Identification of Novel Mutations and Expressions of EPAS1 in Phaeochromocytomas and Paragangliomas



Abstract
:1. Introduction
2. Materials and Methods
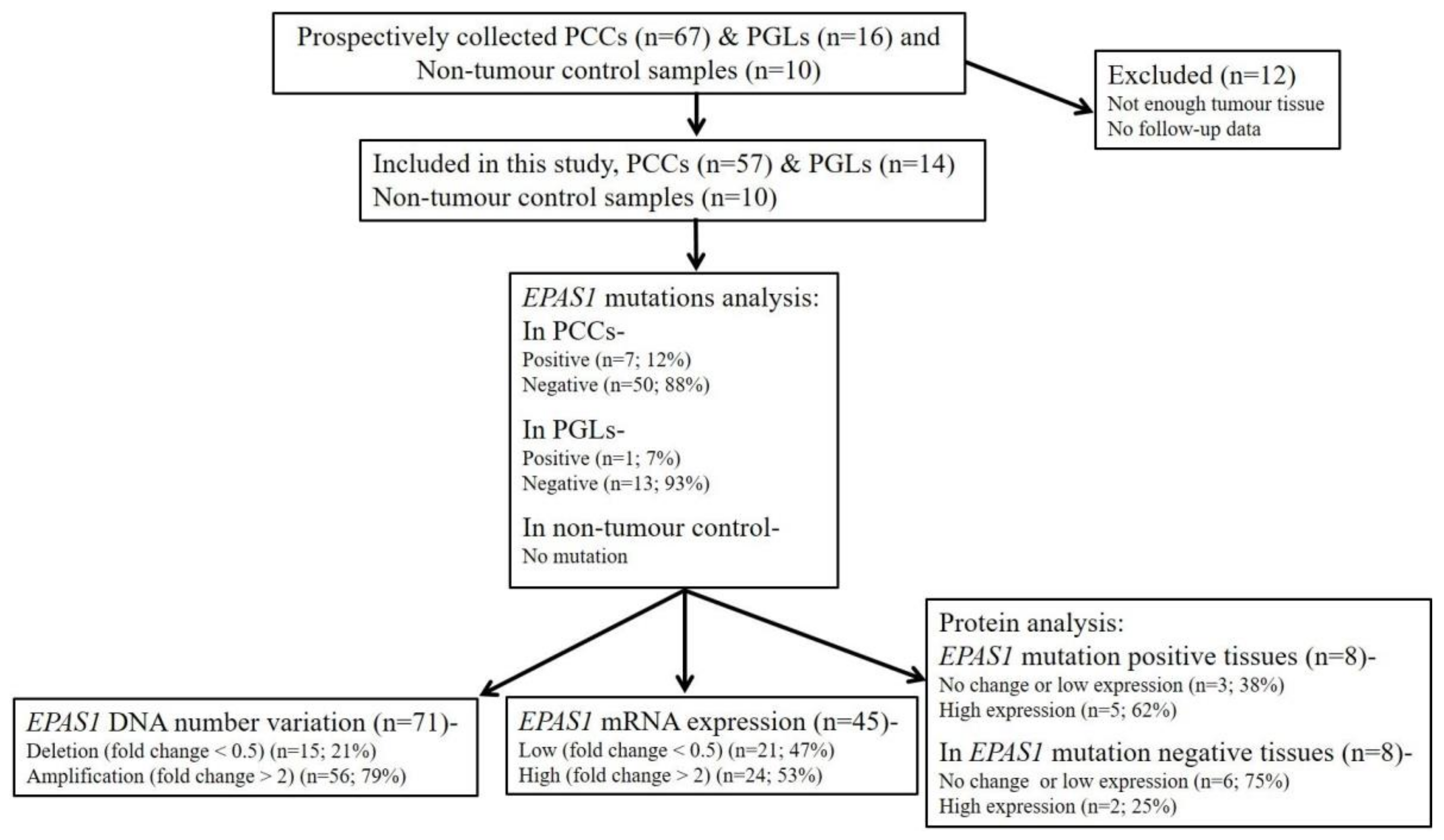
2.1. Recruitments of Tissues and Sample Selection
2.2. Clinical Data of the Selected Cases
2.3. Extractions of DNA and RNA
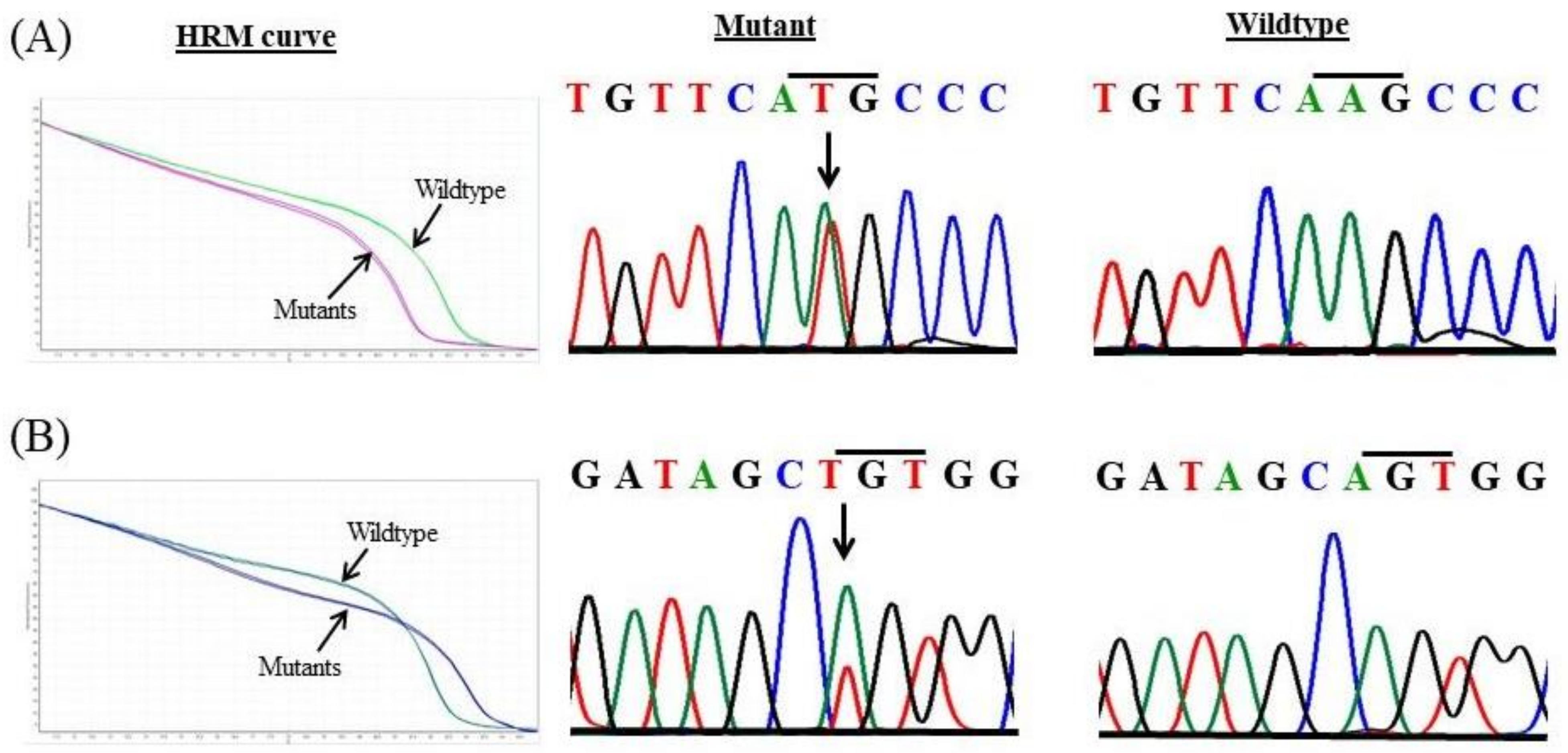
2.4. High-Resolution Melt (HRM) Curve Analysis
2.5. Confirmation of Mutations by Sanger Sequencing
2.6. In Silico Analysis
2.7. Quantitative Real-Time PCR (qPCR) Analysis
2.8. Immunofluorescence
2.9. Statistical Analysis
3. Results
3.1. Identification of Novel EPAS1 Mutations in Phaeochromocytomas/Paragangliomas
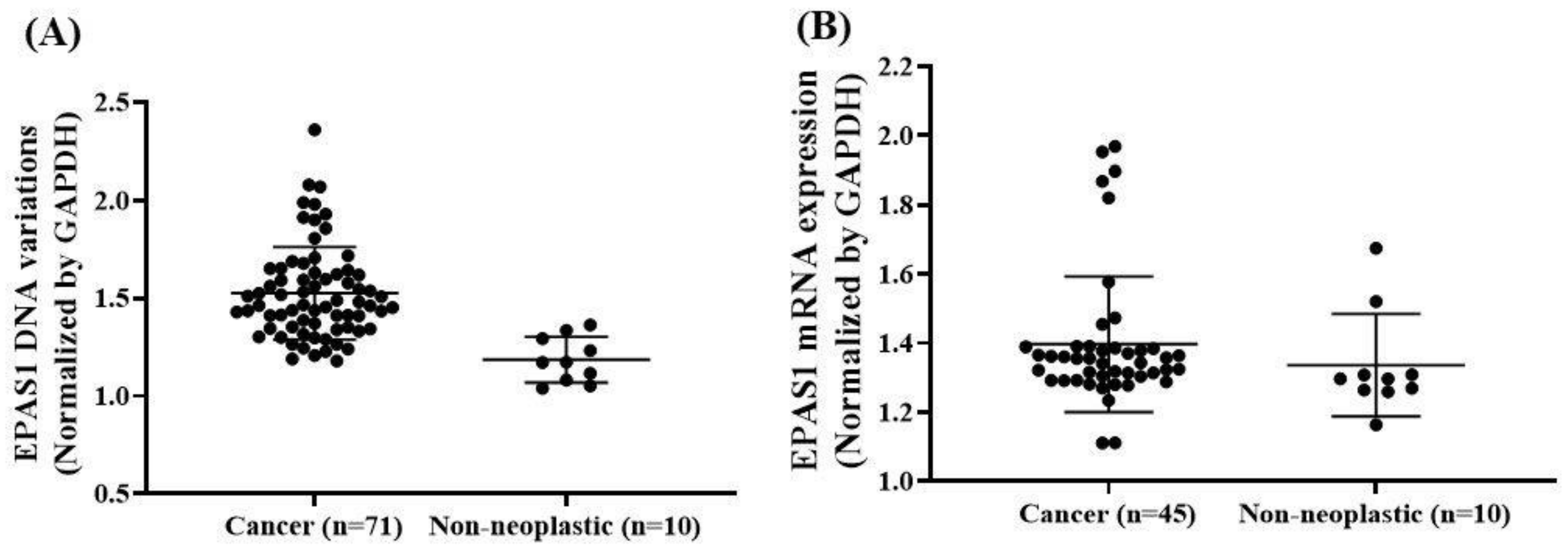
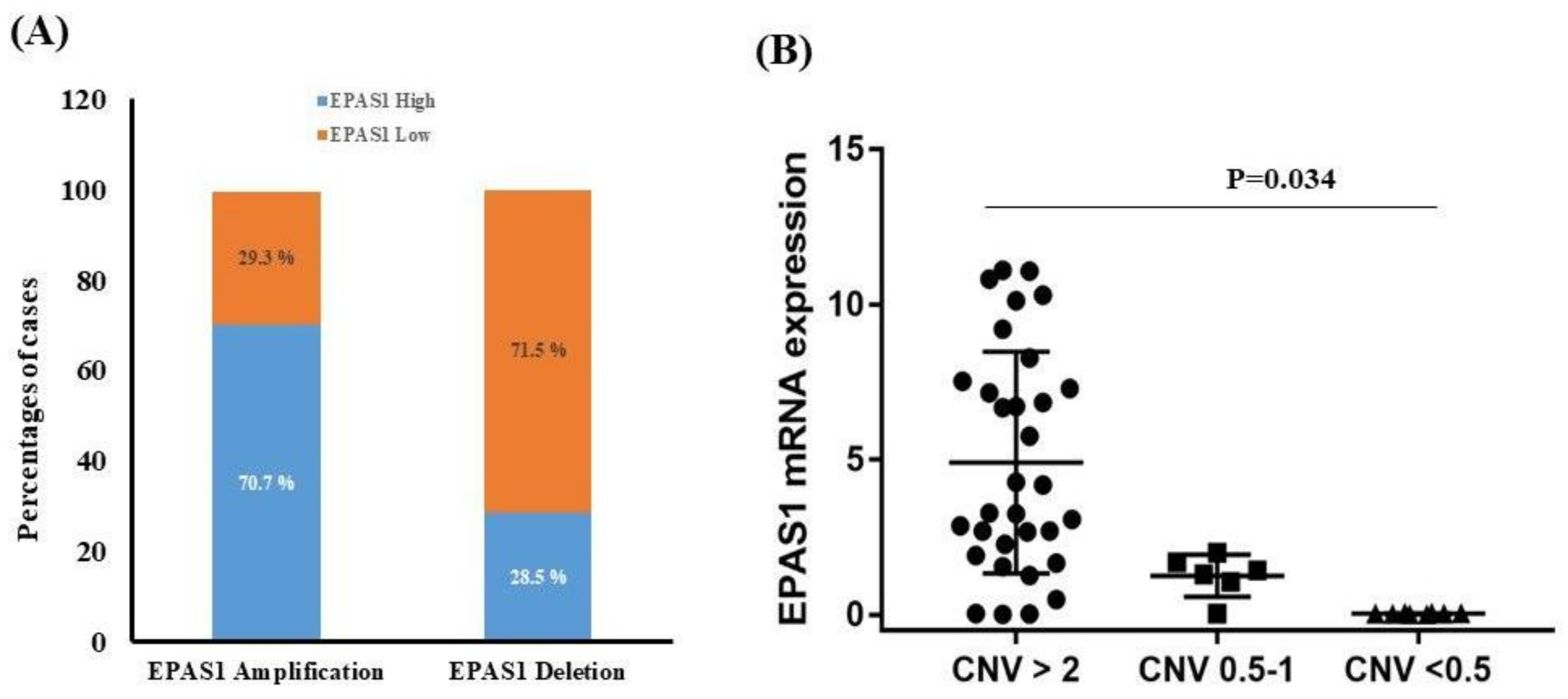
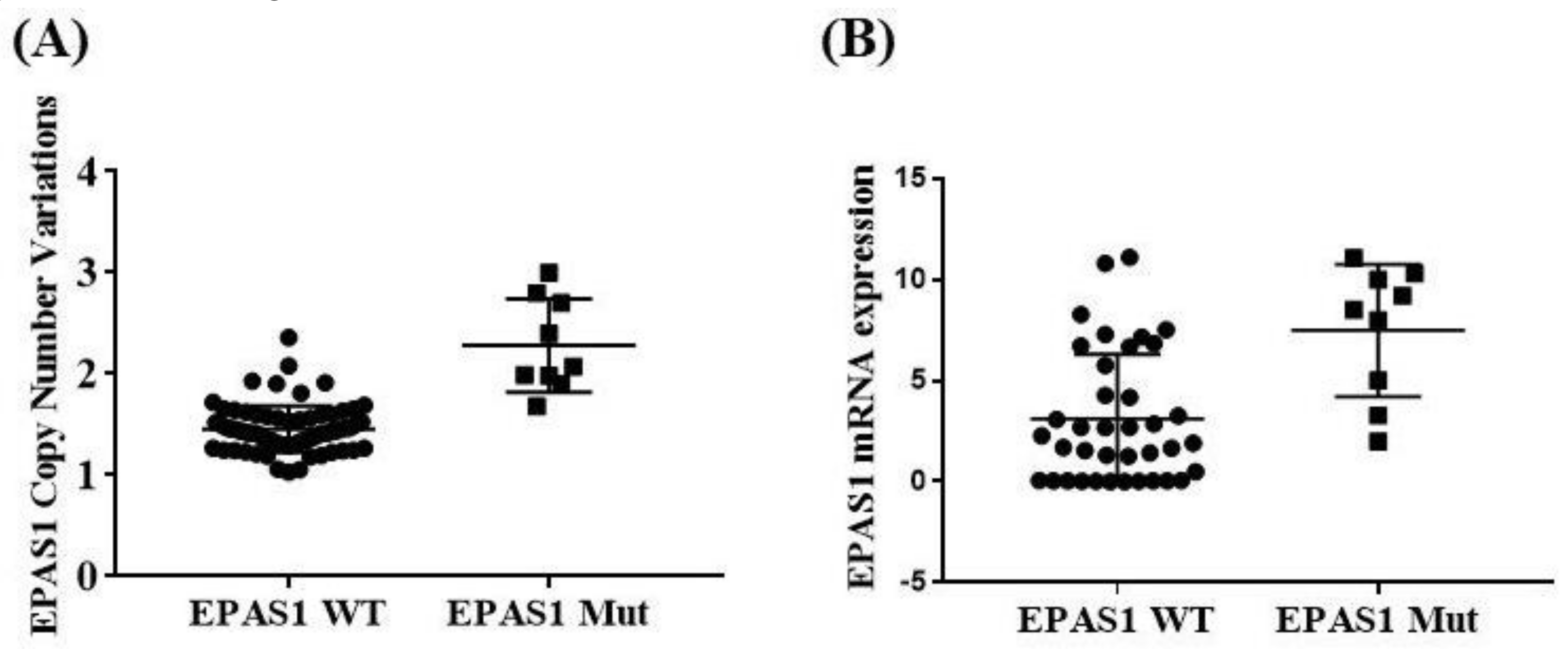
3.2. EPAS1 DNA Number Variations in Phaeochromocytomas/Paragangliomas
3.3. EPAS1 mRNA Expressions in Phaeochromocytomas/Paragangliomas
3.4. EPAS1 Protein Expression in Phaeochromocytomas/Paragangliomas
3.5. Association of EPAS1 DNA Number Variation, mRNA Expression, Protein Expression and Mutations in Phaeochromocytomas
4. Discussion
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Alaikov, T.; Ivanova, M.; Shivarov, V. EPAS1 p.M535T mutation in a Bulgarian family with congenital erythrocytosis. Hematology 2016, 21, 619–622. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alonso, M.H.; Aussó, S.; Lopez-Doriga, A.; Cordero, D.; Guinó, E.; Solé, X.; Barenys, M.; De Oca, J.; Capella, G.; Salazar, R.; et al. Comprehensive analysis of copy number aberrations in microsatellite stable colon cancer in view of stromal component. Br. J. Cancer 2017, 117, 421–431. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Burnichon, N.; Buffet, A.; Parfait, B.; Letouzé, E.; Laurendeau, I.; Loriot, C.; Pasmant, E.; Abermil, N.; Valeyrie-Allanore, L.; Bertherat, J.; et al. Somatic NF1 inactivation is a frequent event in sporadic pheochromocytoma. Hum. Mol. Genet. 2012, 21, 5397–5405. [Google Scholar] [CrossRef] [Green Version]
