATK version 4 [30] (MarkDuplicates)
- Recalibrated base quality scores using GATK version 4 (BaseRecalibrator)
- Called GVCF in 10Mb windows using GATK version 4 (HaplotypeCaller -ERC GVCF)
- Called variants in the same 10Mb windows as above using the GATK version 4 (GenotypeGVCFs)
- Filtered single nucleotide polymorphism calls using GATK version 4 (VariantFiltration) with the filter string ‘QD < 2.0 || FS > 60.0 || MQ < 40.0 || MQRankSum <−12.5 || ReadPosRankSum < −8.0′
- Filtered indel calls using GATK version 4 (VariantFiltration) with the filter string ‘QD < 2.0 || FS > 200.0 || ReadPosRankSum < -20.0 || SOR > 10.0′
- Refined variant calls using BEAGLE v4 [31]
- Phased variant calls using BEAGLE v5 [32]
- Filtered indels and multi-allelic sites.
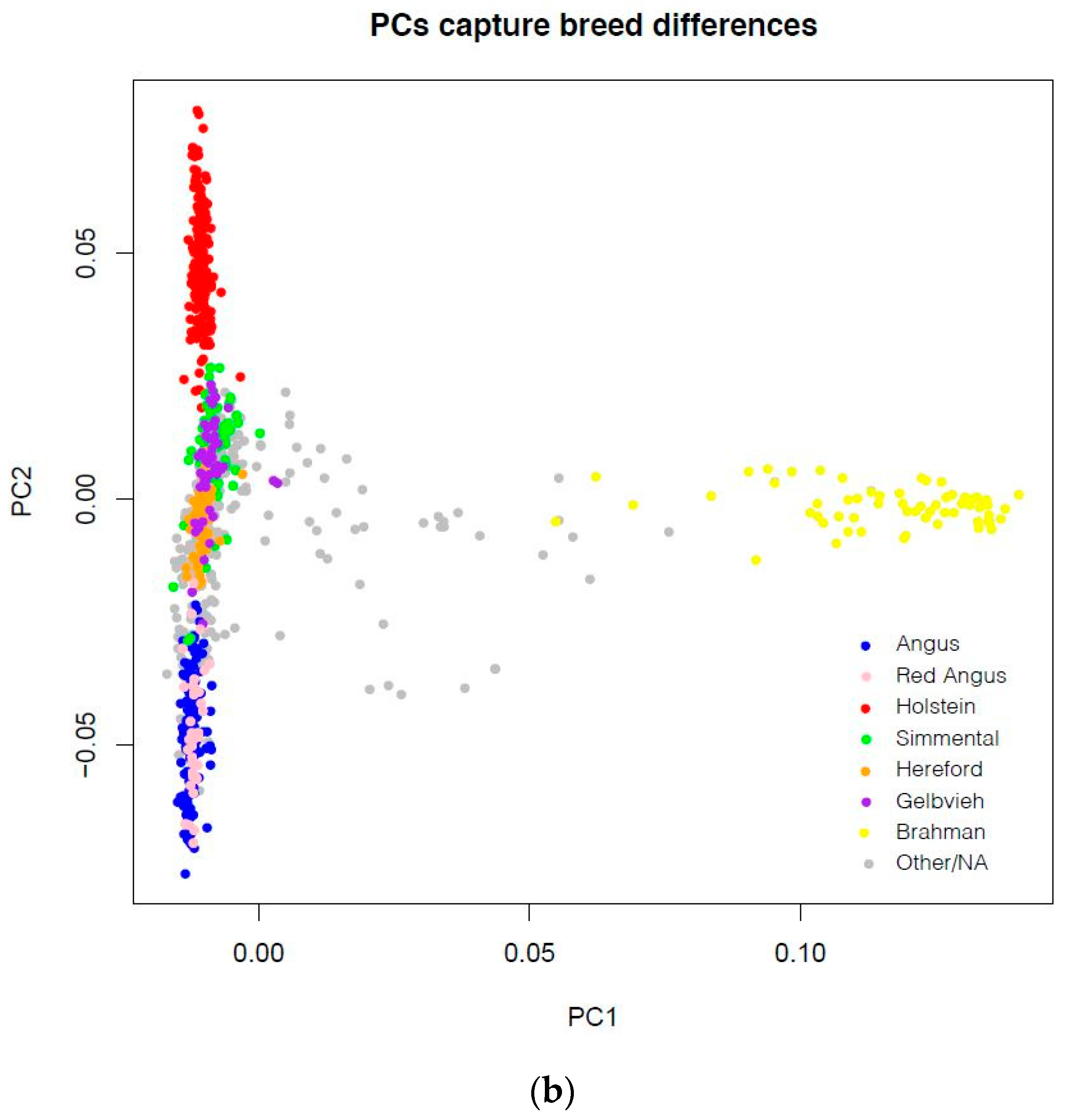
- Principal components were generated using plink 1.9 [33] restricted to 150,000 randomly-chosen bi-allelic SNPs with minor allele frequency (across the entire panel) above 5%.
