GJB2 and GJB6 Genetic Variant Curation in an Argentinean Non-Syndromic Hearing-Impaired Cohort

 ,
,  ,
, 
 and
and 
Abstract
1. Introduction
2. Materials and Methods
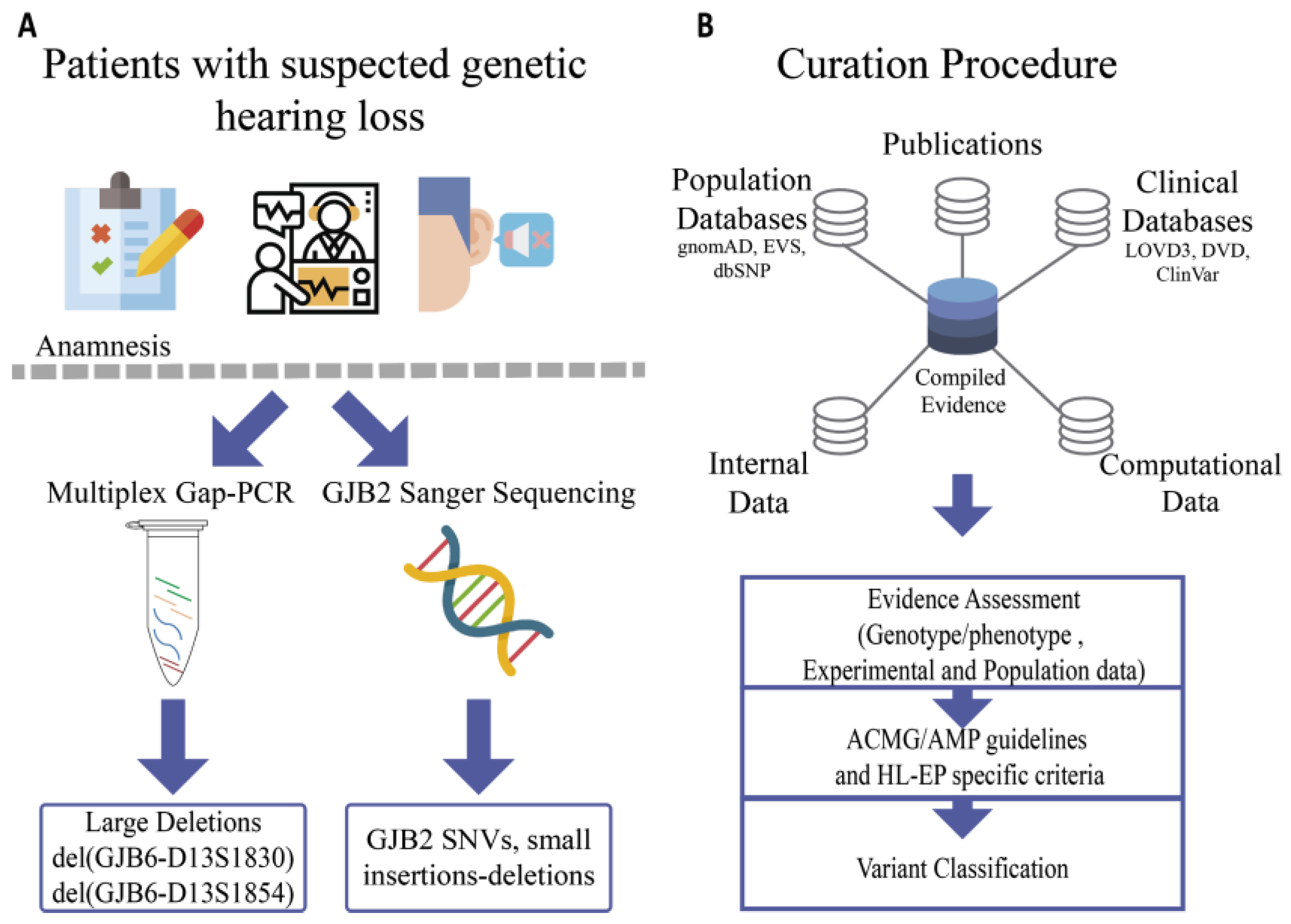
2.1. Part A: Identification of Variants in an Argentinean Cohort
2.1.1. Patients
2.1.2. Samples
2.1.3. GJB2/GJB6 Molecular Studies
2.1.4. Data Analysis
2.2. Part B: Curation of Variants
GJB2-GJB6 Variant Curation
3. Results
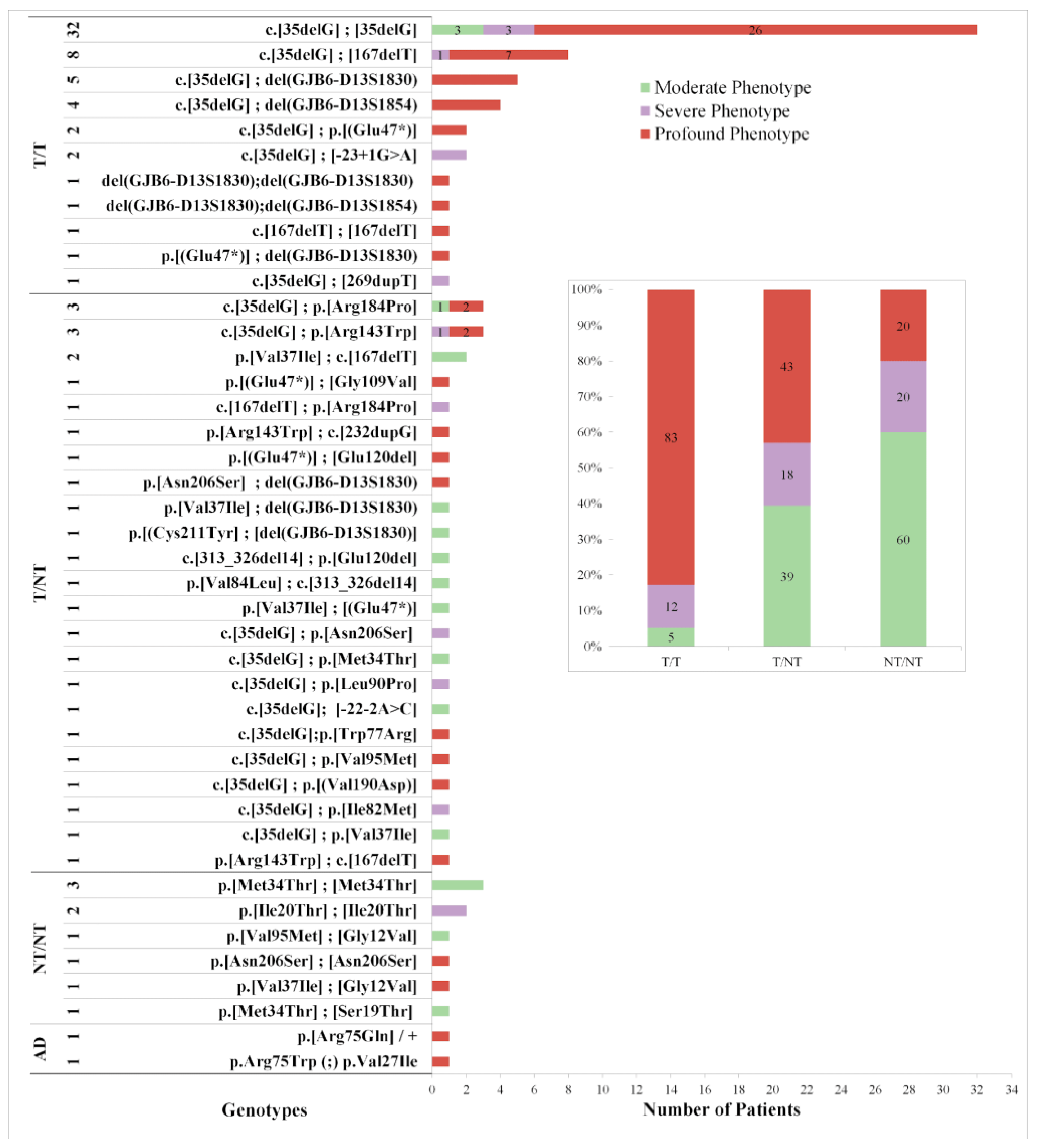
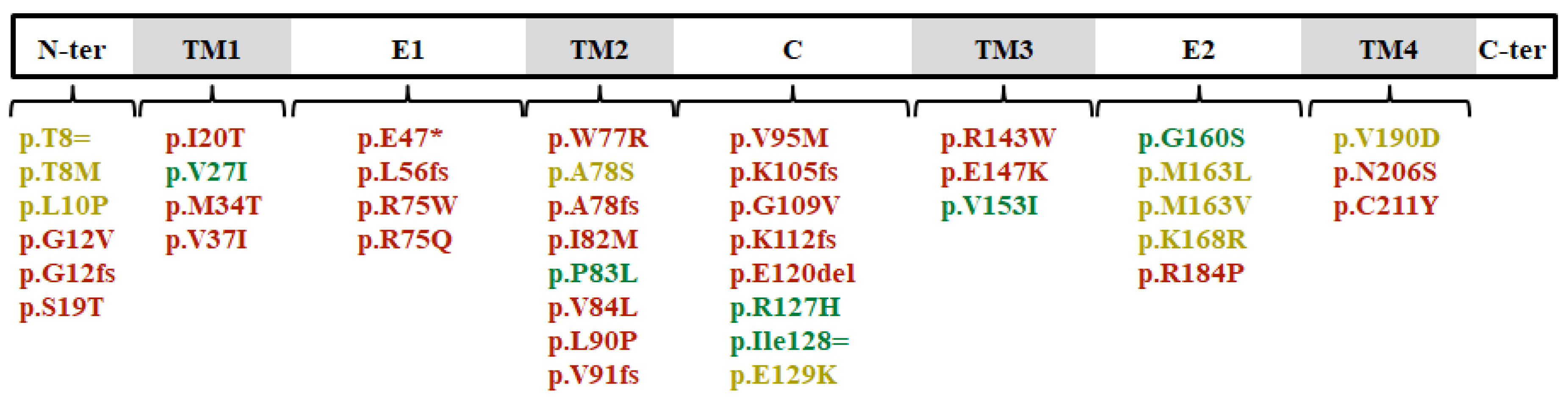
3.1. General Genetic Findings
3.2. Genotype-Phenotype Characterization
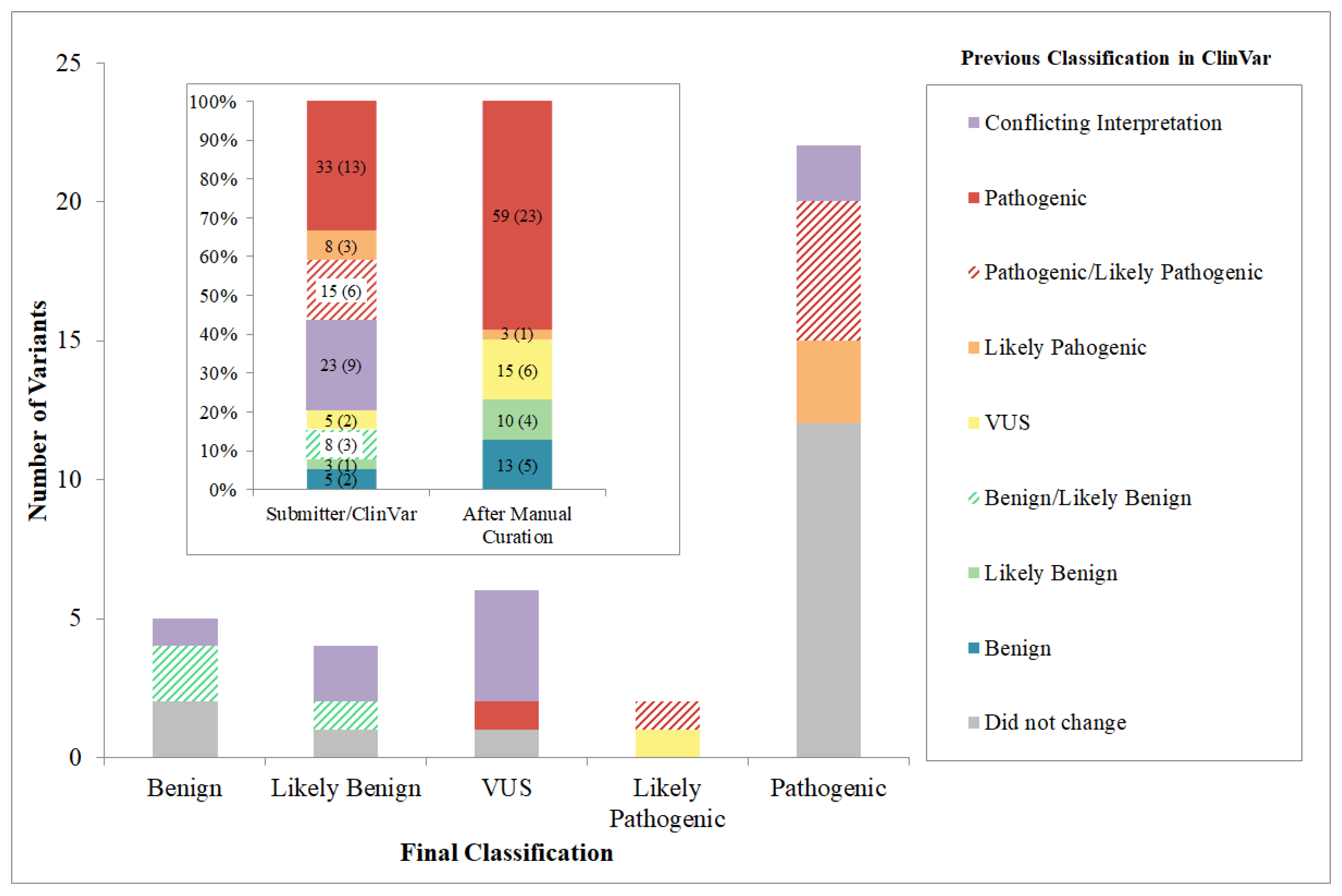
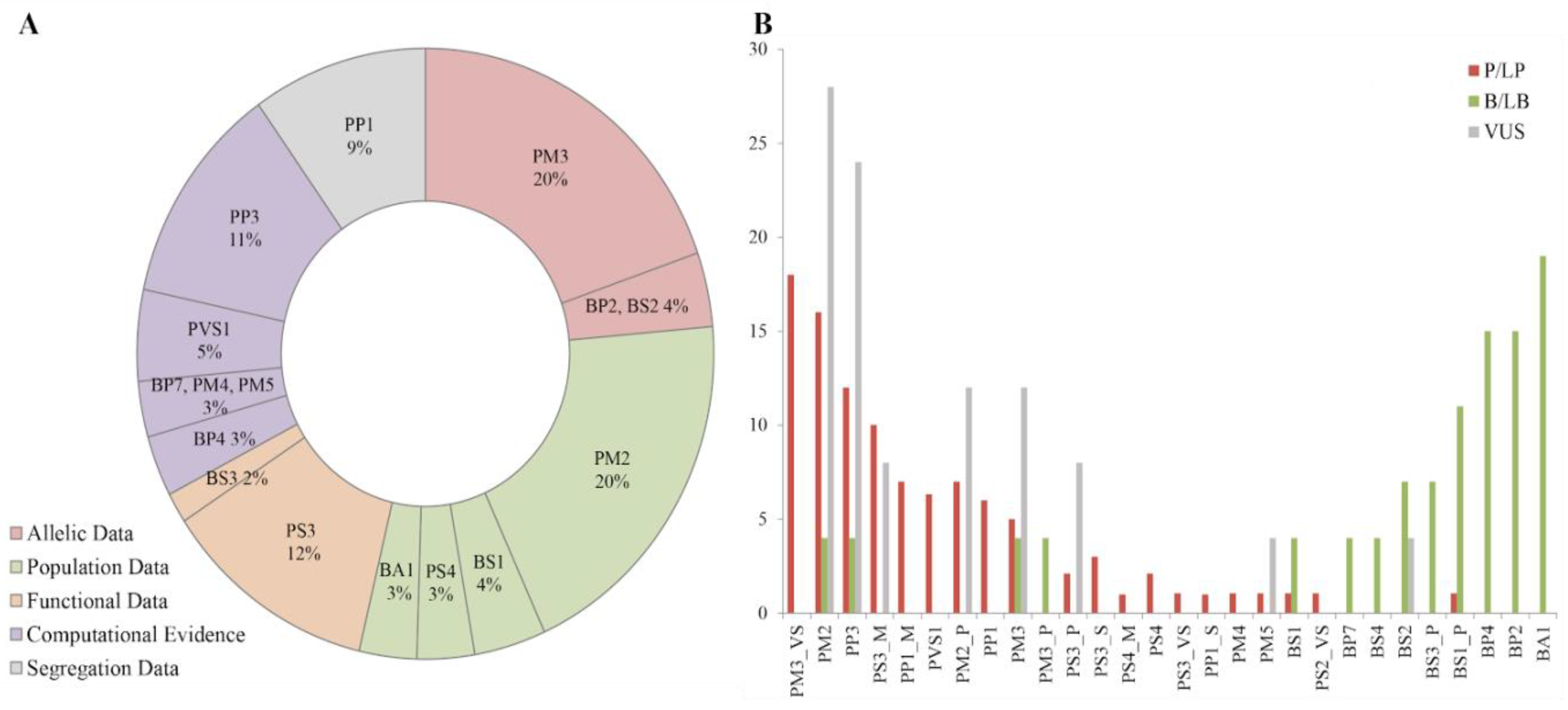
3.3. Variant Curation
4. Discussion
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Morton, C.C.; Nance, W.E. Newborn hearing screening—A silent revolution. N. Engl. J. Med. 2006, 354, 2151–2164. [Google Scholar] [CrossRef]
- Morton, N.E. Genetic Epidemiology of Hearing Impairment. Ann. N. Y. Acad. Sci. 1991, 630, 16–31. [Google Scholar] [CrossRef] [PubMed]
- Shearer, A.E.; Hildebrand, M.S.; Smith, R.J.H. Hereditary Hearing Loss and Deafness Overview. In GeneReviews; Adam, M.P., Ardinger, H.H., Pagon, R.A., Wallace, S.E., Bean, L.J.H., Stephens, K., Amemiya, A., Eds.; University of Washington, Seattle: Seattle, WA, USA, 1999. [Google Scholar]
- Van Camp, G.; Smith, R.J.H. Hereditary Hearing Loss Homepage. Available online: https://hereditaryhearingloss.org (accessed on 16 October 2020).
- Richardson, G.P.; de Monvel, J.B.; Petit, C. How the Genetics of Deafness Illuminates Auditory Physiology. Annu. Rev. Physiol. 2011, 73, 311–334. [Google Scholar] [CrossRef] [PubMed]
- Cama, E.; Melchionda, S.; Palladino, T.; Carella, M.; Santarelli, R.; Genovese, E.; Benettazzo, F.; Zelante, L.; Arslan, E. Hearing loss features in GJB2 biallelic mutations and GJB2/GJB6 digenic inheritance in a large Italian cohort. Int. J. Audiol. 2009, 48, 12–17. [Google Scholar] [CrossRef] [PubMed]
- Del Castillo, I.; Moreno-Pelayo, M.A.; Del Castillo, F.J.; Brownstein, Z.; Marlin, S.; Adina, Q.; Cockburn, D.J.; Pandya, A.; Siemering, K.R.; Chamberlin, G.P.; et al. Prevalence and evolutionary origins of the del(GJB6-D13S1830) mutation in the DFNB1 locus in hearing-impaired subjects: A multicenter study. Am. J. Hum. Genet. 2003, 73, 1452–1458. [Google Scholar] [CrossRef]
- Feldmann, D.; Denoyelle, F.; Chauvin, P.; Garabédian, E.-N.; Couderc, R.; Odent, S.; Joannard, A.; Schmerber, S.; Delobel, B.; Leman, J.; et al. Large deletion of the GJB6 gene in deaf patients heterozygous for the GJB2 gene mutation: Genotypic and phenotypic analysis. Am. J. Med. Genet. A 2004, 127A, 263–267. [Google Scholar] [CrossRef] [PubMed]
- Pampanos, A.; Economides, J.; Iliadou, V.; Neou, P.; Leotsakos, P.; Voyiatzis, N.; Eleftheriades, N.; Tsakanikos, M.; Antoniadi, T.; Hatzaki, A.; et al. Prevalence of GJB2 mutations in prelingual deafness in the Greek population. Int. J. Pediatr. Otorhinolaryngol. 2002, 65, 101–108. [Google Scholar] [CrossRef]
