Existence of Bov-B LINE Retrotransposons in Snake Lineages Reveals Recent Multiple Horizontal Gene Transfers with Copy Number Variation


 ,
,  ,
, 
Abstract
:1. Introduction
2. Material and Methods
2.1. Specimen Collection and DNA Extraction
2.2. Polymerase Chain Reaction Amplification and Molecular Cloning of Bov-B LINE
2.3. Survey of Bov-B LINE Copies in Publicly Released Snake Genome Assemblies
2.4. Sequence Analysis
2.5. Divergence Time Estimation and Mutation Rate between Bov-B LINE, COI, and BDNF
2.6. Quantification of Variation in Bov-B LINE Copy Number Based on Quantitative Real-Time Polymerase Chain Reaction (qPCR)
3. Results
3.1. Characterization of Bov-B LINE
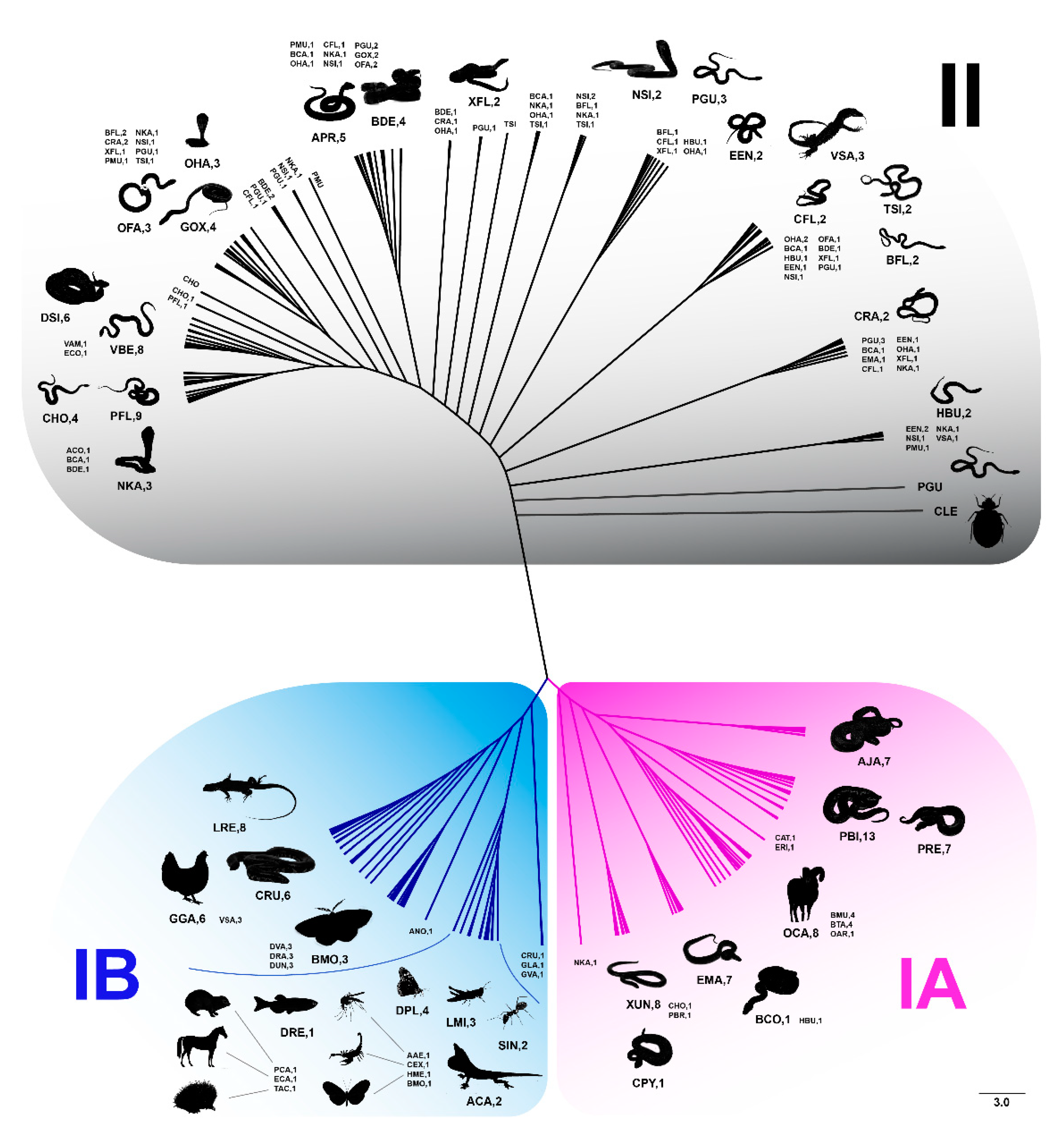
3.2. Sequence Variability of Bov-B LINE within and between Snake Species
3.3. Sequence Variability within and between Bov-B LINE Groups
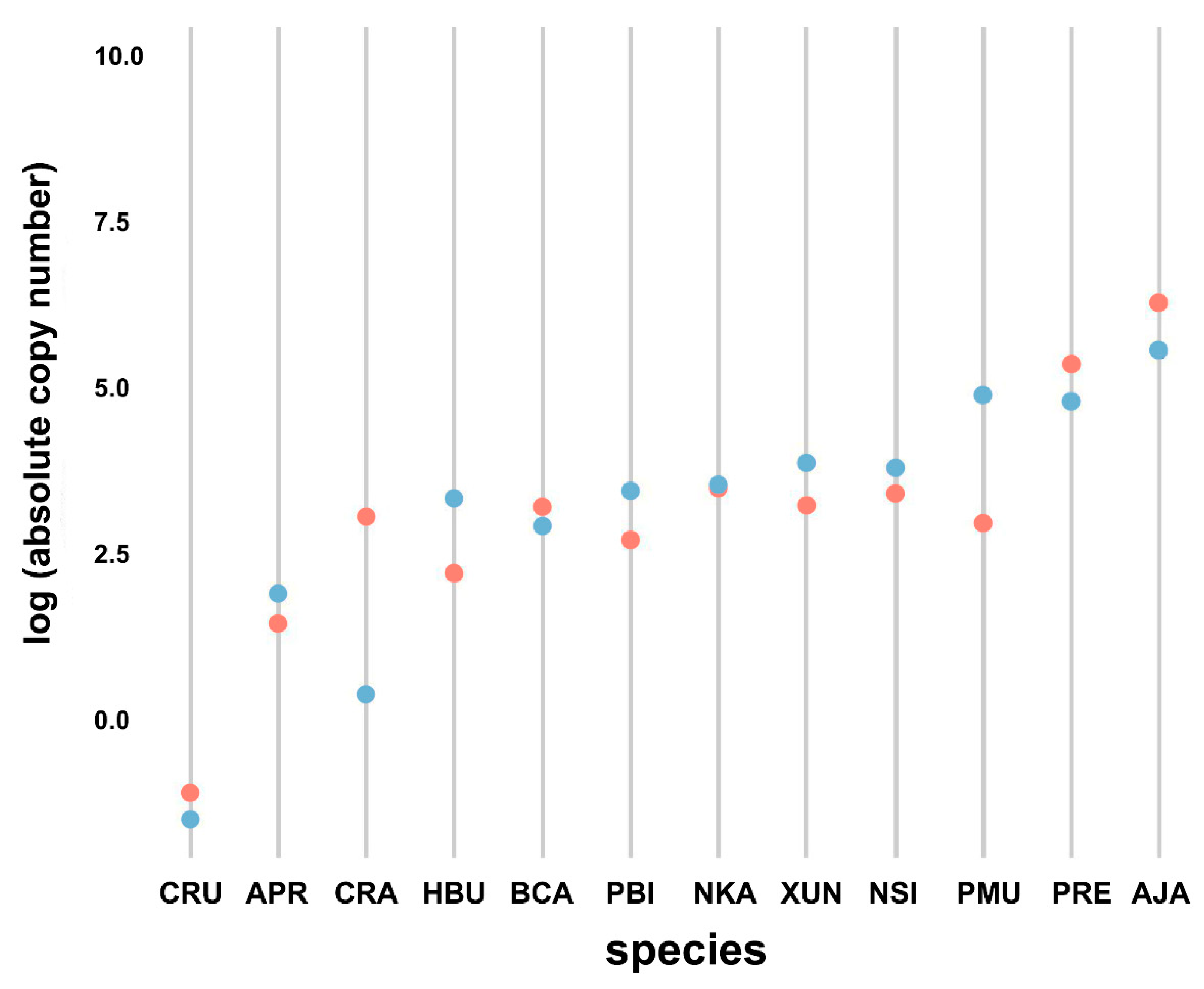
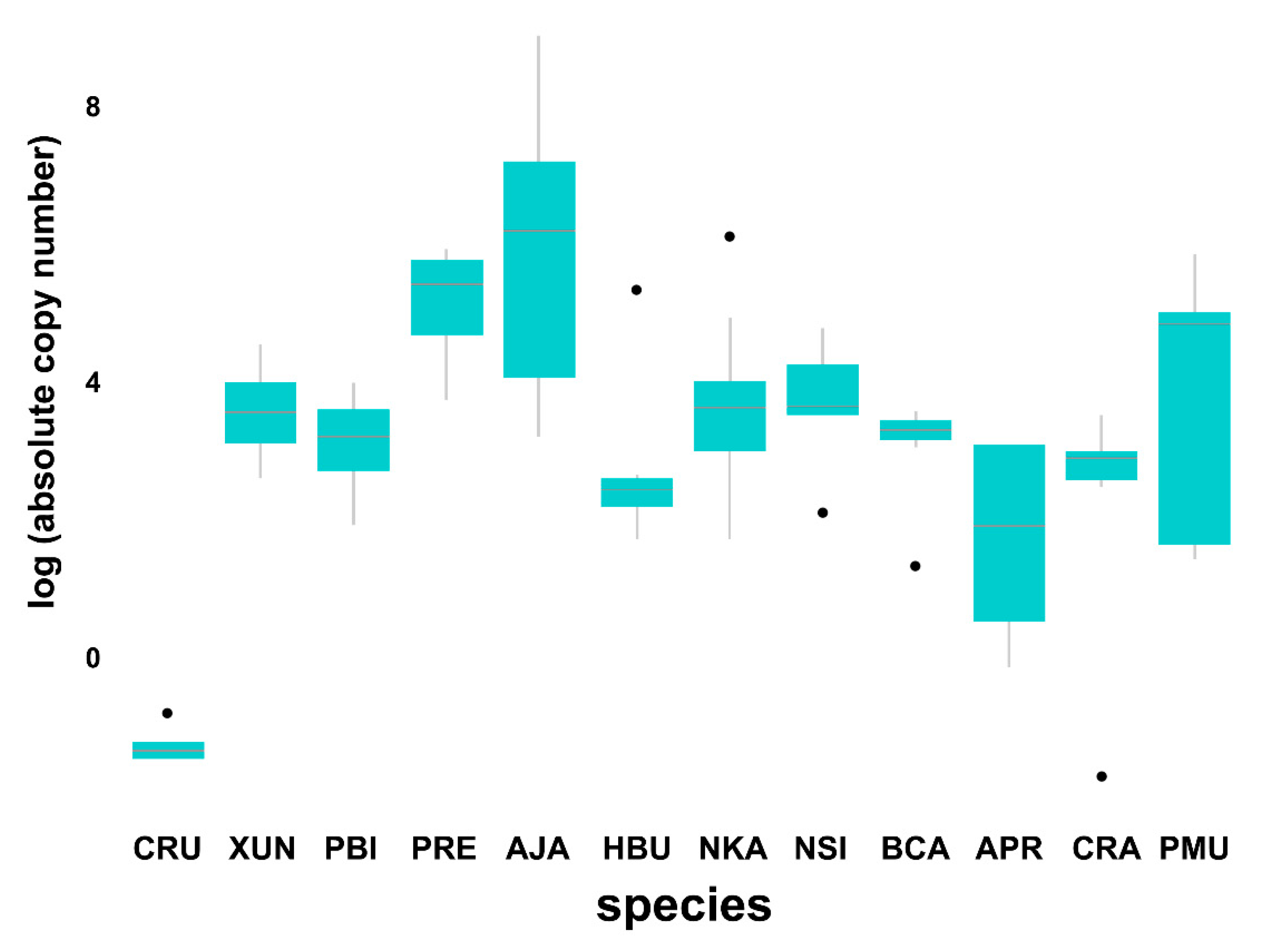
3.4. Comparison of Bov-B LINE Copy Numbers among Snake Species
3.5. Comparison of Bov-B LINE Copy Numbers between Males and Females
3.6. Divergence Time and Mutation Rate
4. Discussion
4.1. Evolutionary History of BOV-B LINE in Snakes
4.2. Independent Copy Number Variation among Snake Species
4.3. Bov-B LINE Copy Number Variation in Males and Females
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Kidwell, M.G. Review Transposable elements and the evolution of genome size in eukaryotes. Genetica 2002, 115, 49–63. [Google Scholar] [CrossRef] [PubMed]
- Chénais, B.; Caruso, A.; Hiard, S.; Casse, N. The impact of transposable elements on eukaryotic genomes: From genome size increase to genetic adaptation to stressful environments. Gene 2012, 509, 7–15. [Google Scholar] [CrossRef] [PubMed]
- Tolli, M.; Boissinot, S. The evolutionary dynamics of transposable elements in eukaryote genomes. Genome Dyn. 2012, 7, 68–91. [Google Scholar]
- Biémont, C.; Vieira, C. Genetics: Junk DNA as an evolutionary force. Nature 2006, 443, 521–524. [Google Scholar] [CrossRef]
- Lynch, M. The Origins of Genome Architecture; Sinauer Associates Inc.: Sunderland, MA, USA, 2007. [Google Scholar]
- Schaack, S.; Gilbert, C.; Feschotte, C. Promiscuous DNA: Horizontal transfer of transposable elements and why it matters for eukaryotic evolution. Trends Ecol. Evol. 2010, 25, 537–546. [Google Scholar] [CrossRef] [Green Version]
- Pace, J.K.; Gilbert, C.; Clark, M.S.; Feschotte, C. Repeated horizontal transfer of a DNA transposon in mammals and other tetrapods. Proc. Natl. Acad. Sci. USA 2008, 105, 17023–17028. [Google Scholar] [CrossRef] [Green Version]
- Gilbert, C.; Schaack, S.; Pace, J.K., II; Brindley, P.J.; Feschotte, C. A role for host parasite interactions in the horizontal transfer of transposons across phyla. Nature 2010, 464, 1347–1350. [Google Scholar] [CrossRef] [Green Version]
- Suntronpong, A.; Thapana, W.; Twilprawat, P.; Prakhongcheep, O.; Somyong, S.; Muangmai, N.; Peyachoknagul, S.; Srikulnath, K. Karyological characterization and identification of four repetitive element groups (the 18S–28S rRNA gene, telomeric sequences, microsatellite repeat motifs, Rex retroelements) of the Asian swamp eel (Monopterus albus). Comp. Cytogenet. 2017, 11, 435–462. [Google Scholar] [CrossRef] [Green Version]