3. Results
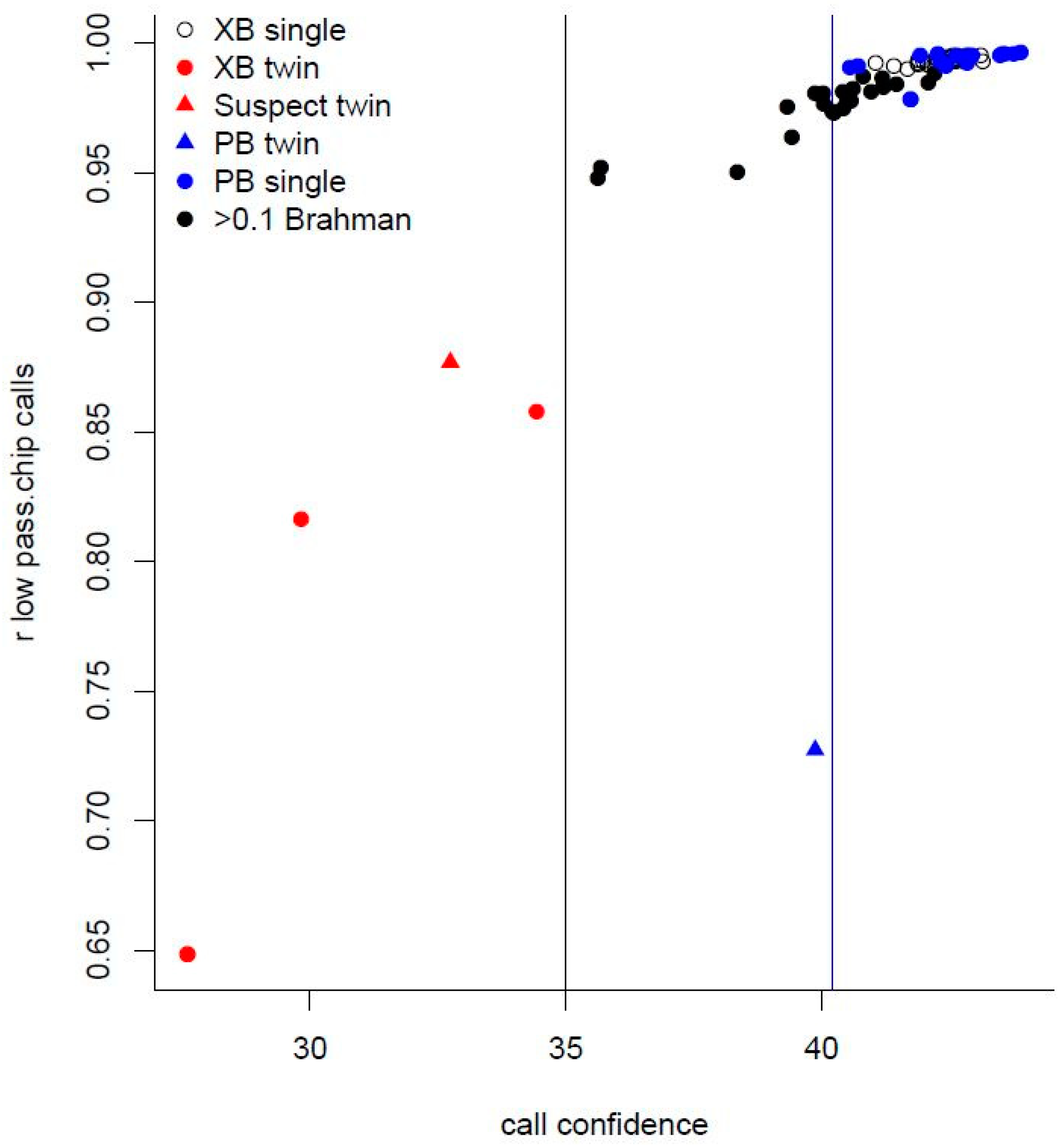
3.1. Cattle Haplotype Reference Panel
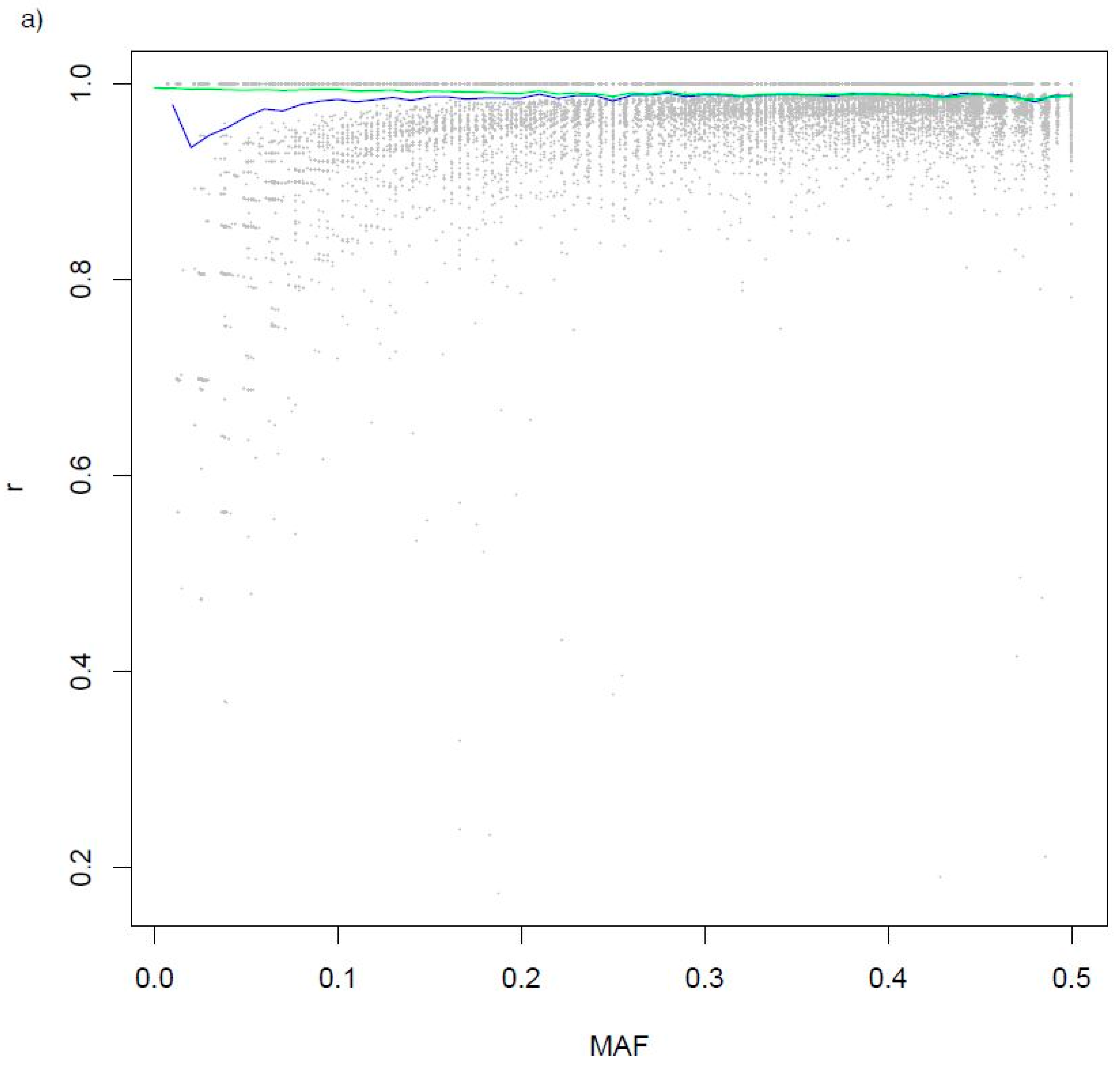
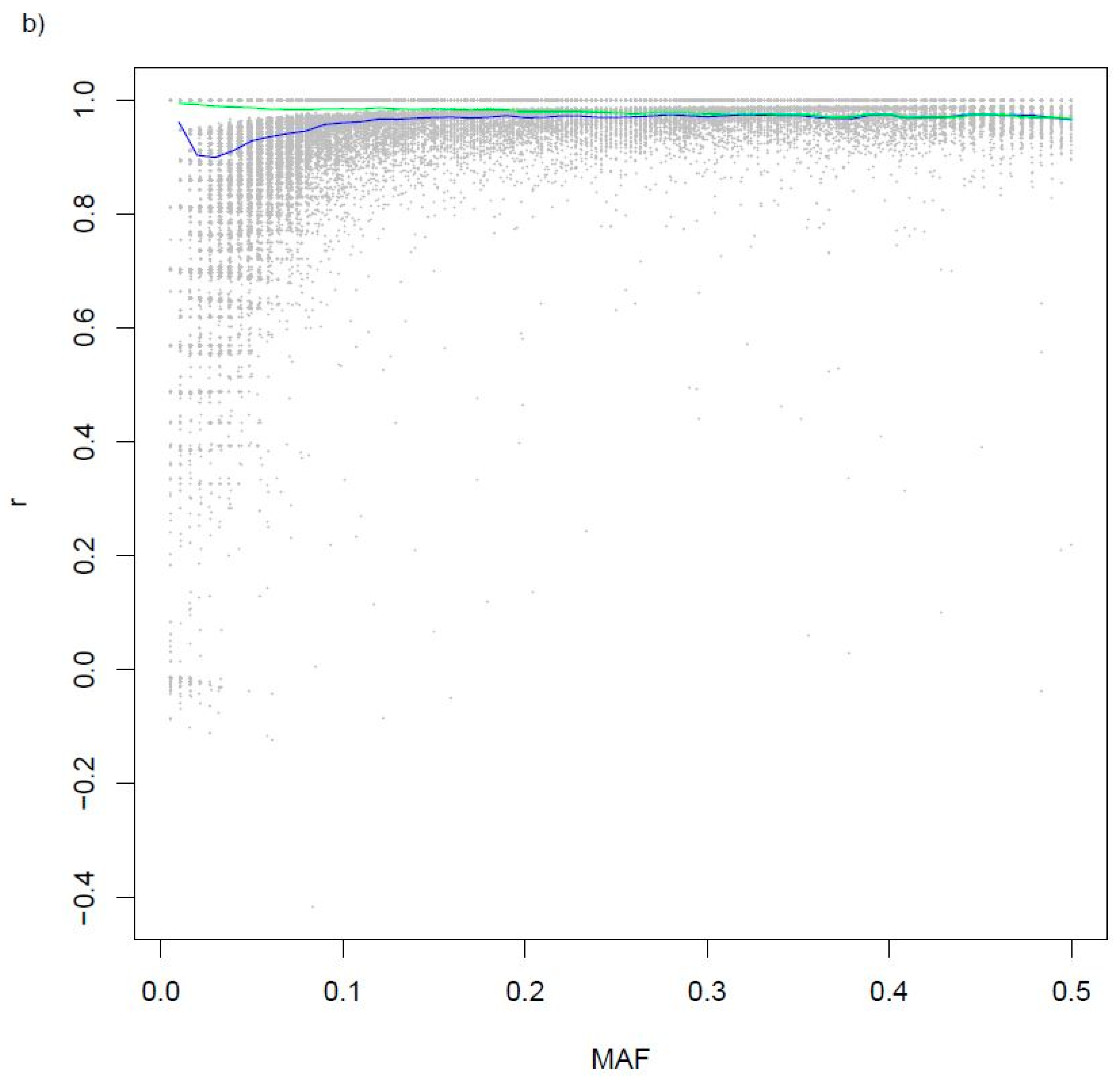
3.2. Variants Imputed from Low-Pass Sequence
3.3. Genomic Prediction
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Moghaddar, N.; Khansefid, M.; van der Werf, J.H.J.; Bolormaa, S.; Duijvesteijn, N.; Clark, S.A.; Swan, A.A.; Daetwyler, H.D.; MacLeod, I.M. Genomic prediction based on selected variants from imputed whole-genome sequence data in Australian sheep populations. Genet. Sel. Evol. 2019, 51, 72. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- MacLeod, I.M.; Bowman, P.J.; vander Jagt, C.J.; Haile-Mariam, M.; Kemper, K.E.; Chamberlain, A.J.; Schrooten, C.; Hayes, B.J.; Goddard, M.E. Exploiting biological priors and sequence variants enhances QTL discovery and genomic prediction of complex traits. BMC Genom. 2016, 17, 144. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xiang, R.; Berg, I.v.d.; MacLeod, I.M.; Hayes, B.J.; Prowse-Wilkins, C.P.; Wang, M.; Bolormaa, S.; Liu, Z.; Rochfort, S.J.; Reich, C.M.; et al. Quantifying the contribution of sequence variants with regulatory and evolutionary significance to 34 bovine complex traits. PNAS 2019, 116, 19398–19408. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rowan, T.N.; Hoff, J.L.; Crum, T.E.; Taylor, J.F.; Schnabel, R.D.; Decker, J.E. A multi-breed reference panel and additional rare variants maximize imputation accuracy in cattle. GSE 2019, 51, 77. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Snelling, W.M.; Bennett, G.L.; Keele, J.W.; Kuehn, L.A.; McDaneld, T.G.; Smith, T.P.; Thallman, R.M.; Kalbfleisch, T.S.; Pollak, E.J. A survey of polymorphisms detected from sequences of popular beef breeds. Anim. Sci. J. 2015, 93, 5128–5143. [Google Scholar] [CrossRef]
- Snelling, W.M.; Kuehn, L.A.; Keel, B.N.; Thallman, R.M.; Bennett, G.L. Linkage disequilibrium among commonly genotyped SNP variants detected from bull sequence. Anim. Genet 2017, 48, 516–522. [Google Scholar] [CrossRef]
- Wasik, K.; Berisa, T.; Pickrell, J.K.; Li, J.H.; Fraser, D.J.; King, K.; Cox, C. Comparing low-pass sequencing and genotyping for trait mapping in pharmacogenetics. bioRxiv 2019, 632141. [Google Scholar] [CrossRef] [Green Version]
- Davies, R.W.; Flint, J.; Myers, S.; Mott, R. Rapid genotype imputation from sequence without reference panels. Nat. Genet 2016, 48, 965–969. [Google Scholar] [CrossRef] [PubMed]
- DNA Sequencing Costs: Data. Available online: https://www.genome.gov/about-genomics/fact-sheets/DNA-Sequencing-Costs-Data (accessed on 26 October 2020).