- del Castillo, I.; Villamar, M.; Moreno-Pelayo, M.A.; del Castillo, F.J.; Alvarez, A.; Tellería, D.; Menéndez, I.; Moreno, F. A deletion involving the connexin 30 gene in nonsyndromic hearing impairment. N. Engl. J. Med. 2002, 346, 243–249. [Google Scholar] [CrossRef]
- Hilgert, N.; Smith, R.J.H.; Van Camp, G. Forty-six genes causing nonsyndromic hearing impairment: Which ones should be analyzed in DNA diagnostics? Mutat. Res. 2009, 681, 189–196. [Google Scholar] [CrossRef]
- Rabionet, R.; Gasparini, P.; Estivill, X. Molecular genetics of hearing impairment due to mutations in gap junction genes encoding beta connexins. Hum. Mutat. 2000, 16, 190–202. [Google Scholar] [CrossRef]
- Meşe, G.; Richard, G.; White, T.W. Gap Junctions: Basic Structure and Function. J. Investig. Dermatol. 2007, 127, 2516–2524. [Google Scholar] [CrossRef] [PubMed]
- Bruzzone, R.; Veronesi, V.; Gomès, D.; Bicego, M.; Duval, N.; Marlin, S.; Petit, C.; D’Andrea, P.; White, T.W. Loss-of-function and residual channel activity of connexin26 mutations associated with non-syndromic deafness. FEBS Lett. 2003, 533, 79–88. [Google Scholar] [CrossRef]
- Palmada, M.; Schmalisch, K.; Böhmer, C.; Schug, N.; Pfister, M.; Lang, F.; Blin, N. Loss of function mutations of the GJB2 gene detected in patients with DFNB1-associated hearing impairment. Neurobiol. Dis. 2006, 22, 112–118. [Google Scholar] [CrossRef] [PubMed]
- White, T.W. Functional analysis of human Cx26 mutations associated with deafness. Brain Res. Rev. 2000, 32, 181–183. [Google Scholar] [CrossRef]
- Gasparini, P.; the Genetic Analysis Consortium of GJB235delG; Rabionet, R.; Barbujani, G.; Melchionda, S.; Petersen, M.; Brøndum-Nielsen, K.; Metspalu, A.; Oitmaa, E.; Pisano, M.; et al. High carrier frequency of the 35delG deafness mutation in European populations. Eur. J. Hum. Genet. 2000, 8, 19–23. [Google Scholar] [CrossRef]
- Cryns, K. A genotype-phenotype correlation for GJB2 (connexin 26) deafness. J. Med. Genet. 2004, 41, 147–154. [Google Scholar] [CrossRef]
- Lucotte, G. High prevalences of carriers of the 35delG mutation of connexin 26 in the Mediterranean area. Int. J. Pediatri. Otorhinolaryngol. 2007, 71, 741–746. [Google Scholar] [CrossRef]
- Azaiez, H.; Booth, K.T.; Ephraim, S.S.; Crone, B.; Black-Ziegelbein, E.A.; Marini, R.J.; Eliot Shearer, A.; Sloan-Heggen, C.M.; Kolbe, D.; Casavant, T.; et al. Genomic Landscape and Mutational Signatures of Deafness-Associated Genes. Am. J. Hum. Genet. 2018, 103, 484–497. [Google Scholar] [CrossRef]
- del Castillo, F.J. A novel deletion involving the connexin-30 gene, del(GJB6-d13s1854), found in trans with mutations in the GJB2 gene (connexin-26) in subjects with DFNB1 non-syndromic hearing impairment. J. Med. Genet. 2005, 42, 588–594. [Google Scholar] [CrossRef]
- Wilch, E.; Azaiez, H.; Fisher, R.A.; Elfenbein, J.; Murgia, A.; Birkenhäger, R.; Bolz, H.; Da Silva-Costa, S.M.; Del Castillo, I.; Haaf, T.; et al. A novel DFNB1 deletion allele supports the existence of a distant cis-regulatory region that controls GJB2 and GJB6 expression. Clin. Genet. 2010, 78, 267–274. [Google Scholar] [CrossRef] [PubMed]