- Burnichon, N.; Vescovo, L.; Amar, L.; Libé, R.; De Reynies, A.; Venisse, A.; Jouanno, E.; Laurendeau, I.; Parfait, B.; Bertherat, J.; et al. Integrative genomic analysis reveals somatic mutations in pheochromocytoma and paraganglioma. Hum. Mol. Genet. 2011, 20, 3974–3985. [Google Scholar] [CrossRef] [PubMed]
- Comino-Méndez, I.; De Cubas, A.A.; Bernal, C.; Álvarez-Escolá, C.; Sánchez-Malo, C.; Ramírez-Tortosa, C.L.; Pedrinaci, S.; Rapizzi, E.; Ercolino, T.; Bernini, G.; et al. Tumoral EPAS1 (HIF2A) mutations explain sporadic pheochromocytoma and paraganglioma in the absence of erythrocytosis. Hum. Mol. Genet. 2013, 22, 2169–2176. [Google Scholar] [CrossRef] [PubMed]
- Comino-Méndez, I.; Gracia-Aznárez, F.J.; Schiavi, F.; Landa, I.; Leandro-García, L.J.; Letón, R.; Honrado, E.; Ramos-Medina, R.; Caronia, D.; Pita, G.; et al. Exome sequencing identifies MAX mutations as a cause of hereditary pheochromocytoma. Nat. Genet. 2011, 43, 663–667. [Google Scholar] [CrossRef] [PubMed]
- Cui, J.; Duan, B.; Zhao, X.; Chen, Y.; Sun, S.; Deng, W.; Zhang, Y.; Du, J.; Chen, Y.; Gu, L. MBD3 mediates epigenetic regulation on EPAS1 promoter in cancer. Tumor Biol. 2016, 37, 13455–13467. [Google Scholar] [CrossRef]
- Dahia, P.L.M.; Ross, K.N.; Wright, M.E.; Hayashida, C.Y.; Santagata, S.; Barontini, M.; Kung, A.L.; Sanso, G.; Powers, J.F.; Tischler, A.S.; et al. A HIF1α Regulatory Loop Links Hypoxia and Mitochondrial Signals in Pheochromocytomas. PLoS Genet. 2005, 1, e8. [Google Scholar] [CrossRef] [Green Version]
- Dahia, P.L.M. Pheochromocytoma and paraganglioma pathogenesis: Learning from genetic heterogeneity. Nat. Rev. Cancer 2014, 14, 108–119. [Google Scholar] [CrossRef]
- Favier, J.; Buffet, A.; Gimenez-Roqueplo, A.-P. HIF2A mutations in paraganglioma with polycythemia. N. Engl. J. Med. 2012, 367, 2161–2162. [Google Scholar]
- Gopalan, V.; Islam, F.; Pillai, S.; Tang, J.C.-O.; Tong, D.K.-H.; Law, S.; Chan, K.-W.; Lam, A.K.-Y. Overexpression of microRNA-1288 in oesophageal squamous cell carcinoma. Exp. Cell Res. 2016, 348, 146–154. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Holmquist-Mengelbier, L.; Fredlund, E.; Löfstedt, T.; Noguera, R.; Navarro, S.; Nilsson, H.; Pietras, A.; Vallon-Christersson, J.; Borg, Å.; Gradin, K.; et al. Recruitment of HIF-1α and HIF-2α to common target genes is differentially regulated in neuroblastoma: HIF-2α promotes an aggressive phenotype. Cancer Cell 2006, 10, 413–423. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Islam, F.; Gopalan, V.; Law, S.; Tang, J.C.-O.; Lam, A.K.-Y. FAM134B promotes esophageal squamous cell carcinoma in vitro and its correlations with clinicopathologic features. Hum. Pathol. 2019, 87, 1–10. [Google Scholar] [CrossRef] [Green Version]
- Islam, F.; Gopalan, V.; Vider, J.; Lu, C.-T.; Lam, A.K.-Y. MiR-142-5p act as an oncogenic microRNA in colorectal cancer: Clinicopathological and functional insights. Exp. Mol. Pathol. 2018, 104, 98–107. [Google Scholar] [CrossRef] [Green Version]
- Islam, F.; Gopalan, V.; Wahab, R.; Lee, K.T.-W.; Haque, H.; Mamoori, A.; Lu, C.-T.; Smith, R.A.; Lam, A.K.Y. Novel FAM134B mutations and their clinicopathological significance in colorectal cancer. Qual. Life Res. 2017, 136, 321–337. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jochmanova, I.; Pacak, K. Genomic Landscape of Pheochromocytoma and Paraganglioma. Trends Cancer 2017, 4, 6–9. [Google Scholar] [CrossRef] [PubMed]
- Kasem, K.; Gopalan, V.; Salajegheh, A.; Lu, C.-T.; Smith, R.A.; Lam, A.K.Y. JK1 (FAM134B) gene and colorectal cancer: A pilot study on the gene copy number alterations and correlations with clinicopathological parameters. Exp. Mol. Pathol. 2014, 97, 31–36. [Google Scholar] [CrossRef]
- Lam, A.K.-Y. Update on Adrenal Tumours in 2017 World Health Organization (WHO) of Endocrine Tumours. Endocr. Pathol. 2017, 28, 213–227. [Google Scholar] [CrossRef]
- Lam, A.K.-Y. Update on paragangliomas and pheochromocytomas. Turk. J. Pathol. 2015, 31, 105–112. [Google Scholar] [CrossRef] [Green Version]
- Lam, A.K.-Y.; Lo, C.Y.; Wat, N.M.S.; Luk, J.M. The clinicopathological features and importance of p53, Rb, and mdm2 expression in phaeochromocytomas and paragangliomas. J. Clin. Pathol. 2001, 54, 443–448. [Google Scholar] [CrossRef] [Green Version]
- Lorenzo, F.R.; Yang, C.; Fui, M.N.T.; Vankayalapati, H.; Zhuang, Z.; Huynh, T.; Prchal, J.T. A novel EPAS1/HIF2A germline mutation in a congenital polycythemia with paraganglioma. J. Mol. Med. 2013, 91, 507–512. [Google Scholar] [CrossRef] [Green Version]