- Tang, W.W.; Dietmann, S.; Irie, N.; Leitch, H.G.; Floros, V.I.; Bradshaw, C.R.; Hackett, J.A.; Chinnery, P.F.; Surani, M.A. A unique gene regulatory network resets the human germline epigenome for development. Cell 2015, 161, 1453–1467. [Google Scholar] [CrossRef] [Green Version]
- Oliver, K.R.; Greene, W.K. Mobile DNA and the TE-Thrust hypothesis: Supporting evidence from the primates. Mobile DNA 2011, 2, 8. [Google Scholar] [CrossRef] [Green Version]
- Schmitz, R.J.; Schultz, M.D.; Urich, M.A.; Nery, J.R.; Pelizzola, M.; Libiger, O.; Alix, A.; McCosh, R.B.; Chen, H.; Schork, N.J.; et al. Patterns of population epigenomic diversity. Nature 2013, 495, 193–198. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kidwell, M.G.; Lisch, D.R. Transposable elements and host genome evolution. Trends Ecol. Evol. 2000, 15, 95–99. [Google Scholar] [CrossRef]
- Feschotte, C. Transposable elements and the evolution of regulatory networks. Nat. Rev. Genet. 2008, 9, 397–405. [Google Scholar] [CrossRef] [Green Version]
- Piskurek, O.; Jackson, D.J. Transposable elements: From DNA parasites to architects of metazoan evolution. Genes 2012, 3, 409–422. [Google Scholar] [CrossRef] [Green Version]
- Buckley, R.M.; Adelson, D.L. Mammalian genome evolution as a result of epigenetic regulation of transposable elements. Biomol. Concepts 2014, 5, 183–194. [Google Scholar] [CrossRef]
- Walsh, A.M.; Kortschak, R.D.; Gardner, M.G.; Bertozzi, T.; Adelson, D.L. Widespread horizontal transfer of retrotransposons. Proc. Natl. Acad. Sci. USA 2013, 110, 1012–1016. [Google Scholar] [CrossRef] [Green Version]
- Richardson, S.R.; Doucet, A.J.; Kopera, H.C.; Moldovan, J.B.; Garcia-Perez, J.L.; Moran, J.V. The Influence of LINE-1 and SINE Retrotransposons on Mammalian Genomes. Microbiol. Spectr. 2015, 3, 1165–1208. [Google Scholar]
- Ivancevic, A.M.; Kortschak, R.D.; Bertozzi, T.; Adelson, D.L. Horizontal transfer of BovB and L1 retrotransposons in eukaryotes. Genome Biol. 2018, 19, 85. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Szemraj, J.; Plucienniczak, G.; Jaworski, J.; Plucienniczak, A. Bovine Alu-like sequences mediate transposition of a new sitespecific retroelement. Gene 1995, 152, 261–264. [Google Scholar] [CrossRef]
- Modi, W.S.; Gallagher, D.S.; Womack, J.E. Evolutionary histories of highly repeated DNA families among the Artiodactyla (Mammalia). J. Mol. Evol. 1996, 42, 337–349. [Google Scholar] [CrossRef]
- Smit, A.F. The origin of interspersed repeats in the human genome. Curr. Opin. Genet. Dev. 1996, 6, 743–748. [Google Scholar] [CrossRef]
- Okada, N.; Hamada, M. The 3′ ends of tRNA-derived SINEs originated from the 3′ ends of LINEs: A new example from the bovine genome. J. Mol. Evol. 1997, 44, S52–S56. [Google Scholar] [CrossRef]
- Kordiš, D.; Gubenšek, F. Unusual horizontal transfer of a long interspersed nuclear element between distant vertebrate classes. Proc. Natl. Acad. Sci. USA 1998, 95, 10704–10709. [Google Scholar] [CrossRef] [Green Version]