- Feldmann, D.; Le Maréchal, C.; Jonard, L.; Thierry, P.; Czajka, C.; Couderc, R.; Ferec, C.; Denoyelle, F.; Marlin, S.; Fellmann, F. A new large deletion in the DFNB1 locus causes nonsyndromic hearing loss. Eur. J. Med. Genet. 2009, 52, 195–200. [Google Scholar] [CrossRef]
- Bliznetz, E.A.; Makienko, O.N.; Okuneva, E.G.; Markova, T.G.; Polyakov, A.V. New recurrent large deletion, encompassing both GJB2 and GB6 genes, results in isolated sensorineural hearing impairment with autosomal recessive mode of inheritance. Russ. J. Genet. 2014, 50, 415–420. [Google Scholar] [CrossRef]
- Tayoun, A.N.A.; Abou Tayoun, A.N.; Mason-Suares, H.; Frisella, A.L.; Bowser, M.; Duffy, E.; Mahanta, L.; Funke, B.; Rehm, H.L.; Amr, S.S. Targeted Droplet-Digital PCR as a Tool for Novel Deletion Discovery at the DFNB1 Locus. Hum. Mutat. 2016, 37, 119–126. [Google Scholar] [CrossRef] [PubMed]
- Dalamón, V.; Lotersztein, V.; Béhèran, A.; Lipovsek, M.; Diamante, F.; Pallares, N.; Francipane, L.; Frechtel, G.; Paoli, B.; Mansilla, E.; et al. GJB2 and GJB6 Genes: Molecular Study and Identification of Novel GJB2 Mutations in the Hearing-Impaired Argentinean Population. Audiol. Neurotol. 2010, 15, 194–202. [Google Scholar] [CrossRef] [PubMed]
- Dalamón, V.; Béhèran, A.; Diamante, F.; Pallares, N.; Diamante, V.; Elgoyhen, A.B. Prevalence of GJB2 mutations and the del(GJB6-D13S1830) in Argentinean non-syndromic deaf patients. Hear. Res. 2005, 207, 43–49. [Google Scholar] [CrossRef] [PubMed]
- Dalamón, V.; Florencia Wernert, M.; Lotersztein, V.; Craig, P.O.; Diamante, R.R.; Barteik, M.E.; Curet, C.; Paoli, B.; Mansilla, E.; Elgoyhen, A.B. Identification of four novel connexin 26 mutations in non-syndromic deaf patients: Genotype-phenotype analysis in moderate cases. Mol. Biol. Rep. 2013, 40, 6945–6955. [Google Scholar] [CrossRef] [PubMed]
- Gravina, L.P.; Foncuberta, M.E.; Estrada, R.C.; Barreiro, C.; Chertkoff, L. Carrier frequency of the 35delG and A1555G deafness mutations in the Argentinean population. Int. J. Pediatri. Otorhinolaryngol. 2007, 71, 639–643. [Google Scholar] [CrossRef]
- Gravina, L.P.; Foncuberta, M.E.; Prieto, M.E.; Garrido, J.; Barreiro, C.; Chertkoff, L. Prevalence of DFNB1 mutations in Argentinean children with non-syndromic deafness. Report of a novel mutation in GJB2. Int. J. Pediatr. Otorhinolaryngol. 2010, 74, 250–254. [Google Scholar] [CrossRef]
- Reynoso, R.A.; Hendl, S.; Barteik, M.E.; Curet, C.A.; Nicemboin, L.; Moreno Barral, J.; Rodríguez Ballesteros, M.; Del Castillo, I.; Moreno, F. Genetic study of hearing loss in families from Argentina. Rev. Fac. Cien. Med. Univ. Nac. Cordoba 2004, 61, 13–19. [Google Scholar]
- Smith, R.J.H.; Jones, M.K.N. Nonsyndromic Hearing Loss and Deafness, DFNB1. 28 September 1998 [Updated 18 August 2016]. In GeneReviews®; Adam, M.P., Ardinger, H.H., Pagon, R.A., Wallace, S.E., Bean, L.J.H., Stephens, K., Amemiya, A., Eds.; University of Washington, Seattle: Seattle, WA, USA, 1993. [Google Scholar] [PubMed]