- Maher, E.R. HIF2 and endocrine neoplasia: An evolving story. Endocr. Relat. Cancer 2013, 20, C5–C7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maxwell, P.H. The HIF pathway in cancer. Semin. Cell Dev. Biol. 2005, 16, 523–530. [Google Scholar] [CrossRef] [PubMed]
- Pan, H.; Chen, Q.; Qi, S.; Li, T.; Liu, B.; Liu, S.; Ma, X.; Wang, B. Mutations in EPAS1 in congenital heart disease in Tibetans. Biosci. Rep. 2018, 38, BSR20181389. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Percy, M.J.; Furlow, P.W.; Lucas, G.S.; Li, X.; Lappin, T.R.; McMullin, M.F.; Lee, F.S. A Gain-of-Function Mutation in theHIF2AGene in Familial Erythrocytosis. N. Engl. J. Med. 2008, 358, 162–168. [Google Scholar] [CrossRef] [Green Version]
- Pillai, S.; Gopalan, V.; Smith, R.A.; Lam, A.K.-Y. Updates on the genetics and the clinical impacts on phaeochromocytoma and paraganglioma in the new era. Crit. Rev. Oncol. 2016, 100, 190–208. [Google Scholar] [CrossRef] [Green Version]
- Pillai, S.; Gopalan, V.; Lo, C.Y.; Liew, V.; Smith, R.A.; Lam, A.K.-Y. Silent genetic alterations identified by targeted next-generation sequencing in pheochromocytoma/paraganglioma: A clinicopathological correlations. Exp. Mol. Pathol. 2017, 102, 41–46. [Google Scholar] [CrossRef]
- Pollard, P.J.; El-Bahrawy, M.; Poulsom, R.; Elia, G.; Killick, P.; Kelly, G.; Hunt, T.; Jeffery, R.; Seedhar, P.; Barwell, J.; et al. Expression of HIF-1α, HIF-2α (EPAS1), and Their Target Genes in Paraganglioma and Pheochromocytoma with VHL and SDH Mutations. J. Clin. Endocrinol. Metab. 2006, 91, 4593–4598. [Google Scholar] [CrossRef] [Green Version]
- Qin, Y.; Yao, L.; King, E.E.; Buddavarapu, K.; Lenci, R.E.; Chocron, E.S.; Lechleiter, J.D.; Sass, M.; Aronin, N.; Schiavi, F.; et al. Germline mutations in TMEM127 confer susceptibility to pheochromocytoma. Nat. Genet. 2010, 42, 229–233. [Google Scholar] [CrossRef] [Green Version]
- Salo-Mullen, E.E.; Lynn, P.B.; Wang, L.; Walsh, M.; Gopalan, A.; Shia, J.; Tran, C.; Man, F.Y.; McBride, S.; Schattner, M.; et al. Contiguous gene deletion of chromosome 2p16.3-p21 as a cause of Lynch syndrome. Fam. Cancer 2017, 17, 71–77. [Google Scholar] [CrossRef]