- Castoe, T.A.; Hall, K.T.; Guibotsy Mboulas, M.L.; Gu, W.; de Koning, A.P.; Fox, S.E.; Poole, A.W.; Vemulapalli, V.; Daza, J.M.; Mockler, T.; et al. Discovery of highly divergent repeat landscapes in snake genomes using high throughput sequencing. Genome Biol. Evol. 2011, 3, 641–653. [Google Scholar] [CrossRef] [Green Version]
- Castoe, T.A.; de Koning, A.P.; Hall, K.T.; Card, D.C.; Schield, D.R.; Fujita, M.K.; Ruggiero, R.P.; Degner, J.F.; Daza, J.M.; Gu, W.; et al. The Burmese python genome reveals the molecular basis for extreme adaptation in snakes. Proc. Natl. Acad. Sci. USA 2013, 110, 20645–20650. [Google Scholar] [CrossRef] [Green Version]
- Vonk, F.J.; Casewell, N.R.; Henkel, C.V.; Heimberg, A.M.; Jansen, H.J.; McCleary, R.J.; Kerkkamp, H.M.; Vos, R.A.; Guerreiro, I.; Calvete, J.J.; et al. The king cobra genome reveals dynamic gene evolution and adaptation in the snake venom system. Proc. Natl. Acad. Sci. USA 2013, 110, 20651–20656. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gilbert, C.; Meik, J.M.; Dashevsky, D.; Card, D.C.; Castoe, T.A.; Schaack, S. Endogenous hepadnaviruses, bornaviruses and circoviruses in snakes. Proc. Biol. Sci. 2014, 281, 20141122. [Google Scholar] [CrossRef]
- Ullate-Agote, A.; Milinkovitch, M.C.; Tzika, A.C. The genome sequence of the corn snake (Pantherophis guttatus), a valuable resource for EvoDevo studies in squamates. Int. J. Dev. Biol. 2015, 58, 881–888. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sanders, W.S.; Arick, M.A., Jr.; Thrash, A.; Rhoads, D.; Pummill, J.; Beaupre, S.; Nanduri, B.; Perkins, A.; Peterson, D.G.; Steele, R.D. A Draft Genome Sequence of the Timber Rattlesnake, Crotalus horridus. Available online: https://www.ncbi.nlm.nih.gov/nuccore/LVCR00000000.1 (accessed on 17 May 2019).
- Shibata, H.; Chijiwa, T.; Oda-Ueda, N.; Nakamura, H.; Yamaguchi, K.; Hattori, S.; Matsubara, K.; Matsuda, Y.; Yamashita, A.; Isomoto, A.; et al. The habu genome reveals accelerated evolution of venom protein genes. Sci. Rep. 2018, 8, 11300. [Google Scholar] [CrossRef] [PubMed]
- Thongchum, R.; Singchat, W.; Laopichienpong, N.; Tawichasri, P.; Kraichak, E.; Prakhongcheep, O.; Sillapaprayoon, S.; Muangmai, N.; Baicharoen, S.; Suntrarachun, S.; et al. Diversity of PBI-DdeI satellite DNA in snakes correlates with rapid independent evolution and different functional roles. Sci. Rep. 2019, 9, 15459. [Google Scholar] [CrossRef] [Green Version]
- Perry, B.W.; Card, D.C.; McGlothlin, J.W.; Pasquesi, G.I.M.; Adams, R.H.; Schield, D.R.; Hales, N.R.; Corbin, A.B.; Demuth, J.P.; Hoffmann, F.G.; et al. Molecular Adaptations for Sensing and Securing Prey and Insight into Amniote Genome Diversity from the Garter Snake Genome. Genome Biol. Evol. 2018, 10, 2110–2129. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kordiš, D. Transposable elements in reptilian and avian (sauropsida) genomes. Cytogenet. Genome Res. 2009, 127, 94–111. [Google Scholar] [CrossRef] [PubMed]
- Mezzasalma, M.; Visone, V.; Petraccioli, A.; Odierna, G.; Capriglione, T.; Guarino, F.M. Non-random accumulation of LINE1-like sequences on differentiated snake W chromosomes. J. Zool. 2016, 300, 67–75. [Google Scholar] [CrossRef]