- D’Andrea, P.; Veronesi, V.; Bicego, M.; Melchionda, S.; Zelante, L.; Di Iorio, E.; Bruzzone, R.; Gasparini, P. Hearing loss: Frequency and functional studies of the most common connexin26 alleles. Biochem. Biophys. Res. Commun. 2002, 296, 685–691. [Google Scholar] [CrossRef]
- Azaiez, H.; Parker Chamberlin, G.; Fischer, S.M.; Welp, C.L.; Prasad, S.D.; Thomas Taggart, R.; del Castillo, I.; Van Camp, G.; Smith, R.J.H. GJB2: The spectrum of deafness-causing allele variants and their phenotype. Hum. Mutat. 2004, 24, 305–311. [Google Scholar] [CrossRef]
- Green, G.E.; Scott, D.A.; McDonald, J.M.; Teagle, H.F.B.; Tomblin, B.J.; Spencer, L.J.; Woodworth, G.G.; Knutson, J.F.; Gantz, B.J.; Sheffield, V.C.; et al. Performance of cochlear implant recipients withGJB2-related deafness. Am. J. Med. Genet. 2002, 109, 167–170. [Google Scholar] [CrossRef] [PubMed]
- Bauer, P.W.; Geers, A.E.; Brenner, C.; Moog, J.S.; Smith, R.J.H. The Effect of GJB2 Allele Variants on Performance After Cochlear Implantation. Laryngoscope 2003, 113, 2135–2140. [Google Scholar] [CrossRef]
- Usami, S.-I.; Nishio, S.-Y.; Moteki, H.; Miyagawa, M.; Yoshimura, H. Cochlear Implantation from the Perspective of Genetic Background. Anat. Rec. 2020, 303, 563–593. [Google Scholar] [CrossRef]
- Amendola, L.M.; Jarvik, G.P.; Leo, M.C.; McLaughlin, H.M.; Akkari, Y.; Amaral, M.D.; Berg, J.S.; Biswas, S.; Bowling, K.M.; Conlin, L.K.; et al. Performance of ACMG-AMP Variant-Interpretation Guidelines among Nine Laboratories in the Clinical Sequencing Exploratory Research Consortium. Am. J. Hum. Genet. 2016, 99, 247. [Google Scholar] [CrossRef] [PubMed]
- Booth, K.T.; Kahrizi, K.; Babanejad, M.; Daghagh, H.; Bademci, G.; Arzhangi, S.; Zareabdollahi, D.; Duman, D.; El-Amraoui, A.; Tekin, M.; et al. Variants in CIB2 cause DFNB48 and not USH1J. Clin. Genet. 2018, 93, 812–821. [Google Scholar] [CrossRef]
- Harrison, S.M.; Dolinsky, J.S.; Knight Johnson, A.E.; Pesaran, T.; Azzariti, D.R.; Bale, S.; Chao, E.C.; Das, S.; Vincent, L.; Rehm, H.L. Clinical laboratories collaborate to resolve differences in variant interpretations submitted to ClinVar. Genet. Med. 2017, 19, 1096–1104. [Google Scholar] [CrossRef]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 2015, 17, 405–423. [Google Scholar] [CrossRef]
- Oza, A.M.; DiStefano, M.T.; Hemphill, S.E.; Cushman, B.J.; Grant, A.R.; Siegert, R.K.; Shen, J.; Chapin, A.; Boczek, N.J.; Schimmenti, L.A.; et al. Expert specification of the ACMG/AMP variant interpretation guidelines for genetic hearing loss. Hum. Mutat. 2018, 39, 1593–1613. [Google Scholar] [CrossRef]
- Murray, M.G.; Thompson, W.F. Rapid isolation of high molecular weight plant DNA. Nucleic Acids Res. 1980, 8, 4321–4326. [Google Scholar] [CrossRef]
- Sequence Assembly and Alignment Software-CodonCode. Available online: https://www.codoncode.com/ (accessed on 16 October 2020).