- Baym, M.; Kryazhimskiy, S.; Lieberman, T.D.; Chung, H.; Desai, M.M.; Kishony, R. Inexpensive Multiplexed Library Preparation for Megabase-Sized Genomes. PLoS ONE 2015, 10, e0128036. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Matukumalli, L.K.; Lawley, C.T.; Schnabel, R.D.; Taylor, J.F.; Allan, M.F.; Heaton, M.P.; O’Connell, J.; Moore, S.S.; Smith, T.P.L.; Sonstegard, T.S.; et al. Development and Characterization of a High Density SNP Genotyping Assay for Cattle. PLoS ONE 2009, 4, e5350. [Google Scholar] [CrossRef] [Green Version]
- FASS. Guide for the Care and Use of Agrictultural Animals in Research and Teaching, 3rd ed.; FASS: Champaign, IL, USA, 2010. [Google Scholar]
- Keel, B.N.; Zarek, C.M.; Keele, J.W.; A Kuehn, L.; Snelling, W.M.; Oliver, W.T.; Freetly, H.C.; Lindholm-Perry, A.K. RNA-Seq Meta-analysis identifies genes in skeletal muscle associated with gain and intake across a multi-season study of crossbred beef steers. BMC Genom. 2018, 19, 430. [Google Scholar] [CrossRef] [PubMed]
- E Gregory, K.; Cundiff, L.V.; Koch, R.M. Breed effects and heterosis in advanced generations of composite populations for preweaning traits of beef cattle. J. Anim. Sci. 1991, 69, 947–960. [Google Scholar] [CrossRef] [PubMed]
- Wheeler, T.L.; Cundiff, L.V.; Shackelford, S.D.; Koohmaraie, M. Characterization of biological types of cattle (Cycle VI): Carcass, yield, and longissimus palatability traits12. J. Anim. Sci. 2004, 82, 1177–1189. [Google Scholar] [CrossRef]
- Ahlberg, C.M.; A Kuehn, L.; Thallman, R.M.; Kachman, S.D.; Snelling, W.M.; Spangler, M.L. Breed effects and genetic parameter estimates for calving difficulty and birth weight in a multibreed population1. J. Anim. Sci. 2016, 94, 1857–1864. [Google Scholar] [CrossRef] [Green Version]
- E Gregory, K.; E Echternkamp, S.; E Dickerson, G.; Cundiff, L.V.; Koch, R.M.; Van Vleck, L.D. Twinning in cattle: I. Foundation animals and genetic and environmental effects on twinning rate. J. Anim. Sci. 1990, 68, 1867–1876. [Google Scholar] [CrossRef] [Green Version]
- VanRaden, P.; Null, D.; Sargolzaei, M.; Wiggans, G.; Tooker, M.; Cole, J.; Sonstegard, T.; Connor, E.; Winters, M.; Van Kaam, J.; et al. Genomic imputation and evaluation using high-density Holstein genotypes. J. Dairy Sci. 2013, 96, 668–678. [Google Scholar] [CrossRef] [Green Version]
- Rosen, B.; Bickhart, D.; Schnabel, R.; Koren, S.; Elsik, C.; Zimin, A.; Dreischer, C.; Schultheiss, S.; Hall, R.; Schroeder, S.; et al. Modernizing the Bovine Reference Genome Assembly. In Proceedings of the World Congress on Genetics Applied to Livestock Production 2018, Molecular Genetics 3, 802, Auckland, New Zealand, 11–16 February 2018. [Google Scholar]
- Schnabel, R. NAGRP Community Data Repository. Available online: https://www.animalgenome.org/repository/cattle/UMC_bovine_coordinates/ (accessed on 11 February 2020).
- VanRaden, P.M. Efficient Methods to Compute Genomic Predictions. J. Dairy Sci. 2008, 91, 4414–4423. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Snelling, W.M.; Allan, M.F.; Keele, J.W.; A Kuehn, L.; Thallman, R.M.; Bennett, G.L.; Ferrell, C.L.; Jenkins, T.G.; Freetly, H.C.; Nielsen, M.K.; et al. Partial-genome evaluation of postweaning feed intake and efficiency of crossbred beef cattle1,2. J. Anim. Sci. 2011, 89, 1731–1741. [Google Scholar] [CrossRef] [Green Version]
- Meyer, K. WOMBAT—A tool for mixed model analyses in quantitative genetics by restricted maximum likelihood (REML). J. Zhejiang Univ. Sci. B 2007, 8, 815–821. [Google Scholar] [CrossRef] [Green Version]
- Snelling, W.M.; Kachman, S.D.; Bennett, G.L.; Spangler, M.L.; A Kuehn, L.; Thallman, R.M. 197 Functional SNP associated with birth weight in independent populations identified with a permutation step added to GBLUP-GWAS. J. Anim. Sci. 2017, 95, 97–98. [Google Scholar] [CrossRef] [Green Version]
- Strandén, I.; Garrick, D. Technical note: Derivation of equivalent computing algorithms for genomic predictions and reliabilities of animal merit. J. Dairy Sci. 2009, 92, 2971–2975. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Seqtk. Available online: https://github.com/lh3/seqtk (accessed on 25 October 2020).
- Loimpute-Public. Available online: https://gitlab.com/gencove/loimpute-public (accessed on 25 October 2020).
- Li, H.; Durbin, R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 2009, 25, 1754–1760. [Google Scholar] [CrossRef] [Green Version]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R. The Sequence Alignment/Map format and SAMtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Der Auwera, G.A.; Carneiro, M.O.; Hartl, C.; Poplin, R.; Del Angel, G.; Levy-Moonshine, A.; Jordan, T.; Shakir, K.; Roazen, D.; Thibault, J.; et al. From FastQ Data to High-Confidence Variant Calls: The Genome Analysis Toolkit Best Practices Pipeline. Curr. Protoc. Bioinform. 2013, 43, 11.10.1–11.10.33. [Google Scholar] [CrossRef]
- Browning, S.R.; Browning, B.L. Rapid and Accurate Haplotype Phasing and Missing-Data Inference for Whole-Genome Association Studies By Use of Localized Haplotype Clustering. Am. J. Hum. Genet. 2007, 81, 1084–1097. [Google Scholar] [CrossRef] [Green Version]
- Browning, B.L.; Zhou, Y.; Browning, S.R. A One-Penny Imputed Genome from Next-Generation Reference Panels. Am. J. Hum. Genet. 2018, 103, 338–348. [Google Scholar] [CrossRef] [Green Version]
- Chang, C.C.; Chow, C.C.; Tellier, L.C.A.M.; Vattikuti, S.; Purcell, S.M.; Lee, J.J. Second-generation PLINK: Rising to the challenge of larger and richer datasets. GigaScience 2015, 4, 7. [Google Scholar] [CrossRef] [PubMed]
- Cingolani, P.; Platts, A.; Wang, L.; Coon, M.; Nguyen, T.; Wang, L.; Land, S.; Lu, X.; Ruden, D.M. A program for annotating and predicting the effects of single nucleotide polymorphisms, SnpEff: SNPs in the genome of Drosophila melanogaster strain w1118; iso-2; iso-3. Fly (Austin) 2012, 6, 80–92. [Google Scholar] [CrossRef] [Green Version]
- Ensembl No Title. Available online: ftp://ftp.ensembl.org/pub/release-96/gtf/bos_taurus/Bos_taurus.ARS-UCD1.2.96.gtf.gz (accessed on 14 April 2019).