- Matsubara, K.; Nishida, C.; Matsuda, Y.; Kumazawa, Y. Sex chromosome evolution in snakes inferred from divergence patterns of two gametologous genes and chromosome distribution of sex chromosome linked repetitive sequences. Zool. Lett. 2016, 2, 19. [Google Scholar] [CrossRef] [Green Version]
- Singchat, W.; O’Connor, R.E.; Tawichasri, P.; Suntronpong, A.; Sillapaprayoon, S.; Suntrarachun, S.; Muangmai, N.; Baicharoen, S.; Peyachoknagul, S.; Chanhome, L.; et al. Chromosome map of the Siamese cobra: Did partial synteny of sex chromosomes in the amniote represent “a hypothetical ancestral super-sex chromosome” or random distribution? BMC Genom. 2018, 19, 939. [Google Scholar] [CrossRef]
- Singchat, W.; Sillapaprayoon, S.; Muangmai, N.; Baicharoen, S.; Indananda, C.; Duengkae, P.; Peyachoknagul, S.; O’Connor, R.E.; Griffin, D.K.; Srikulnath, K. Do sex chromosomes of snakes, monitor lizards, and iguanian lizards result from multiple fission of an “ancestral amniote super-sex chromosome”? Chromosome Res. 2020, 28, 209–228. [Google Scholar] [CrossRef]
- Singchat, W.; Ahmad, S.F.; Sillapaprayoon, S.; Muangmai, N.; Duengkae, P.; Peyachoknagul, S.; O’Connor, R.E.; Griffin, D.K.; Srikulnath, K. Partial Amniote Sex Chromosomal Linkage Homologies Shared on Snake W Sex Chromosomes Support the Ancestral Super-Sex Chromosome Evolution in Amniotes. Front. Genet. 2020, 11, 948. [Google Scholar] [CrossRef]
- Ahmad, S.F.; Singchat, W.; Jehangir, M.; Panthum, T.; Srikulnath, K. Consequence of Paradigm Shift with Repeat Landscapes in Reptiles: Powerful Facilitators of Chromosomal Rearrangements for Diversity and Evolution (Running Title: Genomic Impact of Repeatson Chromosomal Dynamics in Reptiles). Genes 2020, 11, 827. [Google Scholar] [CrossRef]
- Ezaz, T.; Srikulnath, K.; Graves, J.A.M. Origin of amniote sex chromosomes: An ancestral super-sex chromosome, or common requirements? J Hered. 2017, 108, 94–105. [Google Scholar] [CrossRef]
- Kerkkamp, H.M.; Kini, R.M.; Pospelov, A.S.; Vonk, F.J.; Henkel, C.V.; Richardson, M.K. Snake Genome Sequencing: Results and Future Prospects. Toxins 2016, 8, 360. [Google Scholar] [CrossRef] [Green Version]
- Eickbush, T.H.; Malik, H.S. Origins and evolution of retrotransposons. Mobile DNA 2002, 2, 1111–1144. [Google Scholar]
- Kapitonov, V.V.; Tempel, S.; Jurka, J. Simple and fast classification of non-LTR retrotransposons based on phylogeny of their RT domain protein sequences. Gene 2009, 448, 207–213. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Laopichienpong, N.; Muangmai, N.; Chanhome, L.; Suntrarachun, S.; Twilprawat, P.; Peyachoknagul, S.; Srikulnath, K. Evolutionary dynamics of the gametologous CTNNB1 gene on the Z and W chromosomes of snakes. J. Hered. 2017, 108, 142–151. [Google Scholar] [PubMed]
- Laopichienpong, N.; Tawichasri, P.; Chanhome, L.; Phatcharakullawarawat, R.; Singchat, W.; Kantachumpoo, A.; Muangmai, N.; Suntrarachun, S.; Matsubara, K.; Peyachoknagul, S.; et al. A novel method of caenophidian snake sex identification using molecular markers based on two gametologous genes. Ecol. Evol. 2017, 7, 4661–4669. [Google Scholar] [CrossRef]