- Altschul, S.F.; Gish, W.; Miller, W.; Myers, E.W.; Lipman, D.J. Basic local alignment search tool. J. Mol. Biol. 1990, 215, 403–410. [Google Scholar] [CrossRef]
- Snoeckx, R.L.; Huygen, P.L.M.; Feldmann, D.; Marlin, S.; Denoyelle, F.; Waligora, J.; Mueller-Malesinska, M.; Pollak, A.; Ploski, R.; Murgia, A.; et al. GJB2 Mutations and Degree of Hearing Loss: A Multicenter Study. Am. J. Hum. Genet. 2005, 77, 945–957. [Google Scholar] [CrossRef] [PubMed]
- Shahin, H.; Walsh, T.; Sobe, T.; Lynch, E.; King, M.-C.; Avraham, K.B.; Kanaan, M. Genetics of congenital deafness in the Palestinian population: Multiple connexin 26 alleles with shared origins in the Middle East. Hum. Genet. 2002, 110, 284–289. [Google Scholar] [CrossRef] [PubMed]
- Del Castillo, F.J.; Del Castillo, I. DFNB1 Non-syndromic Hearing Impairment: Diversity of Mutations and Associated Phenotypes. Front. Mol. Neurosci. 2017, 10, 428. [Google Scholar] [CrossRef]
- Gandía, M.; Del Castillo, F.J.; Rodríguez-Álvarez, F.J.; Garrido, G.; Villamar, M.; Calderón, M.; Moreno-Pelayo, M.A.; Moreno, F.; del Castillo, I. A novel splice-site mutation in the GJB2 gene causing mild postlingual hearing impairment. PLoS ONE 2013, 8, e73566. [Google Scholar] [CrossRef] [PubMed]
- den Dunnen, J.T.; Dalgleish, R.; Maglott, D.R.; Hart, R.K.; Greenblatt, M.S.; McGowan-Jordan, J.; Roux, A.-F.; Smith, T.; Antonarakis, S.E.; Taschner, P.E.M. HGVS Recommendations for the Description of Sequence Variants: 2016 Update. Hum. Mutat. 2016, 37, 564–569. [Google Scholar] [CrossRef]
- Wildeman, M.; van Ophuizen, E.; den Dunnen, J.T.; Taschner, P.E.M. Improving sequence variant descriptions in mutation databases and literature using the Mutalyzer sequence variation nomenclature checker. Hum. Mutat. 2008, 29, 6–13. [Google Scholar] [CrossRef]
- Ioannidis, N.M.; Rothstein, J.H.; Pejaver, V.; Middha, S.; McDonnell, S.K.; Baheti, S.; Musolf, A.; Li, Q.; Holzinger, E.; Karyadi, D.; et al. REVEL: An Ensemble Method for Predicting the Pathogenicity of Rare Missense Variants. Am. J. Hum. Genet. 2016, 99, 877–885. [Google Scholar] [CrossRef]
- Rentzsch, P.; Witten, D.; Cooper, G.M.; Shendure, J.; Kircher, M. CADD: Predicting the deleteriousness of variants throughout the human genome. Nucleic Acids Res. 2019, 47, D886–D894. [Google Scholar] [CrossRef]
- Desmet, F.-O.; Hamroun, D.; Lalande, M.; Collod-Béroud, G.; Claustres, M.; Béroud, C. Human Splicing Finder: An online bioinformatics tool to predict splicing signals. Nucleic Acids Res. 2009, 37, e67. [Google Scholar] [CrossRef] [PubMed]
- Yeo, G.; Burge, C.B. Maximum entropy modeling of short sequence motifs with applications to RNA splicing signals. Burge. J. Comput. Biol. 2004, 11, 377–394. [Google Scholar]
- Abou Tayoun, A.N.; Pesaran, T.; DiStefano, M.T.; Oza, A.; Rehm, H.L.; Biesecker, L.G.; Harrison, S.M.; ClinGen Sequence Variant Interpretation Working Group (ClinGen SVI). Recommendations for interpreting the loss of function PVS1 ACMG/AMP variant criterion. Hum. Mutat. 2018, 39, 1517–1524. [Google Scholar] [CrossRef]
- Shamsani, J.; Kazakoff, S.H.; Armean, I.M.; McLaren, W.; Parsons, M.T.; Thompson, B.A.; O’Mara, T.A.; Hunt, S.E.; Waddell, N.; Spurdle, A.B. A plugin for the Ensembl Variant Effect Predictor that uses MaxEntScan to predict variant spliceogenicity. Bioinformatics 2019, 35, 2315–2317. [Google Scholar] [CrossRef] [PubMed]
- Karczewski, K.J.; Francioli, L.C.; Tiao, G.; Cummings, B.B.; Alföldi, J.; Wang, Q.; Collins, R.L.; Laricchia, K.M.; Ganna, A.; Birnbaum, D.P.; et al. The mutational constraint spectrum quantified from variation in 141,456 humans. Nature 2020, 581, 434–443. [Google Scholar] [CrossRef] [PubMed]
- Sherry, S.T. dbSNP: The NCBI database of genetic variation. Nucleic Acids Res. 2001, 29, 308–311. [Google Scholar] [CrossRef]
- Exome Variant Server, NHLBI GO Exome Sequencing Project (ESP), Seattle, WA. Available online: http://evs.gs.washington.edu/EVS/ (accessed on 16 October 2020).
- Landrum, M.J.; Lee, J.M.; Benson, M.; Brown, G.R.; Chao, C.; Chitipiralla, S.; Gu, B.; Hart, J.; Hoffman, D.; Jang, W.; et al. ClinVar: Improving access to variant interpretations and supporting evidence. Nucleic Acids Res. 2018, 46, D1062–D1067. [Google Scholar] [CrossRef]
- Fokkema, I.F.A.C.; Ivo, F.A.; Taschner, P.E.M.; Schaafsma, G.C.P.; Celli, J.; Laros, J.F.J.; den Dunnen, J.T. LOVD v.2.0: The next generation in gene variant databases. Hum. Mutat. 2011, 32, 557–563. [Google Scholar] [CrossRef]
- MacDonald, J.R.; Ziman, R.; Yuen, R.K.C.; Feuk, L.; Scherer, S.W. The Database of Genomic Variants: A curated collection of structural variation in the human genome. Nucleic Acids Res. 2014, 42, D986–D992. [Google Scholar] [CrossRef]
- EMBL-EBI Database of Genomic Variants Archive. Available online: https://www.ebi.ac.uk/dgva/ (accessed on 16 October 2020).
- Frequency Filter. Available online: https://www.cardiodb.org/allelefrequencyapp/ (accessed on 16 October 2020).
- Kopanos, C.; Tsiolkas, V.; Kouris, A.; Chapple, C.E.; Aguilera, M.A.; Meyer, R.; Massouras, A. VarSome: The human genomic variant search engine. Bioinformatics 2019, 35, 1978–1980. [Google Scholar] [CrossRef]
- 6601 Free Vector Icons of Webpage. Available online: https://www.flaticon.com/free-icons/webpage (accessed on 16 October 2020).