- Zorc, M.; Ogorevc, J.; Dovc, P. The new bovine reference genome assembly provides new insight into genomic organization of the bovine major histocompatibility complex. J. Central Eur. Agric. 2019, 20, 1111–1115. [Google Scholar] [CrossRef]
- Lillie, F.R. The Theory of the Free-Martin. Science 1916, 43, 611–613. [Google Scholar] [CrossRef] [Green Version]
- Owen, R.D.; Miller, N.E.; Bailey, C.J.; Stevenson, J.A.F. Immunogenetic consequences of vascular anastomoses between bovine twins. Science 1945, 102, 400–401. [Google Scholar] [CrossRef]
- López-Gatius, F.; Hunter, R. Spontaneous reduction of advanced twin embryos: Its occurrence and clinical relevance in dairy cattle. Theriogenology 2005, 63, 118–125. [Google Scholar] [CrossRef]
- Padula, A.M. The freemartin syndrome: An update. Anim. Reprod. Sci. 2005, 87, 93–109. [Google Scholar] [CrossRef]
- McDaneld, T.G.; Kuehn, L.A.; Thomas, M.G.; Snelling, W.M.; Sonstegard, T.S.; Matukumalli, L.K.; Smith, T.P.L.; Pollak, E.J.; Keele, J.W. Y are you not pregnant: Identification of Y chromosome segments in female cattle with decreased reproductive efficiency. J. Anim. Sci. 2012, 90, 2142–2151. [Google Scholar] [CrossRef] [Green Version]
- Lasater Beefmasters, Lasater Philosophy, Composite Cattle. Available online: https://isabeefmasters.com/about-us/beefmasters-history/ (accessed on 1 March 2020).
- Breed History—Santa Gertrudis Breeders International. Available online: https://santagertrudis.com/sgbi/santa-gertrudis-breed-history/ (accessed on 1 March 2020).
- Koren, S.; Rhie, A.; Walenz, B.P.; Dilthey, A.T.; Bickhart, D.M.; Kingan, S.B.; Hiendleder, S.; Williams, J.L.; Smith, S.R.; Phillippy, A.M. De novo assembly of haplotype-resolved genomes with trio binning. Nat. Biotechnol. 2018, 36, 1174–1182. [Google Scholar] [CrossRef] [PubMed]
- Ros-Freixedes, R.; Gonen, S.; Gorjanc, G.; Hickey, J.M. A method for allocating low-coverage sequencing resources by targeting haplotypes rather than individuals. Genet. Sel. Evol. 2017, 49, 78. [Google Scholar] [CrossRef] [Green Version]