- Tawichasri, P.; Laopichienpong, N.; Chanhome, L.; Phatcharakullawarawat, R.; Singchat, W.; Koomgun, T.; Prasongmaneerut, T.; Rerkamnuaychoke, W.; Sillapaprayoon, S.; Muangmai, N.; et al. Using blood and non-invasive shed skin samples to identify sex of caenophidian snakes based on multiplex PCR assay. Zool. Anz. 2017, 271, 6–14. [Google Scholar] [CrossRef]
- Supikamolseni, A.; Ngaoburanawit, N.; Sumontha, M.; Chanhome, L.; Suntrarachun, S.; Peyachoknagul, S.; Srikulnath, K. Molecular barcoding of venomous snakes and species-specific multiplex PCR assay to identify snake groups for which antivenom is available in Thailand. Genet. Mol. Res. 2015, 14, 13981–13997. [Google Scholar] [CrossRef]
- Godakova, S.A.; Sevast’yanova, G.A.; Semyenova, S.K. Structure and distribution of the retrotransposon Bov-B LINE. Mol. Gen. Mikrobiol. Virusol. 2016, 34, 9–12. [Google Scholar] [CrossRef]
- Jurka, J.; Kapitonov, V.V.; Pavlicek, A.; Klonowski, P.; Kohany, O.; Walichiewicz, J. Repbase Update, a database of eukaryotic repetitive elements. Cytogenet. Genome Res. 2005, 110, 462–467. [Google Scholar] [CrossRef]
- Federhen, S. The NCBI Taxonomy database. Nucleic Acids Res. 2012, 40, D136–D143. [Google Scholar] [CrossRef] [Green Version]
- Edgar, R.C. MUSCLE: Multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 2004, 32, 1792–1797. [Google Scholar] [CrossRef] [Green Version]
- Kumar, S.; Stecher, G.; Li, M.; Knyaz, C.; Tamura, K. MEGA X: Molecular Evolutionary Genetics Analysis across computing platforms. Mol. Biol. Evol. 2018, 35, 1547–1549. [Google Scholar] [CrossRef]
- Nguyen, L.T.; Schmidt, H.A.; von Haeseler, A.; Minh, B.Q. IQ-TREE: A fast and effective stochastic algorithm for estimating maximum likelihood phylogenies. Mol. Biol. Evol. 2015, 32, 268–274. [Google Scholar] [CrossRef] [PubMed]
- Trifinopoulos, J.; Nguyen, L.T.; von Haeseler, A.; Minh, B.Q. W-IQ-TREE: A fast online phylogenetic tool for maximum likelihood analysis. Nucleic Acids Res. 2016, 44, W232–W235. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bao, W.; Kojima, K.K.; Kohany, O. Repbase Update, a database of repetitive elements in eukaryotic genomes. Mob. DNA 2015, 6, 11. [Google Scholar] [CrossRef] [Green Version]
- Clark, K.; Karsch-Mizrachi, I.; Lipman, D.J.; Ostell, J.; Sayers, E.W. GenBank. Nucleic Acids Res. 2016, 44, D67–D72. [Google Scholar] [CrossRef] [Green Version]
- Rozas, J.; Ferrer-Mata, A.; Sánchez-DelBarrio, J.C.; Guirao-Rico, S.; Librado, P.; Ramos-Onsins, S.E.; Sánchez-Gracia, A. DnaSP 6: DNA Sequence Polymorphism Analysis of Large Datasets. Mol. Biol. Evol. 2017, 34, 3299–3302. [Google Scholar] [CrossRef]
- R Core Team. R: A Language and Environment for Statistical Computing; R Foundation for Statistical Computing: Vienna, Austria, 2019; Available online: https://www.r-project.org/ (accessed on 5 February 2019).
- Blaser, N. Rdist: Calculate Pairwise Distances; R Package Version 0.0.3. 2018. Available online: https://CRAN.R-project.org/package=rdist (accessed on 5 February 2019).