- Denoyelle, F.; Marlin, S.; Weil, D.; Moatti, L.; Chauvin, P.; Garabédian, E.N.; Petit, C. Clinical features of the prevalent form of childhood deafness, DFNB1, due to a connexin-26 gene defect: Implications for genetic counselling. Lancet 1999, 353, 1298–1303. [Google Scholar] [CrossRef]
- Gasmelseed, N.M.A.; Schmidt, M.; Magzoub, M.M.A.; Macharia, M.; Elmustafa, O.M.; Ototo, B.; Winkler, E.; Ruge, G.; Horstmann, R.D.; Meyer, C.G. Low frequency of deafness-associated GJB2 variants in Kenya and Sudan and novel GJB2 variants. Hum. Mutat. 2004, 23, 206–207. [Google Scholar] [CrossRef] [PubMed]
- Sansović, I.; Knezević, J.; Musani, V.; Seeman, P.; Barisić, I.; Pavelić, J. GJB2 mutations in patients with nonsyndromic hearing loss from Croatia. Genet. Test. Mol. Biomark. 2009, 13, 693–699. [Google Scholar] [CrossRef]
- Kenna, M.A.; Wu, B.L.; Cotanche, D.A.; Korf, B.R.; Rehm, H.L. Connexin 26 studies in patients with sensorineural hearing loss. Arch. Otolaryngol. Head Neck Surg. 2001, 127, 1037–1042. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Matos, T.D.; Simões-Teixeira, H.; Caria, H.; Gonçalves, A.C.; Chora, J.; Correia, M.D.C.; Moura, C.; Rosa, H.; Monteiro, L.; O’Neill, A.; et al. Spectrum and frequency of GJB2 mutations in a cohort of 264 Portuguese nonsyndromic sensorineural hearing loss patients. Int. J. Audiol. 2013, 52, 466–471. [Google Scholar] [CrossRef][Green Version]
- Pandya, A.; Arnos, K.S.; Xia, X.J.; Welch, K.O.; Blanton, S.H.; Friedman, T.B.; Garcia Sanchez, G.; Liu MD, X.Z.; Morell, R.; Nance, W.E. Frequency and distribution of GJB2 (connexin 26) and GJB6 (connexin 30) mutations in a large North American repository of deaf probands. Genet. Med. 2003, 5, 295–303. [Google Scholar] [CrossRef] [PubMed]
- Rabionet, R.; Zelante, L.; López-Bigas, N.; D’Agruma, L.; Melchionda, S.; Restagno, G.; Arbonés, M.L.; Gasparini, P.; Estivill, X. Molecular basis of childhood deafness resulting from mutations in the GJB2 (connexin 26) gene. Hum. Genet. 2000, 106, 40–44. [Google Scholar]
- Zelante, L.; Gasparini, P.; Estivill, X.; Melchionda, S.; D’Agruma, L.; Govea, N.; Milá, M.; Monica, M.D.; Lutfi, J.; Shohat, M.; et al. Connexin26 mutations associated with the most common form of non-syndromic neurosensory autosomal recessive deafness (DFNB1) in Mediterraneans. Hum. Mol. Genet. 1997, 6, 1605–1609. [Google Scholar] [CrossRef]
- Löffler, J.; Nekahm, D.; Hirst-Stadlmann, A.; Günther, B.; Menzel, H.J.; Utermann, G.; Janecke, A.R. Sensorineural hearing loss and the incidence of Cx26 mutations in Austria. Eur. J. Hum. Genet. 2001, 9, 226–230. [Google Scholar] [CrossRef]
- Kelley, P.M.; Harris, D.J.; Comer, B.C.; Askew, J.W.; Fowler, T.; Smith, S.D.; Kimberling, W.J. Novel mutations in the connexin 26 gene (GJB2) that cause autosomal recessive (DFNB1) hearing loss. Am. J. Hum. Genet. 1998, 62, 792–799. [Google Scholar] [CrossRef]
- Kelsell, D.P.; Dunlop, J.; Stevens, H.P.; Lench, N.J.; Liang, J.N.; Parry, G.; Mueller, R.F.; Leigh, I.M. Connexin 26 mutations in hereditary non-syndromic sensorineural deafness. Nature 1997, 387, 80–83. [Google Scholar] [CrossRef]
- Denoyelle, F.; Weil, D.; Maw, M.A.; Wilcox, S.A.; Lench, N.J.; Allen-Powell, D.R.; Osborn, A.H.; Dahl, H.H.; Middleton, A.; Houseman, M.J.; et al. Prelingual deafness: High prevalence of a 30delG mutation in the connexin 26 gene. Hum. Mol. Genet. 1997, 6, 2173–2177. [Google Scholar] [CrossRef]
- Richard, G.; White, T.W.; Smith, L.E.; Bailey, R.A.; Compton, J.G.; Paul, D.L.; Bale, S.J. Functional defects of Cx26 resulting from a heterozygous missense mutation in a family with dominant deaf-mutism and palmoplantar keratoderma. Hum. Genet. 1998, 103, 393–399. [Google Scholar] [CrossRef] [PubMed]
- Uyguner, O.; Tukel, T.; Baykal, C.; Eris, H.; Emiroglu, M.; Hafiz, G.; Ghanbari, A.; Baserer, N.; Yuksel-Apak, M.; Wollnik, B. The novel R75Q mutation in the GJB2 gene causes autosomal dominant hearing loss and palmoplantar keratoderma in a Turkish family. Clin. Genet. 2002, 62, 306–309. [Google Scholar] [CrossRef] [PubMed]
- Carrasquillo, M.M.; Zlotogora, J.; Barges, S.; Chakravarti, A. Two different connexin 26 mutations in an inbred kindred segregating non-syndromic recessive deafness: Implications for genetic studies in isolated populations. Hum. Mol. Genet. 1997, 6, 2163–2172. [Google Scholar] [CrossRef][Green Version]
- Putcha, G.V.; Bejjani, B.A.; Bleoo, S.; Booker, J.K.; Carey, J.C.; Carson, N.; Das, S.; Dempsey, M.A.; Gastier-Foster, J.M.; Greinwald, J.H., Jr.; et al. A multicenter study of the frequency and distribution of GJB2 and GJB6 mutations in a large North American cohort. Genet. Med. 2007, 9, 413–426. [Google Scholar] [CrossRef]
- Kupka, S.; Braun, S.; Aberle, S.; Haack, B.; Ebauer, M.; Zeissler, U.; Zenner, H.-P.; Blin, N.; Pfister, M. Frequencies of GJB2 mutations in German control individuals and patients showing sporadic non-syndromic hearing impairment. Hum. Mutat. 2002, 20, 77–78. [Google Scholar] [CrossRef] [PubMed]
- Scott, D.A.; Kraft, M.L.; Carmi, R.; Ramesh, A.; Elbedour, K.; Yairi, Y.; Srisailapathy, C.R.; Rosengren, S.S.; Markham, A.F.; Mueller, R.F.; et al. Identification of mutations in the connexin 26 gene that cause autosomal recessive nonsyndromic hearing loss. Hum. Mutat. 1998, 11, 387–394. [Google Scholar] [CrossRef]
- Dalamón, V.; Lotersztein, V.; Lipovsek, M.; Bèherán, A.; Mondino, M.E.; Diamante, F.; Pallares, N.; Diamante, V.; Elgoyhen, A.B. Performance of speech perception after cochlear implantation in DFNB1 patients. Acta Otolaryngol. 2009, 129, 395–398. [Google Scholar] [CrossRef]
- Estivill, X.; Fortina, P.; Surrey, S.; Rabionet, R.; Melchionda, S.; D’Agruma, L.; Mansfield, E.; Rappaport, E.; Govea, N.; Milà, M.; et al. Connexin-26 mutations in sporadic and inherited sensorineural deafness. Lancet 1998, 351, 394–398. [Google Scholar] [CrossRef]