- Snelling, W.M.; Cushman, R.A.; Keele, J.W.; Maltecca, C.; Thomas, M.G.; Fortes, M.R.S.; Reverter, A. BREEDING AND GENETICS SYMPOSIUM: Networks and pathways to guide genomic selection. J. Anim. Sci. 2013, 91, 537–552. [Google Scholar] [CrossRef] [Green Version]
- Saatchi, M.; Schnabel, R.D.; Taylor, J.F.; Garrick, D.J. Large-effect pleiotropic or closely linked QTL segregate within and across ten US cattle breeds. BMC Genom. 2014, 15, 1–17. [Google Scholar] [CrossRef] [Green Version]
- Saatchi, M.; Garrick, D.J. Developing a Reduced SNP Panel for Low-cost Genotyping in Beef Cattle. Anim. Sci. Pap. Rep. 2014, 660. [Google Scholar] [CrossRef]

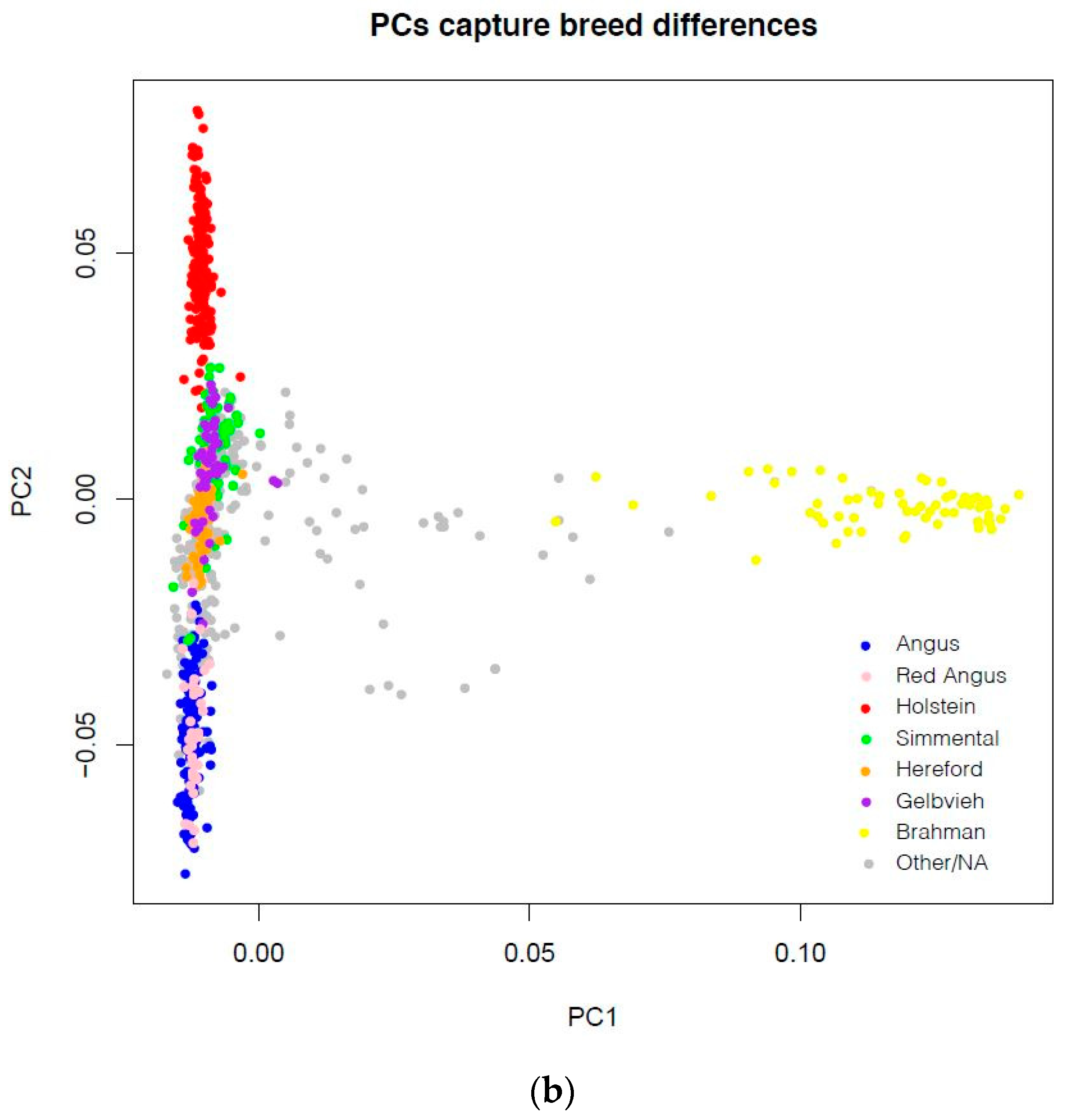

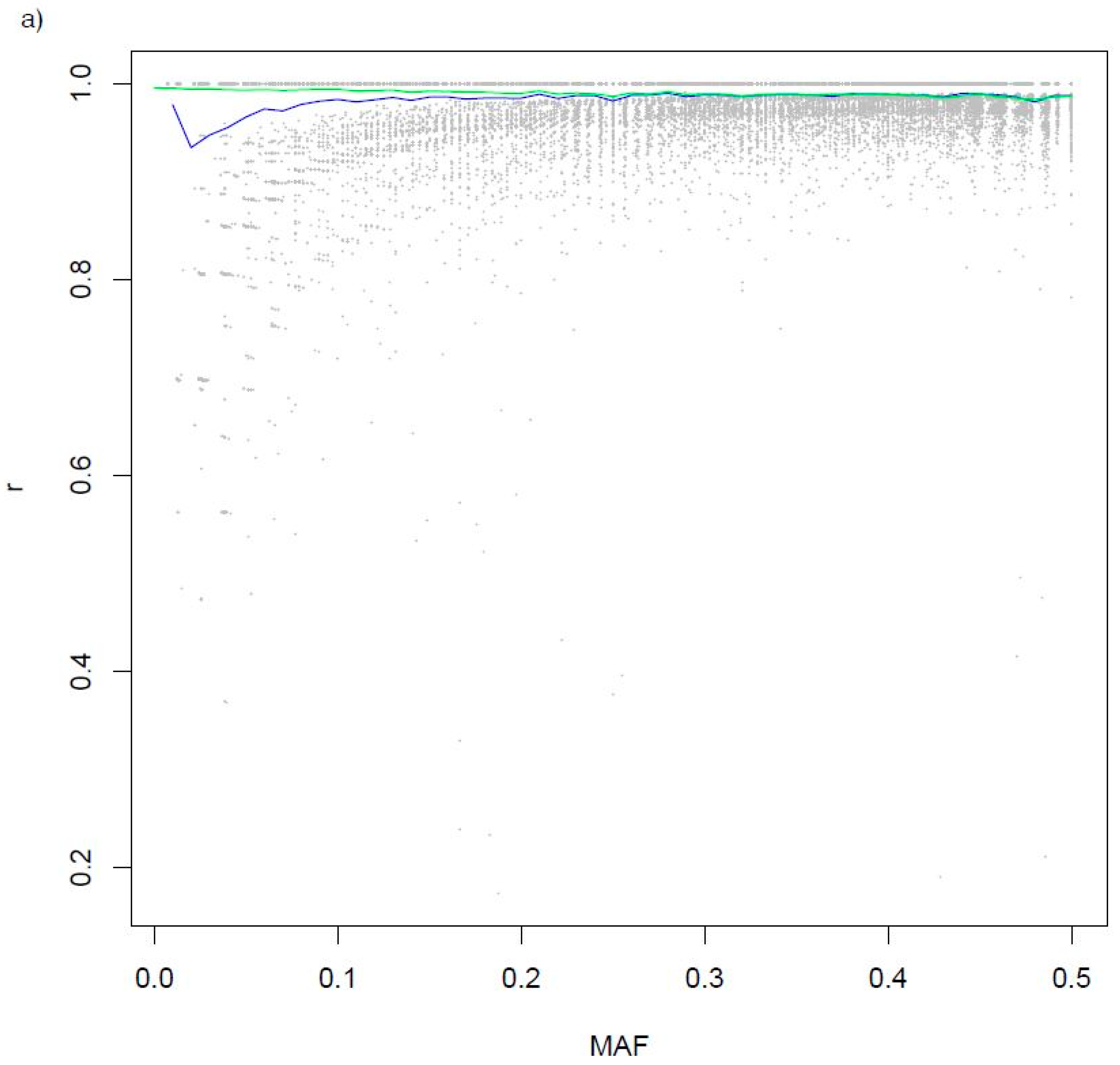
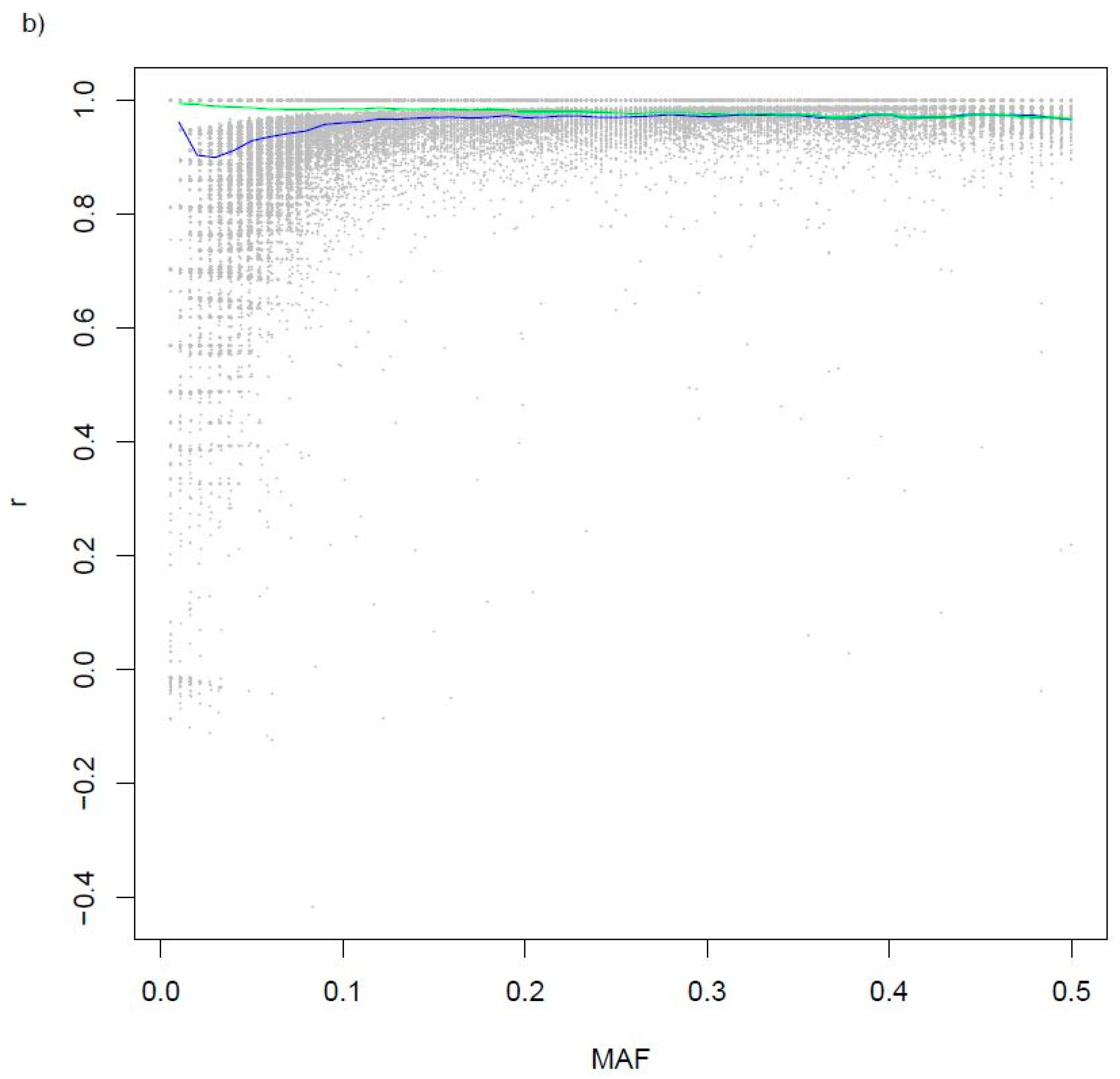

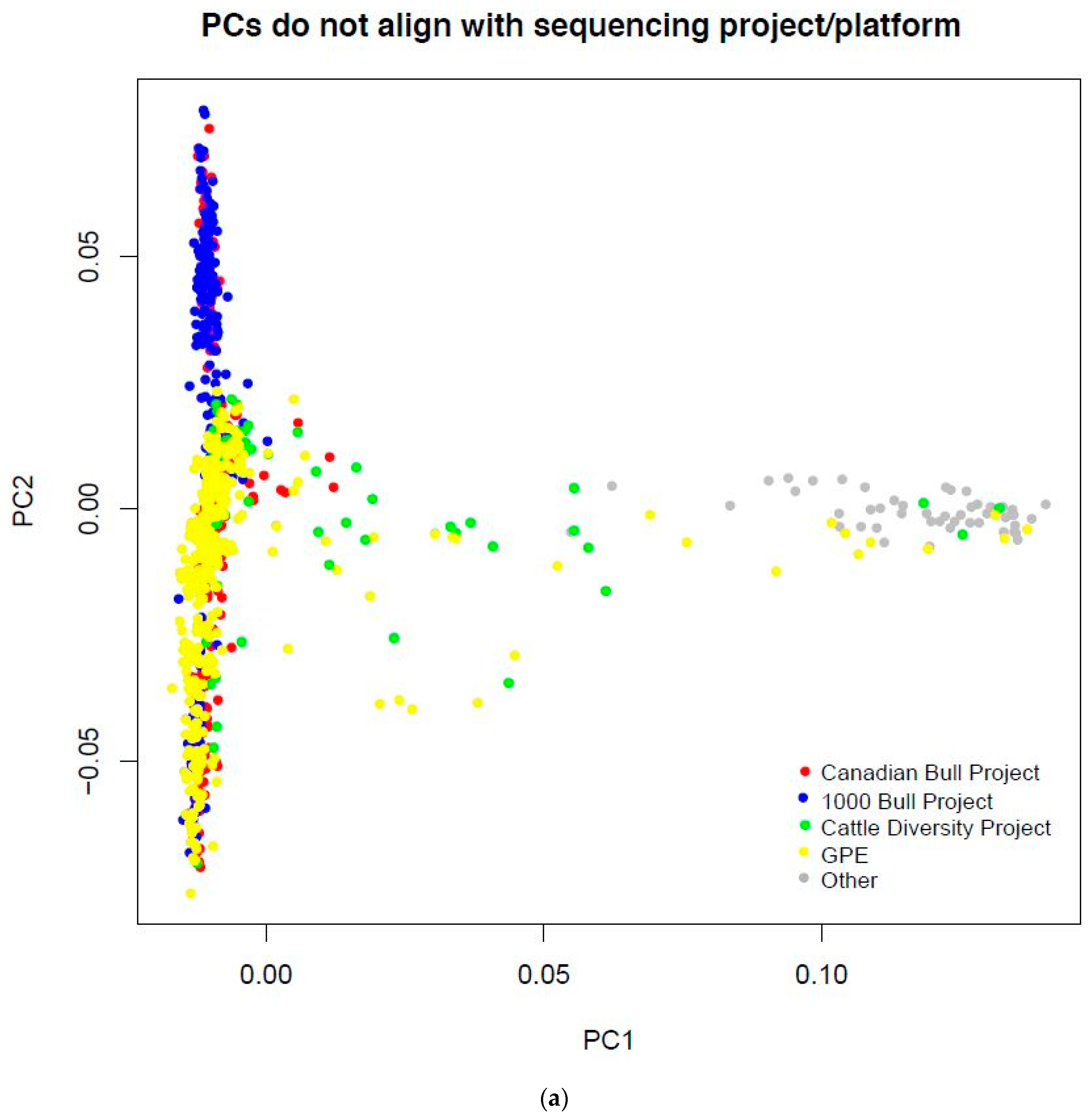{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| SNP Array | N |
|---|---|
| BovineSNP50 a | 9930 |
| BovineHD b | 1547 |
| GGP c -F250 | 2339 |
| GGP-50K | 3068 |
| GGP d | 5083 |
| Reference b | SNP Array c | |||
|---|---|---|---|---|
| Classification a | Variants | Genes | Variants | Genes |
| Protein-changing | 332,714 | 21,066 | 29,519 | 10,673 |
| High impact | 14,773 | 9084 | 545 | 509 |
| Non-synonymous SNP | 318,269 | 20,978 | 29,011 | 10,576 |
| Potentially regulatory | 327,357 | 18,110 | 13,072 | 8076 |
| Untranslated region (UTR) | 318,495 | 15,288 | 12,447 | 7557 |
| Non-coding RNA | 8940 | 2822 | 627 | 519 |
| Intergenic | 38,694,029 | 396,306 | ||
| Intronic | 19,533,912 | 272,510 | ||
| Total | 59,198,026 | 21,334 | 715,402 | 10,683 |
| Correlation (r) Scale | −log(1−r) Scale | |||||
|---|---|---|---|---|---|---|
| Sire Breed | Effect a | SE | p Value | Effect a | SE | p Value |
| Red Angus | 4.60 × 10−4 | 4.92 × 10−3 | 9.26 × 10−1 | −0.07 | 0.21 | 7.52 × 10−1 |
| Brahman | −2.79 × 10−2 | 3.81 × 10−3 | 1.59 × 10−9 | 1.86 | 0.16 | 8.17 × 10−16 |
| Beefmaster | −2.08 × 10−2 | 3.36 × 10−3 | 1.01 × 10−7 | 1.60 | 0.14 | 1.78 × 10−15 |