- Bauer, D.F. Constructing confidence sets using rank statistics. J Am. Stat. Assoc. 1972, 67, 687–690. [Google Scholar] [CrossRef]
- Yandell, B.S. Practical Data Analysis for Designed Experiments; CRC Press: Boca Raton, FL, USA, 1997; Volume 39. [Google Scholar]
- Excoffier, L.; Smouse, P.E.; Quattro, J.M. Analysis of molecular variance inferred from metric distance among DNA haplotypes: Application to human mitochondrial DNA restriction data. Genetics 1992, 131, 479–491. [Google Scholar]
- Schneider, S.; Roessli, D.; Excoffier, L. ARLEQUIN: A Software for Population Genetics Data Analysis, version 2.000; University of Geneva, Genetics and Biometry Laboratory: Geneva, Switzerland, 2000. [Google Scholar]
- Hall, T.A. BioEdit: A user-friendly biological sequence alignment editor and analysis program for windows 95/98/NT. Nucleic Acids Symp. Ser. 1999, 41, 95–98. [Google Scholar]
- Nei, M.; Gojobori, T. Simple methods for estimating the numbers of synonymous and nonsynonymous nucleotide substitutions. Mol. Biol. Evol. 1986, 3, 418–426. [Google Scholar]
- Jukes, T.H.; Cantor, C.R. Evolution of Protein Molecules; Academic Press: New York, NY, USA, 1969; pp. 21–132. [Google Scholar]
- Makowsky, R.; Marshall, J.C., Jr.; McVay, J.; Chippindale, P.T.; Rissler, L.J. Phylogeographic analysis and environmental niche modeling of the plain-bellied watersnake (Nerodia erythrogaster) reveals low levels of genetic and ecological differentiation. Mol. Phylogenet. Evol. 2010, 55, 985–995. [Google Scholar] [CrossRef] [Green Version]
- Drummond, A.J.; Suchard, M.A.; Xie, D.; Rambaut, A. Bayesian phylogenetics with BEAUti and the BEAST 1.7. Mol. Biol. Evol. 2012, 29, 1969–1973. [Google Scholar] [CrossRef] [Green Version]
- Vidal, N.; Rage, J.C.; Couloux, A.; Hedges, S.B. Snake (Serpentes). In The Time Tree of Life; Hedges, S.B., Kumar, S., Eds.; Oxford University Press: New York, NY, USA, 2009; pp. 390–397. [Google Scholar]
- Rambaut, A.; Suchard, M.A.; Xie, D.; Drummond, A.J. Tracer v1.5. 2014. Available online: http://beast.bio.ed.ac.uk/Tracer (accessed on 5 February 2019).
- Rambaut, A. FigTree Tree Figure Drawing Tool, Version 1.3.1. 2009. Available online: http://tree.bio.ed.ac.uk/software/figtree/ (accessed on 5 February 2019).
- Huemer, P.; Mutanen, M.; Sefc, K.M.; Hebert, P.D.N. Testing DNA Barcode Performance in 1000 Species of European Lepidoptera: Large Geographic Distances Have Small Genetic Impacts. PLoS ONE 2014, 9, e115774. [Google Scholar] [CrossRef] [Green Version]
- DiDomenico, A.; Hedin, M. New species in the Sitalcina sura species group (Opiliones, Laniatores, Phalangodidae), with evidence for a biogeographic link between California desert canyons and Arizona sky islands. ZooKeys 2016, 586, 1–36. [Google Scholar]
- Chaves, R.; Ferreira, D.; Mendes-da-Silva, A.; Meles, S.; Adega, F. FA-SAT Is an Old Satellite DNA Frozen in Several Bilateria Genomes. Genome Biol. Evol. 2017, 9, 3073–3087. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schultheiss, O.C.; Stanton, S.J. Assessment of salivary hormones. In Methods in Social Neuroscience; Harmon-Jones, E., Beer, J.S., Eds.; Guilford Press: New York, NY, USA, 2009. [Google Scholar]
- Hanneman, S.K.; Cox, C.D.; Green, K.E.; Kang, D.H. Estimating intra- and inter-assay variability in salivary cortisol. Biol. Res. Nurs. 2011, 13, 243–250. [Google Scholar] [CrossRef] [PubMed]
- Singchat, W.; Kraichak, E.; Tawichasri, P.; Tawan, T.; Suntronpong, A.; Sillapaprayoon, S.; Phatcharakullawarawat, R.; Muangmai, N.; Suntrarachun, S.; Baicharoen, S.; et al. Dynamics of telomere length in captive Siamese cobra (Naja kaouthia) related to age and sex. Ecol. Evol. 2019, 9, 6366–6377. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zupunski, V.; Gubensek, F.; Kordis, D. Evolutionary dynamics and evolutionary history in the RTE clade of non-LTR retrotransposons. Mol. Biol. Evol. 2001, 18, 1849–1863. [Google Scholar] [CrossRef] [Green Version]
- Pyron, R.A.; Burbrink, F.T.; Wiens, J.J. A phylogeny and revised classification of Squamata, including 4161 species of lizards and snakes. BMC Evol. Biol. 2013, 13, 93. [Google Scholar] [CrossRef] [Green Version]
- Laopichienpong, N.; Muangmai, N.; Supikamolseni, A.; Twilprawat, P.; Chanhome, L.; Suntrarachun, S.; Peyachoknagul, S.; Srikulnath, K. Assessment of snake DNA barcodes based onmitochondrial COI and Cytb genes revealed multiple putative cryptic species in Thailand. Gene 2016, 594, 238–247. [Google Scholar] [CrossRef]
- Singchat, W.; Areesirisuk, P.; Sillapaprayoon, S.; Muangmai, N.; Baicharoen, S.; Suntrarachun, S.; Chanhome, L.; Peyachoknagul, S.; Srikulnath, K. Complete mitochondrial genome of Siamese cobra (Naja kaouthia) determined using next-generation sequencing. Mitochondrial DNA B 2019, 4, 577–578. [Google Scholar] [CrossRef]
- Cutter, A.D. Multilocus patterns of polymorphism and selection across the X-chromosome of Caenorhabditis remanei. Genetics 2008, 178, 1661–1672. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- González, J.; Petrov, D.A. The adaptive role of transposable elements in the Drosophila genome. Gene 2009, 448, 124–133. [Google Scholar] [CrossRef] [Green Version]