- Schwarz, J.M.; Rödelsperger, C.; Schuelke, M.; Seelow, D. MutationTaster evaluates disease-causing potential of sequence alterations. Nat. Methods 2010, 7, 575–576. [Google Scholar] [CrossRef] [PubMed]
- Takeda, N.; Maemura, K.; Imai, Y.; Harada, T.; Kawanami, D.; Nojiri, T.; Manabe, I.; Nagai, R. Endothelial PAS Domain Protein 1 Gene Promotes Angiogenesis through the Transactivation of Both Vascular Endothelial Growth Factor and Its Receptor, Flt-1. Circ. Res. 2004, 95, 146–153. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tian, H.; McKnight, S.L.; Russell, D.W. Endothelial PAS domain protein 1 (EPAS1), a transcription factor selectively expressed in endothelial cells. Genes Dev. 1997, 11, 72–82. [Google Scholar] [CrossRef] [Green Version]
- Toledo, R.A.; Qin, Y.; Srikantan, S.; Morales, N.P.; Li, Q.; Deng, Y.; Kim, S.-W.; Pereira, M.A.A.; Toledo, S.P.; Su, X.; et al. In vivo and in vitro oncogenic effects of HIF2A mutations in pheochromocytomas and paragangliomas. Endocr. Relat. Cancer 2013, 20, 349–359. [Google Scholar] [CrossRef] [Green Version]
- Wahab, R.; Gopalan, V.; Islam, F.; Smith, R.A.; Qiao, B.; Lam, A.K.-Y. Cellular expression, in-vitro and in-vivo confirmation of GAEC1 oncogenic properties in colon cancer. Eur. J. Cell Biol. 2017, 96, 487–495. [Google Scholar] [CrossRef] [PubMed]
- Welander, J.; Andreasson, A.; Brauckhoff, M.; Bäckdahl, M.; Larsson, C.; Gimm, O.; Söderkvist, P. Frequent, EPAS1/HIF2α exons 9 and 12 mutations in non-familial pheochromocytoma. Endocr. Relat. Cancer 2014, 21, 495–504. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Welander, J.; Larsson, C.; Bäckdahl, M.; Hareni, N.; Sivlér, T.; Brauckhoff, M.; Söderkvist, P.; Gimm, O. Integrative genomics reveals frequent somatic NF1 mutations in sporadic pheochromocytomas. Hum. Mol. Genet. 2012, 21, 5406–5416. [Google Scholar] [CrossRef] [Green Version]
- Welander, J.; Söderkvist, P.; Gimm, O. Genetics and clinical characteristics of hereditary pheochromocytomas and paragangliomas. Endocr. Relat. Cancer 2011, 18, R253–R276. [Google Scholar] [CrossRef] [Green Version]