| Brangus | −1.05 × 10−2 | 3.11 × 10−3 | 1.37 × 10−3 | 1.15 | 0.13 | 1.25 × 10−11 |
| Charolais | −2.02 × 10−3 | 3.36 × 10−3 | 5.50 × 10−1 | 0.36 | 0.14 | 1.55 × 10−2 |
| ChiAngus | −2.48 × 10−3 | 4.92 × 10−3 | 6.16 × 10−1 | 0.40 | 0.21 | 6.38 × 10−2 |
| South Devon | −2.54 × 10−3 | 6.60 × 10−3 | 7.02 × 10−1 | 0.44 | 0.28 | 1.22 × 10−1 |
| Gelbvieh | −1.63 × 10−3 | 3.81 × 10−3 | 6.71 × 10−1 | 0.29 | 0.16 | 7.83 × 10−2 |
| Hereford | −6.70 × 10−4 | 3.55 × 10−3 | 8.51 × 10−1 | 0.13 | 0.15 | 4.10 × 10−1 |
| Limousin | −1.80 × 10−3 | 6.60 × 10−3 | 7.86 × 10−1 | 0.34 | 0.28 | 2.34 × 10−1 |
| Maine-Anjou | −3.20 × 10−4 | 4.92 × 10−3 | 9.48 × 10−1 | 0.09 | 0.21 | 6.72 × 10−1 |
| Salers | −2.75 × 10−3 | 3.81 × 10−3 | 4.74 × 10−1 | 0.47 | 0.16 | 5.95 × 10−3 |
| Braunveih | −3.89 × 10−3 | 4.92 × 10−3 | 4.33 × 10−1 | 0.61 | 0.21 | 5.66 × 10−3 |
| Simmental | −2.57 × 10−4 | 4.21 × 10−3 | 9.52 × 10−1 | 0.07 | 0.18 | 6.79 × 10−1 |
| Shorthorn | −1.25 × 10−3 | 3.55 × 10−3 | 7.62 × 10−1 | 0.24 | 0.15 | 1.22 × 10−1 |
| Santa Gertrudis | −2.21 × 10−2 | 3.36 × 10−3 | 2.34 × 10−8 | 1.66 | 0.14 | 5.55 × 10−15 |
| Birth Weight | Postweaning Gain | Marbling Score | ||||
|---|---|---|---|---|---|---|
| Relationship a | h2 (SE) | N | h2 | n | h2 | n |
| Pedigree a | 0.595 (0.008) | 78,625 | 0.526 (0.010) | 68,846 | 0.538 (0.018) | 33,850 |
| Gall b | 0.573 (0.011) | 16,512 | 0.474 (0.013) | 16,144 | 0.508 (0.017) | 10,898 |
| GF250 c | 0.545 (0.011) | 16,440 | 0.442 (0.012) | 16,068 | 0.471 (0.016) | 10,822 |
| GF250s d | 0.380 (0.023) | 16,440 | 0.270 (0.019) | 16,068 | 0.342 (0.021) | 10,822 |
| GF250r e | 0.066 (0.007) | 16,440 | 0.062 (0.007) | 16,068 | 0.105 (0.009) | 10,822 |
| G50K f | 0.519 (0.011) | 16,440 | 0.437 (0.012) | 16,068 | 0.466 (0.016) | 10,822 |
| Birth Weight | Postweaning Gain | Marbling Score | ||||
|---|---|---|---|---|---|---|
| Predictions Using Imputed SNP Array Genotypes | ||||||
| Pedigree a | Gall b | Pedigree | Gall | Pedigree | Gall | |