- González, J.; Lenkov, K.; Lipatov, M.; Macpherson, J.M.; Petrov, D.A. High Rate of Recent Transposable Element–Induced Adaptation in Drosophila melanogaster. PLoS Biol. 2008, 6, e251. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Petrov, D.A.; Fiston-Lavier, A.S.; Lipatov, M.; Lenkov, K.; González, J. Population genomics of transposable elements in Drosophila melanogaster. Mol. Biol. Evol. 2011, 28, 1633–1644. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guio, L.; González, J. New insights on the evolution of genome content: Population dynamics of transposable elements in flies and humans. In Evolutionary Genomics; Anisimova, M., Ed.; Humana: New York, NY, USA, 2019; p. 1910. [Google Scholar]
- Silva, J.C.; Loreto, E.L.S.; Clark, J.B. Factors that affect the horizontal transfer of transposable elements. Curr. Issues Mol. Biol. 2004, 6, 57–71. [Google Scholar] [PubMed]
- Dewannieux, M.; Heidmann, T. L1-mediated retrotransposition of murine B1 and B2 SINEs recapitulated in cultured cells. J Mol. Biol. 2005, 349, 241–247. [Google Scholar] [CrossRef]
- Binkiene, R. Helminth Fauna of Shrews (Sorex spp.) in Lithuania. Acta Zool. Lit. 2006, 16, 241–245. [Google Scholar] [CrossRef]
- Dunemann, S.M.; Wasmuth, J.D. Horizontal transfer of a retrotransposon between parasitic nematodes and the common shrew. Mob. DNA 2019, 10, 24. [Google Scholar] [CrossRef] [Green Version]
- Lai, O.; Ho, D.; Glick, S.; Jagdeo, J. Bed bugs and possible transmission of human pathogens: A systematic review. Arch. Dermatol. Res. 2016, 308, 531–538. [Google Scholar] [CrossRef] [Green Version]
- Kroschel, J.; Mujica, N.; Okonya, J.; Alyokhin, A. Insect pests affecting potatoes in tropical, subtropical, and temperate regions. In The Potato Crop; Campos, H., Ortiz, O., Eds.; Springer: Berlin/Heidelberg, Germany, 2020. [Google Scholar]
- Barker, S.C.; Walker, A.R. Ticks of Australia. The species that infest domestic animals and humans. Zootaxa 2014, 3816, 1–144. [Google Scholar] [CrossRef] [PubMed]
- Adelson, D.L.; Raison, J.M.; Edgar, R.C. Characterization and distribution of retrotransposons and simple sequence repeats in the bovine genome. Proc. Natl. Acad. Sci. USA 2009, 106, 12855–12860. [Google Scholar] [CrossRef] [Green Version]
- Matsubara, K.; Tarui, H.; Toriba, M.; Yamada, K.; Nishida-Umehara, C.; Agata, K.; Matsuda, Y. Evidence for different origin of sex chromosomes in snakes, birds, and mammals and step-wise differentiation of snake sex chromosomes. Proc. Natl. Acad. Sci. USA 2006, 103, 18190–18195. [Google Scholar] [CrossRef] [Green Version]
- Rovatsos, M.; Johnson Pokorná, M.; Kratochvíl, L. Differentiation of sex chromosomes and karyotype characterisation in the dragonsnake Xenodermus javanicus (Squamata: Xenodermatidae). Cytogenet. Genome Res. 2015, 147, 48–54. [Google Scholar] [CrossRef] [PubMed]
- Tollis, M.; Boissinot, S. The transposable element profile of the anolis genome: How a lizard can provide insights into the evolution of vertebrate genome size and structure. Mob. Genet. Elements. 2011, 1, 107–111. [Google Scholar] [CrossRef] [Green Version]
- Caldwell, M.W.; Nydam, R.L.; Palci, A.; Apesteguía, S. The oldest known snakes from the middle Jurassic-Lower Cretaceous provide insights on snake evolution. Nat. Commun. 2015, 6, 5996. [Google Scholar] [CrossRef] [Green Version]

{kind=link}
{kind=link}
{kind=link}
| Species | Abbreviation | Superfamily | Family | Number of Individuals Used (Male + Female) |
|---|---|---|---|---|
| Cylindrophis ruffus | CRU | Henophidia | Cylindrophiidae | 2 + 2 |
| Epicrates maurus | EMA | Henophidia | Boidae | 1 + 1 |
| Xenopeltis unicolor | XUN | Henophidia | Xenopeltidae | 3 + 3 |
| Python bivittatus | PBI | Henophidia | Pythonidae | 2 + 2 |
| Python regius | PRE | Henophidia | Pythonidae | 2 + 2 |
| Acrochordus javanicus | AJA | Caenophidia | Acrochordidae | 3 + 3 |
| Daboia siamensis | DSI | Caenophidia | Viperidae | 4 + 2 |
| Homalopsis buccata | HBU | Caenophidia | Homalopsidae | 3 + 2 |
| Enhydris enhydris | EEN | Caenophidia | Homalopsidae | 1 + 1 |
| Ophiophagus hannah | OHA | Caenophidia | Elapidae | 1 + 1 |
| Naja kaouthia | NKA | Caenophidia | Elapidae | 3 + 3 |
| Naja siamensis | NSI | Caenophidia | Elapidae | 3 + 2 |
| Bungarus candidus | BCA | Caenophidia | Elapidae | 4 + 4 |
| Bungarus flaviceps | BFL | Caenophidia | Elapidae | 1 + 1 |
| Oligodon fasciolatus | OFA | Caenophidia | Colubridae | 1 + 1 |
| Ahaetulla prasina | APR | Caenophidia | Colubridae | 2 + 2 |
| Boiga dendrophila | BDE | Caenophidia | Colubridae | 1 + 1 |
| Gonyosoma oxycephalum | GOX | Caenophidia | Colubridae | 1 + 1 |
| Coelognathus flavolineatus | CFL | Caenophidia | Colubridae | 1 + 1 |
| Coelognathus radiatus | CRA | Caenophidia | Colubridae | 2 + 4 |
| Xenochrophis flavipunctatus | XFL | Caenophidia | Colubridae | 1 + 1 |
| Ptyas mucosa | PMU | Caenophidia | Colubridae | 2 + 3 |
| Pantherophis guttatus | PGU | Caenophidia | Colubridae | 1 + 1 |
| Varanus salvator | VSA | - | Varanidae | 1 + 1 |