- Yang, C.; Hong, C.S.; Prchal, J.T.; Balint, M.T.; Pacak, K.; Zhuang, Z. Somatic mosaicism of EPAS1 mutations in the syndrome of paraganglioma and somatostatinoma associated with polycythemia. Hum. Genome Var. 2015, 2, 15053. [Google Scholar] [CrossRef] [Green Version]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample ID | Type | Change in DNA Sequence | Change in Protein Sequence | DNA Copy Number Change | mRNA Expression | Protein Expression | Effects on Protein Features | In Silico Prediction | ||
|---|---|---|---|---|---|---|---|---|---|---|
| Mutation Taster | PROVEAN | SIFT | ||||||||
| P3 | PCC | c.1091A>T | p.Lys364Met | No change | High | High | Amino acids sequence change Protein structure (might be) affected Splice site changes | Diseases causing | Deleterious | Damaging |
| P33 | PCC | c.1091A>T | p.Lys364Met | Amplification | High | High | Amino acids sequence change Protein structure (might be) affected Splice site changes | Diseases causing | Deleterious | Damaging |
| P78 | PCC | c.1129A>T | P.Ser377Cys | Amplification | High | High | Amino acids sequence changed | Polymorphism | Neutral | Tolerated |
| P81 | PCC | c.1091A>T | p.Lys364Met | Amplification | No Change | No Change | Amino acids sequence change Protein structure (might be) affected Splice site changes | Diseases causing | Deleterious | Damaging |
| P93 | PCC | c.1129A>T | P.Ser377Cys | Amplification | High | High | Amino acids sequence changed | Polymorphism | Neutral | Tolerated |
| P94 * | PCC | c.1091A>T | p.Lys364Met | Amplification | No Change | Low | Amino acids sequence change Protein structure (might be) affected Splice site changes | Diseases causing | Deleterious | Damaging |
| P99 | PCC | c.1091A>T | p.Lys364Met | Amplification | High | High | Amino acids sequence change Protein structure (might be) affected Splice site changes | Diseases causing | Deleterious | Damaging |
| P122 * | PGL | c.1091A>T | p.Lys364Met | Amplification | High | Low | Amino acids sequence change Protein structure (might be) affected Splice site changes | Diseases causing | Deleterious | Damaging |
| Features | Number | Mutation Positive | Mutation Negative | p-Value |
|---|---|---|---|---|