- Brobby, G.W.; Müller-Myhsok, B.; Horstmann, R.D. Connexin 26 R143W mutation associated with recessive nonsyndromic sensorineural deafness in Africa. N. Engl. J. Med. 1998, 338, 548–550. [Google Scholar] [CrossRef]
- Frei, K.; Lucas, T.; Ramsebner, R.; Schöfer, C.; Baumgartner, W.-D.; Weipoltshammer, K.; Erginel-Unaltuna, N.; Wachtler, F.J.; Kirschhofer, K. A novel connexin 26 mutation associated with autosomal recessive sensorineural deafness. Audiol. Neurootol. 2004, 9, 47–50. [Google Scholar] [CrossRef] [PubMed]
- Marlin, S.; Garabédian, E.N.; Roger, G.; Moatti, L.; Matha, N.; Lewin, P.; Petit, C.; Denoyelle, F. Connexin 26 gene mutations in congenitally deaf children: Pitfalls for genetic counseling. Arch. Otolaryngol. Head Neck Surg. 2001, 127, 927–933. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Matos, T.D.; Caria, H.; Simões-Teixeira, H.; Aasen, T.; Dias, O.; Andrea, M.; Kelsell, D.P.; Fialho, G. A novel M163L mutation in connexin 26 causing cell death and associated with autosomal dominant hearing loss. Hear. Res. 2008, 240, 87–92. [Google Scholar] [CrossRef] [PubMed]
- Samanich, J.; Lowes, C.; Burk, R.; Shanske, S.; Lu, J.; Shanske, A.; Morrow, B.E. Mutations in GJB2, GJB6, and mitochondrial DNA are rare in African American and Caribbean Hispanic individuals with hearing impairment. Am. J. Med. Genet. A 2007, 143A, 830–838. [Google Scholar] [CrossRef]
- Frei, K.; Ramsebner, R.; Lucas, T.; Hamader, G.; Szuhai, K.; Weipoltshammer, K.; Baumgartner, W.-D.; Wachtler, F.J.; Kirschhofer, K. GJB2 mutations in hearing impairment: Identification of a broad clinical spectrum for improved genetic counseling. Laryngoscope 2005, 115, 461–465. [Google Scholar] [CrossRef]
- Oliveira, C.A.; Maciel-Guerra, A.T.; Sartorato, E.L. Deafness resulting from mutations in the GJB2 (connexin 26) gene in Brazilian patients. Clin. Genet. 2002, 61, 354–358. [Google Scholar] [CrossRef] [PubMed]
- Morell, R.J.; Kim, H.J.; Hood, L.J.; Goforth, L.; Friderici, K.; Fisher, R.; Van Camp, G.; Berlin, C.I.; Oddoux, C.; Ostrer, H.; et al. Mutations in the Connexin 26 Gene (GJB2) among Ashkenazi Jews with Nonsyndromic Recessive Deafness. N. Engl. J. Med. 1998, 339, 1500–1505. [Google Scholar] [CrossRef]
- Dalamon, V.; Fiori, M.C.; Figueroa, V.A.; Oliva, C.A.; del Rio, R.; Gonzalez, W.; Canan, J.; Elgoyhen, A.B.; Altenberg, G.A.; Retamal, M.A. Gap-junctional channel and hemichannel activity of two recently identified connexin 26 mutants associated with deafness. Pflügers Arch. Eur. J. Physiol. 2016, 468, 909–918. [Google Scholar] [CrossRef]
- Primignani, P.; Trotta, L.; Castorina, P.; Lalatta, F.; Sironi, F.; Radaelli, C.; Degiorgio, D.; Curcio, C.; Travi, M.; Ambrosetti, U.; et al. Analysis of the GJB2 and GJB6 Genes in Italian Patients with Nonsyndromic Hearing Loss: Frequencies, Novel Mutations, Genotypes, and Degree of Hearing Loss. Genet. Test. Mol. Biomark. 2009, 13, 209–217. [Google Scholar] [CrossRef]
- Kudo, T. Transgenic expression of a dominant-negative connexin26 causes degeneration of the organ of Corti and non-syndromic deafness. Hum. Mol. Genet. 2003, 12, 995–1004. [Google Scholar] [CrossRef] [PubMed]
- Marziano, N.K. Mutations in the gene for connexin 26 (GJB2) that cause hearing loss have a dominant negative effect on connexin 30. Hum. Mol. Genet. 2003, 12, 805–812. [Google Scholar] [CrossRef]
- Piazza, V.; Beltramello, M.; Menniti, M.; Colao, E.; Malatesta, P.; Argento, R.; Chiarella, G.; Gallo, L.V.; Catalano, M.; Perrotti, N.; et al. Functional analysis of R75Q mutation in the gene coding for Connexin 26 identified in a family with nonsyndromic hearing loss. Clin. Genet. 2005, 68, 161–166. [Google Scholar] [CrossRef] [PubMed]
- Neocleous, V.; Aspris, A.; Shahpenterian, V.; Nicolaou, V.; Panagi, C.; Ioannou, I.; Kyamides, Y.; Anastasiadou, V.; Phylactou, L.A. High Frequency of 35delG GJB2 Mutation and Absence of del(GJB6-D13S1830) in Greek Cypriot Patients with Nonsyndromic Hearing Loss. Genet. Test. 2006, 10, 285–289. [Google Scholar] [CrossRef] [PubMed]
- Günther, B.; Steiner, A.; Nekahm-Heis, D.; Albegger, K.; Zorowka, P.; Utermann, G.; Janecke, A. The 342-kb deletion inGJB6is not present in patients with non-syndromic hearing loss from Austria. Hum. Mutat. 2003, 22, 180. [Google Scholar] [CrossRef]
- Uyguner, O.; Emiroglu, M.; Uzumcu, A.; Hafiz, G.; Ghanbari, A.; Baserer, N.; Yuksel-Apak, M.; Wollnik, B. Frequencies of gap- and tight-junction mutations in Turkish families with autosomal-recessive non-syndromic hearing loss. Clin. Genet. 2003, 64, 65–69. [Google Scholar] [CrossRef]
- Pandey, K.R.; Maden, N.; Poudel, B.; Pradhananga, S.; Sharma, A.K. The Curation of Genetic Variants: Difficulties and Possible Solutions. Genom. Proteom. Bioinform. 2012, 10, 317–325. [Google Scholar] [CrossRef]
- Fokkema, I.F.A.C.; Ivo, F.A.; Velde, K.J.; Slofstra, M.K.; Ruivenkamp, C.A.L.; Vogel, M.J.; Pfundt, R.; Blok, M.J.; Lekanne Deprez, R.H.; Waisfisz, Q.; et al. Dutch genome diagnostic laboratories accelerated and improved variant interpretation and increased accuracy by sharing data. Hum. Mutat. 2019, 40, 2230–2238. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Nucleotide Change | Protein Variant | Mutated Alleles/ Total Alleles Tested (Percentage) | Reference | ClinVar | Final Classification (ACMG/AMP HL-EP) | Rules Applied |
|---|---|---|---|---|---|---|
| c.-23+1G>A | - | 2/1200 (0.16%) | [68] | Pathogenic | Pathogenic | PM2, PVS1, PM3_VS, PS3_P |
| c.-22-12C>T | - | 1/1200 (0.083%) | [69] | Benign | Benign | BA1, BP4 |
| c.-22-2A>C | - | 1/1200 (0.083%) | [70] | VUS * | Likely Pathogenic | PM3_M, PP1_S, PS3_P, BS1. |
| c.-15C>T | - | 1/1200 (0.083%) | [69] | Benign/ Likely Benign | Benign | BA1, BP4 |
| c.23C>T | p.Thr8Met | 1/1200 (0.083%) | [71] | Conflicting Interpretation | VUS | PM2_P, PS3_P, PM3 |