| Leiolepis reevesii | LRE | - | Agamidae | 1 + 1 |
| Gallus gallus | GGA | - | Phasianidae | 1 + 1 |
| Species | Family | Accession No. | Percentage of Bov-B in Genome |
|---|---|---|---|
| Python bivittatus | Pythonidae | AEQU00000000.2 | 0.00115 |
| Protobothrops flavoviridis | Viperidae | BFFQ00000000.1 | 0.01406 |
| Crotalus Pyrrhus | Viperidae | JPMF00000000.1 | 0.00003 |
| Vipera berus beru | Viperidae | JTGP00000000.1 | 0.01983 |
| Crotalus horridus | Viperidae | LVCR00000000.1 | 0.00137 |
| Ophiophagus hannah | Elapidae | AZIM00000000.1 | 0.00307 |
| Naja kaouthia | Elapidae | PRJNA506318 | 0.00276 |
| Pantherophis guttatus | Colubridae | JTLQ00000000.1 | 0.00121 |
| Thamnophis sirtalis | Colubridae | LFLD00000000.1 | 0.00093 |
| Species | Length (bp) | n | %AT | Nucleotide Diversity (π) * | Accession Number |
|---|---|---|---|---|---|
| Cylindrophis ruffus | 375–379 | 6 | 56.04 | 0.07 ± 0.02 | LC365540-LC365545 |
| Epicrates maurus | 332–380 | 10 | 56.36 | 0.10 ± 0.02 | LC365530-LC365539 |
| Xenopeltis unicolor | 375–381 | 8 | 59.26 | 0.10 ± 0.01 | LC365508-LC365515 |
| Python bivittatus | 377–379 | 7 | 57.25 | 0.04 ± 0.00 | LC365523-LC365529 |
| Python regius | 379 | 7 | 56.84 | 0.04 ± 0.01 | LC365516-LC365522 |
| Acrochordus javanicus | 375–379 | 7 | 58.78 | 0.07 ± 0.01 | LC365546-LC365552 |
| Daboia siamensis | 338–379 | 6 | 59.58 | 0.08 ± 0.01 | LC365592-LC365597 |
| Homalopsis buccata | 364–379 | 5 | 59.57 | 0.13 ± 0.01 | LC365610-LC365614 |
| Enhydris enhydris | 369–380 | 6 | 60.65 | 0.12 ± 0.01 | LC365598-LC365603 |
| Ophiophagus hannah | 369–380 | 4 | 59.59 | 0.08 ± 0.01 | LC365640-LC365643 |
| Naja kaouthia | 371–380 | 5 | 59.91 | 0.07 ± 0.01 | LC365617-LC365621 |
| Naja siamensis | 345–379 | 9 | 59.96 | 0.12 ± 0.01 | LC365623-LC365631 |
| Bungarus candidus | 342–379 | 5 | 59.96 | 0.11 ± 0.01 | LC365559-LC365563 |
| Bungarus flaviceps | 369–377 | 6 | 60.79 | 0.14 ± 0.01 | LC365633-LC365638 |
| Oligodon fasciolatus | 366–385 | 6 | 60.91 | 0.11 ± 0.01 | LC365574-LC365579 |
| Ahaetulla prasina | 348–379 | 5 | 59.66 | 0.07 ± 0.01 | LC365554-LC365558 |
| Boiga dendrophila | 366–381 | 8 | 59.73 | 0.10 ± 0.01 | LC365565-LC365572 |
| Gonyosoma oxycephalum | 363–379 | 6 | 60.91 | 0.10 ± 0.01 | LC365604-LC365609 |
| Coelognathus flavolineatus | 370–381 | 6 | 59.33 | 0.11 ± 0.01 | LC365586-LC365590 |
| Coelognathus radiatus | 369–379 | 6 | 59.56 | 0.09 ± 0.01 | LC384853-LC384858 |
| Xenochrophis flavipunctatus | 366–381 | 5 | 60.96 | 0.13 ± 0.02 | LC365651-LC365655 |
| Ptyas mucosa | 378–379 | 4 | 59.75 | 0.10 ± 0.01 | LC365645-LC365648 |
| Pantherophis guttatus | 359–385 | 8 | 59.52 | 0.08 ± 0.01 | LC384860-LC384867 |
| Varanus salvator | 362–379 | 7 | 57.49 | 0.15 ± 0.02 | LC365672-LC365678 |
| Leiolepis reevesii | 379 | 7 | 55.72 | 0.07 ± 0.01 | LC365663-LC365669 |
| Gallus gallus | 372–379 | 6 | 55.98 | 0.04 ± 0.01 | LC365657-LC365662 |
| Group | n | Nucleotide Diversity (π) |
|---|---|---|
| I | 55 | 0.11 ± 0.01 |
| II | 153 | 0.09 ± 0.00 |
| IA | 49 | 0.10 ± 0.01 |
| IB | 6 | 0.07 ± 0.02 |
| Group | p-Distance |
|---|---|
| I vs. II | 0.09 ± 0.01 |
| IA vs. IB | 0.09 ± 0.02 |
| Group | n | Nucleotide Diversity (π) |
|---|---|---|
| I | 124 | 0.22 ± 0.02 |
| II | 158 | 0.09 ± 0.00 |
| Group | p-Distance |
|---|---|
| I vs. II | 0.18 ± 0.04 |
| IA vs. IB | 0.24 ± 0.04 |
| IA vs. II | 0.12 ± 0.04 |
| IB vs. II | 0.24 ± 0.04 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Puinongpo, W.; Singchat, W.; Petpradub, S.; Kraichak, E.; Nunome, M.; Laopichienpong, N.; Thongchum, R.; Intarasorn, T.; Sillapaprayoon, S.; Indananda, C.; et al. Existence of Bov-B LINE Retrotransposons in Snake Lineages Reveals Recent Multiple Horizontal Gene Transfers with Copy Number Variation. Genes 2020, 11, 1241. https://doi.org/10.3390/genes11111241
Puinongpo W, Singchat W, Petpradub S, Kraichak E, Nunome M, Laopichienpong N, Thongchum R, Intarasorn T, Sillapaprayoon S, Indananda C, et al. Existence of Bov-B LINE Retrotransposons in Snake Lineages Reveals Recent Multiple Horizontal Gene Transfers with Copy Number Variation. Genes. 2020; 11(11):1241. https://doi.org/10.3390/genes11111241
Chicago/Turabian StylePuinongpo, Weerada, Worapong Singchat, Supaporn Petpradub, Ekaphan Kraichak, Mitsuo Nunome, Nararat Laopichienpong, Ratchaphol Thongchum, Thanphong Intarasorn, Siwapech Sillapaprayoon, Chantra Indananda, and et al. 2020. "Existence of Bov-B LINE Retrotransposons in Snake Lineages Reveals Recent Multiple Horizontal Gene Transfers with Copy Number Variation" Genes 11, no. 11: 1241. https://doi.org/10.3390/genes11111241
APA StylePuinongpo, W., Singchat, W., Petpradub, S., Kraichak, E., Nunome, M., Laopichienpong, N., Thongchum, R., Intarasorn, T., Sillapaprayoon, S., Indananda, C., Muangmai, N., Suntrarachun, S., Baicharoen, S., Chanhome, L., Peyachoknagul, S., & Srikulnath, K. (2020). Existence of Bov-B LINE Retrotransposons in Snake Lineages Reveals Recent Multiple Horizontal Gene Transfers with Copy Number Variation. Genes, 11(11), 1241. https://doi.org/10.3390/genes11111241