| Total patients | 71 (100.00%) | 8 (11.26%) | 63 (88.74%) | - |
| Gender | ||||
| Male | 37 (52.11%) | 4 (10.81%) | 33 (89.19%) | 0.596 |
| Female | 34 (47.89%) | 4 (11.76%) | 30 (88.24%) | |
| Age | ||||
| ≤50 | 40 (56.34%) | 6 (15.00%) | 34 (85.00%) | 0.229 |
| >50 | 31 (43.66%) | 2 (6.45%) | 29 (93.55%) | |
| Race | ||||
| Chinese | 56 (78.87%) | 8 (14.29%) | 48 (85.71%) | 0.134 |
| Non-Chinese | 15 (21.13%) | - | 15 (100.00%) | |
| Side | ||||
| Unilateral | 63 (88.73%) | 7 (11.11%) | 56 (88.89%) | 0.636 |
| Bilateral | 8 (11.27%) | 1 (12.50%) | 7 (87.50%) | |
| Tumour location | ||||
| Adrenal gland | 57 (80.28%) | 6 (10.53%) | 51 (89.47%) | 0.497 |
| Carotid body | 14 (19.72%) | 2 (12.29%) | 12 (85.71%) | |
| Tumour size | ||||
| <50 mm | 33 (46.48%) | 1 (3.030%) | 32 (96.97%) | 0.044 |
| ≥50 mm | 38 (53.52%) | 7 (18.42%) | 31 (81.58%) | |
| Tumour weight * | ||||
| ≤50 gm | 17 (45.95%) | - | 17 (100.00%) | 0.000 |
| >50 gm | 20 (54.05%) | 3 (15.00%) | 17 (85.00%) | |
| Tumour types | ||||
| Non-metastasizing | 59 (83.10%) | 5 (8.47%) | 54 (91.53%) | 0.167 |
| Metastasizing | 12 (16.90%) | 3 (25.00%) | 9 (75.00%) |
| Features | Number | DNA Amplification | DNA Deletion | p-Value |
|---|---|---|---|---|
| Total patients | 71 (100.00%) | 8 (11.26%) | 63 (88.74%) | - |
| Gender | ||||
| Male | 37 (52.11%) | 28 (75.68%) | 9 (24.32%) | 0.347 |
| Female | 34 (47.89%) | 28 (82.35%) | 6 (17.65%) | |
| Age | ||||
| ≤50 | 40 (56.34%) | 30 (75.00%) | 10 (25.00%) | 0.271 |
| >50 | 31 (43.66%) | 26 (83.87%) | 5 (16.13%) | |
| Race | ||||
| Chinese | 56 (78.87%) | 45 (80.36%) | 11 (19.64%) | 0.392 |
| Non-Chinese | 15 (21.13%) | 11 (73.33%) | 4 (26.67%) | |
| Side | ||||
| Unilateral | 63 (88.73%) | 51 (80.95%) | 12 (19.05%) | 0.219 |
| Bilateral | 8 (11.27%) | 5 (62.50%) | 3 (37.50%) | |
| Tumour location | ||||
| Adrenal gland | 57 (80.28%) | 48 (84.21%) | 9 (15.79%) | 0.037 |
| Carotid body | 14 (19.72%) | 8 (57.14%) | 6 (15.79%) | |
| Tumour size | ||||
| <50 mm | 33 (46.48%) | 23 (69.70%) | 10 (30.30%) | 0.120 |
| ≥50 mm | 38 (53.52%) | 32 (84.21%) | 6 (15.79%) | |
| Tumour weight * | ||||
| ≤50 gm | 17 (45.95%) | 14 (82.35%) | 3 (17.65%) | 0.857 |