| c.24G>A | p.(Thr8=) | 1/1200 (0.083%) | [72] | Conflicting Interpretation | VUS | PP3, PM2 |
| c.29T>C | p.Leu10Pro | 1/1200 (0.083%) | [73] | absent | VUS | PM2, PP3, PS3_M |
| c.35G>T | p.Gly12Val | 2/1200 (0.16%) | [74] | Pathogenic/ Likely Pathogenic | Pathogenic | PM2_M, PM3_VS, PP3, PS3_M |
| c.35delG | p.Gly12Valfs*2 | 111/1200 (9.25%) | [75] | Pathogenic * | Pathogenic | already curated by HL-EP |
| c.56G>C | p.Ser19Thr | 1/1200 (0.083%) | [74] | Likely Pathogenic | Pathogenic | PM2, PM3_VS, PP1_M, PS3_M |
| c.59T>C | p.Ile20Thr | 4/1200 (0.34%) | [76] | Pathogenic/ Likely Pathogenic | Pathogenic | PM2, PM3, PP1_P, PP3, PS3_M |
| c.79G>A | p.Val27Ile | 100/1200 (8.34%) | [77] | Benign | Benign | BA1, BP2, BS3_P |
| c.101T>C | p.Met34Thr | 19/1200 (1.58%) | [78] | Pathogenic * | Pathogenic | already curated by HL-EP |
| c.109G>A | p.Val37Ile | 10/1200 (0.84%) | [77] | Pathogenic * | Pathogenic | already curated by HL-EP |
| c.139G>T | p.(Glu47*) | 6/1200 (0.5%) | [79] | Pathogenic | Pathogenic | PVS1, PM2_P, PM3_VS |
| c.167delT | p.Leu56Argfs*81 | 14/1200 (1.16%) | [75] | Pathogenic * | Pathogenic | already curated by HL-EP |
| c.223C>T | p.Arg75Trp | 1/1200 (0.083%) | [80] | Pathogenic | Pathogenic | PM2, PS2_VS, PP1_P, PP3, PS3 |
| c.224G>A | p.Arg75Gln | 1/1200 (0.083%) | [81] | Pathogenic | Pathogenic | PM2, PS4_M, PP1_M, PM5, PP3, PS3_M |
| c.229T>C | p.Trp77Arg | 1/1200 (0.083%) | [82] | Pathogenic | Pathogenic | PM2_P, PM3_VS, PP3, PS3_M |
| c.232dupG | p.(Ala78Glyfs*24) | 1/1200 (0.083%) | [83] | Likely Pathogenic | Pathogenic | PM2, PVS1, PM3 |
| c.232G>T | p.(Ala78Ser) | 1/1200 (0.083%) | [28] | absent | VUS | PM2, PP3, PM5 |
| c.246C>G | p.Ile82Met | 1/1200 (0.083%) | [84] | Likely Pathogenic | Pathogenic | PM2, PP1_M, PM3_VS, PP3, PS3_M |
| c.249C>G | p.Phe83Leu | 2/1200 (0.16%) | [85] | Benign/ Likely Benign | Likely Benign | BS1_P, BP2, BS3_P |
| c.250G>C | p.Val84Leu | 1/1200 (0.083%) | [77] | Pathogenic/ Likely Pathogenic | Pathogenic | PM2, PP3, PM3_VS, PP1_P |
| c.269T>C | p.Leu90Pro | 3/1200 (0.25%) | [68] | Conflicting Interpretation | Pathogenic | BS1_P, PP3, PM3_VS, PS3_M |
| c.269dup | p.(Val91Serfs*11) | 1/1200 (0.083%) | [68] | Pathogenic | Pathogenic | PM2, PVS1, PM3_VS, PP1_M |
| c.283G>A | p.(Val95Met) | 2/1200 (0.16%) | [77] | Pathogenic/ Likely Pathogenic | Pathogenic | PM2, PM3_VS, PP1_P, PP3 |
| c.313_326del14 | p.(Lys105Glyfs*5) | 2/1200 (0.16%) | [79] | Pathogenic | Pathogenic | PM2_P, PVS1, PM3_VS. |
| c.326G>T | p.Gly109Val | 1/1200 (0.083%) | [86] | absent | Likely Pathogenic | PM2, PM3, PS3_M |
| c.334_335delAA | p.(Lys112Glufs*2) | 2/1200 (0.16%) | [77] | Pathogenic /Likely Pathogenic | Pathogenic | PM2, PVS1, PM3_VS, PP1_M |
| c.358_360delGAG | p.Glu120del | 2/1200 (0.16%) | [79] | Pathogenic | Pathogenic | PM2_P, PM4, PM3_VS, PS3_M |
| c.380G>A | p.Arg127His | 1/1200 (0.083%) | [87] | Conflicting Interpretation | Benign | BA1, BS2, BS4, PM3_P |
| c.384C>T | p.(Ile128=) | 1/1200 (0.083%) | [75] | Likely Benign | Likely Benign | PM2, BP2, BP4, BP7 |
| c.385G>A | p.(Glu129Lys) | 1/1200 (0.083%) | [71] | Conflicting Interpretation | VUS | PM2, PM3 |
| c.427C>T | p.Arg143Trp | 5/1200 (0.42%) | [88] | Pathogenic | Pathogenic | PM2_P, PM3_VS, PP1_P |
| c.439G>A | p.(Glu147Lys) | 1/1200 (0.083%) | [89] | Pathogenic/ Likely Pathogenic | Pathogenic | PM2, PM3_VS, PP1_Mod, PP3 |
| c.457G>A | p.Val153Ile | 3/1200 (0.25%) | [90] | Benign/ Likely Benign | Benign | BA1, BS2 |
| c.478G>A | p.(Gly160Ser) | 2/1200 (0.16%) | [85] | Conflicting Interpretation | Likely Benign | BS1_P, PP3, BP2, PM3 |
| c.487A>G | p.Met163Val | 2/1200 (0.16%) | [90] | VUS | VUS | PM2_P, PP3, PS3_M, BS2 |
| c.487A>C | p.Met163Leu | 1/1200 (0.083%) | [91] | Pathogenic | VUS | PM2, PS3_P |
| c.503A>G | p.(Lys168Arg) | 5/1200 (0.42%) | [92] | Conflicting Interpretation | VUS | PM2_P, PP3 |
| c.551G>C | p.Arg184Pro | 4/1200 (0.34%) | [79] | Conflicting Interpretation | Pathogenic | PM2, PM3_VS, PP3, PS3_M |
| c.569T>A | p.(Val190Asp) | 1/1200 (0.083%) | [28] | absent | VUS | PM2, PM3, PP3 |
| c.617A>G | p.Asn206Ser | 4/1200 (0.34%) | [90] | Pathogenic | Pathogenic | PM2_P, PP3, PP1_M, PM3_VS, PS3_M |
| c.632G>A | p.(Cys211Tyr) | 1/1200 (0.083%) | [28] | absent | Likely Pathogenic | PM2, PM3,PP3, PP1_P |
| c.*1C>T (3′UTR) | - | 2/1200 (0.16%) | [93] | Conflicting Interpretation | Likely Benign | BS1_P, BP4 |
| del(GJB6-D13S1830) | - | 12/1200 (1%) | [10] | Pathogenic | Pathogenic | PS3, PS4, PM2_P, PM3_VS |
| del(GJB6-D13S1854) | - | 5/1200 (0.42%) | [21] | Pathogenic | Pathogenic | PM2, PS3, PS4, PM3_VS |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Buonfiglio, P.; Bruque, C.D.; Luce, L.; Giliberto, F.; Lotersztein, V.; Menazzi, S.; Paoli, B.; Elgoyhen, A.B.; Dalamón, V. GJB2 and GJB6 Genetic Variant Curation in an Argentinean Non-Syndromic Hearing-Impaired Cohort. Genes 2020, 11, 1233. https://doi.org/10.3390/genes11101233
Buonfiglio P, Bruque CD, Luce L, Giliberto F, Lotersztein V, Menazzi S, Paoli B, Elgoyhen AB, Dalamón V. GJB2 and GJB6 Genetic Variant Curation in an Argentinean Non-Syndromic Hearing-Impaired Cohort. Genes. 2020; 11(10):1233. https://doi.org/10.3390/genes11101233
Chicago/Turabian StyleBuonfiglio, Paula, Carlos D. Bruque, Leonela Luce, Florencia Giliberto, Vanesa Lotersztein, Sebastián Menazzi, Bibiana Paoli, Ana Belén Elgoyhen, and Viviana Dalamón. 2020. "GJB2 and GJB6 Genetic Variant Curation in an Argentinean Non-Syndromic Hearing-Impaired Cohort" Genes 11, no. 10: 1233. https://doi.org/10.3390/genes11101233
APA StyleBuonfiglio, P., Bruque, C. D., Luce, L., Giliberto, F., Lotersztein, V., Menazzi, S., Paoli, B., Elgoyhen, A. B., & Dalamón, V. (2020). GJB2 and GJB6 Genetic Variant Curation in an Argentinean Non-Syndromic Hearing-Impaired Cohort. Genes, 11(10), 1233. https://doi.org/10.3390/genes11101233