| GF250 c | 0.738 (0.061) | 0.904 (0.037) | 0.779 (0.055) | 0.881 (0.041) | 0.770 (0.057) | 0.926 (0.032) |
| GF250s d | 0.555 (0.079) | 0.681 (0.067) | 0.653 (0.069) | 0.714 (0.063) | 0.655 (0.069) | 0.750 (0.059) |
| GF250r e | 0.379 (0.090) | 0.481 (0.083) | 0.344 (0.093) | 0.385 (0.090) | 0.629 (0.070) | 0.741 (0.058) |
| G50K f | 0.710 (0.063) | 0.888 (0.039) | 0.785 (0.055) | 0.886 (0.040) | 0.794 (0.053) | 0.950 (0.026) |
| Predictions Using Genotypes Imputed from Low-Coverage Sequence | ||||||
| GF250 c | 0.680 (0.067) | 0.866 (0.044) | 0.779 (0.055) | 0.887 (0.040) | 0.769 (0.057) | 0.936 (0.030) |
| Gf250s d | 0.531 (0.081) | 0.634 (0.073) | 0.635 (0.071) | 0.722 (0.063) | 0.641 (0.071) | 0.738 (0.062) |
| GF250r e | 0.286 (0.100) | 0.390 (0.094) | 0.332 (0.096) | 0.395 (0.094) | 0.649 (0.071) | 0.776 (0.057) |
| G50K f | 0.676 (0.067) | 0.866 (0.044) | 0.805 (0.052) | 0.903 (0.037) | 0.760 (0.058) | 0.941 (0.029) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Snelling, W.M.; Hoff, J.L.; Li, J.H.; Kuehn, L.A.; Keel, B.N.; Lindholm-Perry, A.K.; Pickrell, J.K. Assessment of Imputation from Low-Pass Sequencing to Predict Merit of Beef Steers. Genes 2020, 11, 1312. https://doi.org/10.3390/genes11111312
Snelling WM, Hoff JL, Li JH, Kuehn LA, Keel BN, Lindholm-Perry AK, Pickrell JK. Assessment of Imputation from Low-Pass Sequencing to Predict Merit of Beef Steers. Genes. 2020; 11(11):1312. https://doi.org/10.3390/genes11111312
Chicago/Turabian StyleSnelling, Warren M., Jesse L. Hoff, Jeremiah H. Li, Larry A. Kuehn, Brittney N. Keel, Amanda K. Lindholm-Perry, and Joseph K. Pickrell. 2020. "Assessment of Imputation from Low-Pass Sequencing to Predict Merit of Beef Steers" Genes 11, no. 11: 1312. https://doi.org/10.3390/genes11111312
APA StyleSnelling, W. M., Hoff, J. L., Li, J. H., Kuehn, L. A., Keel, B. N., Lindholm-Perry, A. K., & Pickrell, J. K. (2020). Assessment of Imputation from Low-Pass Sequencing to Predict Merit of Beef Steers. Genes, 11(11), 1312. https://doi.org/10.3390/genes11111312