| >50 gm | 20 (54.05%) | 17 (85.00%) | 17 (15.00%) | |
| Tumour types | ||||
| Non-metastasizing | 59 (83.10%) | 47 (79.66%) | 12 (20.34%) | 0.852 |
| Metastasizing | 12 (16.90%) | 9 (75.00%) | 3 (25.00%) |
| Features | Number | High Expression | Low Expression | p-Value |
|---|---|---|---|---|
| Total patients | 45 (100.00%) | 24 (53.33%) | 21 (46.67%) | - |
| Gender | ||||
| Male | 25 (55.56%) | 14 (56.00%) | 11 (44.00%) | 0.460 |
| Female | 20 (44.44%) | 10 (50.00%) | 6 (50.00%) | |
| Age | ||||
| ≤50 | 28 (62.22%) | 16 (57.14%) | 12 (42.86%) | 0.363 |
| >50 | 17 (37.78%) | 8 (47.06%) | 9 (52.94%) | |
| Race | ||||
| Chinese | 35 (77.78%) | 18 (51.43%) | 17 (48.57%) | 0.454 |
| Non-Chinese | 10 (22.22%) | 6 (60.00%) | 4 (40.00%) | |
| Side | ||||
| Unilateral | 41 (91.11%) | 23 (56.10%) | 18 (43.90%) | 0.254 |
| Bilateral | 4 (8.89%) | 1 (25.00%) | 3 (75.00%) | |
| Tumour location | ||||
| Adrenal gland | 37 (82.22%) | 24 (64.86%) | 13 (35.14%) | 0.001 |
| Carotid body | 8 (17.78%) | - | 8 (100%) | |
| Tumour size | ||||
| <50 mm | 17 (37.78%) | 7 (41.2%) | 10 (58.8%) | 0.167 |
| ≥50 mm | 28 (62.22%) | 17 (60.71%) | 11 (39.29%) | |
| Tumour weight * | ||||
| ≤50 gm | 6 (35.29%) | 3 (50.0%) | 3 (50.0%) | 0.627 |
| >50 gm | 11 (64.71%) | 5 (45.45%) | 6 (54.55%) | |
| Tumour types | ||||
| Non-metastasizing | 36 (80.0%) | 22 (61.11%) | 14 (38.89%) | 0.057 |
| Metastasizing | 9 (20.0%) | 2 (22.22%) | 7 (77.78%) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Islam, F.; Pillai, S.; Gopalan, V.; Lam, A.K.-Y. Identification of Novel Mutations and Expressions of EPAS1 in Phaeochromocytomas and Paragangliomas. Genes 2020, 11, 1254. https://doi.org/10.3390/genes11111254
Islam F, Pillai S, Gopalan V, Lam AK-Y. Identification of Novel Mutations and Expressions of EPAS1 in Phaeochromocytomas and Paragangliomas. Genes. 2020; 11(11):1254. https://doi.org/10.3390/genes11111254
Chicago/Turabian StyleIslam, Farhadul, Suja Pillai, Vinod Gopalan, and Alfred King-Yin Lam. 2020. "Identification of Novel Mutations and Expressions of EPAS1 in Phaeochromocytomas and Paragangliomas" Genes 11, no. 11: 1254. https://doi.org/10.3390/genes11111254